

Abstract
Myelofibrosis (either primary or post-polycythemia vera/essential thrombocythemia) is a chronic and debilitating myeloproliferative neoplasm for which there is no well-accepted standard of care. Clinical manifestations of this disease (eg, cytopenias, splenomegaly, bone marrow fibrosis) and constitutional symptoms (eg, hypercatabolic state, fatigue, night sweats, fever) create significant treatment challenges. For example, progressive splenomegaly increases the risk for more serious clinical sequelae (eg, portal hypertension, splenic infarction). Myelofibrosis arises from hematopoietic stem cells or early progenitor cells. However, the molecular mechanisms underlying its pathogenesis and clinical presentation are poorly understood, delaying the development of effective and targeted treatments. Recent studies have implicated mutations that directly or indirectly lead to deregulated activation of Janus-activated kinase 2 (JAK2). Appreciation for the activation of JAK2 and the importance of increased levels of circulating proinflammatory cytokines in the pathogenesis and clinical manifestations of myelofibrosis has led to novel therapeutic agents targeting JAKs. This review will briefly discuss the origins of the JAK2 hypothesis, the clinical relevance of JAK2 mutations in myelofibrosis, and recent clinical progress in targeting JAKs as a therapeutic intervention for patients with this chronic and debilitating disease.
Keywords: JAK2V617F, myelofibrosis, myeloproliferative neoplasms, JAK2 inhibitors
Introduction
Philadelphia chromosome–negative myeloproliferative neoplasms (Ph-negative MPNs) include polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF) (1). In addition, myelofibrosis (MF) can develop secondarily in patients with PV and ET (post-PV or -ET MF). These chronic disorders are thought to arise from hematopoietic stem cells or early progenitor cells (2). The recent identification of somatic mutations in the Janus-activated kinase 2 gene (JAK2), as well as in the thrombopoietin receptor that result in exaggerated JAK2 signaling, has increased our understanding of the pathogenesis of Ph-negative MPNs. Although JAK2 signaling appears to be important in each of these 3 neoplasms, they are distinct clinical conditions with different treatment approaches and different expectations regarding potential outcomes. For example, MF is associated with short median survival (approximately 5–7 y) compared with PV and ET (≥20 y) (3–7). Information on the diagnosis, clinical manifestations, and treatment of PV and ET have been reviewed elsewhere (8–11). Although much of our discussion will focus on PMF, patients with post–ET or post–PV MF present with many of the same symptoms and treatment challenges observed in PMF, including constitutional symptoms and splenomegaly as well as similarly shortened survival (12). The diagnosis of PMF requires a finding of increased megakaryocyte proliferation and abnormal megakaryocyte morphology, typically with bone marrow fibrosis and the presence of the JAK2V617F mutation or other clonal marker (eg, MPLW515L>K); in the absence of such a marker, there should be no evidence of an inflammatory state (Table 1) (13). The diagnosis is supported by a finding of anemia of an otherwise unexplained cause, an abnormal blood film, constitutional symptoms and/or evidence of splenomegaly (1, 13). Progressive worsening of symptoms may occur over time, resulting in increased patient morbidity and mortality (13). Multivariate analysis of clinical parameters documented at the time of diagnosis has identified risk factors (age >65 y, presence of constitutional symptoms, hemoglobin level <10 g/dL, leukocyte count >25×109/L, and circulating blasts ≥1%) for decreased survival in PMF (7). Patients with 0 factors are classified as low risk, 1 factor as intermediate-1 risk, 2 factors as intermediate-2 risk, and ≥3 factors as high risk.
Table 1.
Myelofibrosis Diagnostic Criteria
| Criteria for Primary Myelofibrosis* | |
|---|---|
| Major Criteria | |
| 1 | Megakaryocyte proliferation and atypical morphology,† typically accompanied by bone marrow fibrosis (ie, increased collagen or reticulin staining) |
| 2 | Does not meet World Health Organization criteria for polycythemia vera,‡ chronic myelogenous leukemia,§ myelodysplastic syndrome,|| or other myeloid neoplasm |
| 3 | Presence of JAK2 V617F or other clonal marker (eg, MPL W515L>K)¶ |
| Minor Criteria | |
| 1 | Leukoerythroblastosis# |
| 2 | Serum level of lactate dehydrogenase increased# |
| 3 | Anemia# |
| 4 | Palpable splenomegaly# |
JAK=Janus-activated kinase.
Revised World Health Organization criteria for primary myelofibrosis modified from Tefferi A, Thiele J, Orazi A, et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood 2007;110:1092–7.
Dense clustered megakaryocytes with abnormally increased nuclear/cytoplasmic ratio and hyperchromatic and round or irregularly folded nuclei. In the absence of overt reticulin fibrosis, increased megakaryocyte proliferation must be accompanied by granulocyte proliferation, with or without erythropoiesis.
Diagnosis requires a finding of increased hemoglobin level that is corrected by iron replacement therapy in the presence of decreased serum ferritin.
Diagnosis requires the absence of BCR-ABL.
Absence of dyserythropoiesis and dysgranulocytosis is required.
In the absence of clonal marker, requires that bone marrow fibrosis not be secondary to infection, inflammation, exposure to toxic agents, or autoimmune response, or the presence of hematologic (eg, lymphoma, hairy cell leukemia) or metastatic malignancy.
May present as borderline to marked abnormality.
Constitutional symptoms commonly reported include fatigue (84% of patients; n=456), night sweats (56%), pruritus (50%), bone pain (47%), undesired weight loss (20%), fever (18%), and spleen pain (7%) (14). Constitutional symptoms in general and weight loss specifically may carry adverse prognostic value for survival in patients with MF (4, 15, 16). Fatigue occurs in most patients and the severity of fatigue increases with the severity of anemia and the presence of constitutional symptoms (ie, pruritus, fever, weight loss) (1). The biological basis of constitutional symptoms in MF is believed to be related to massive splenomegaly and hypermetabolic state caused by excessive production of inflammatory cytokines. Symptoms such as fatigue, weight loss, night sweats, and fevers improve significantly following splenectomy (17, 18). This means that both mechanical decompression of the gastrointestinal tract with splenectomy (allowing for improvement in splenomegaly-related symptoms and weight gain) and tumor debulking (spleen being the major repository of neoplastic cells in the body) result in significant relief from systemic symptoms. In addition, cytokines such as interleukin-6 (IL-6), the circulating levels of which are significantly higher in MF patients than in healthy subjects (19), may play an important role in the symptomatic burden in MF. It is not known, however, whether the inflammatory cytokine milieu is a direct consequence of or a bystander effect of neoplastic clones in MF (18). Apart from causing a multitude of symptoms, splenomegaly can result in sequestration of red blood cells, granulocytes, and platelets, resulting in anemia with wide variations in white cell and platelet counts. A summary of the pathological features of MF is shown in Figure 1. The presence of marked splenomegaly increases the risk of other serious complications, including portal hypertension (a manifestation of increased portal flow and thrombotic obstruction of portal veins) and splenic infarction, both of which may require splenectomy (18), a procedure associated with high rates of perioperative morbidity (eg, hemorrhage, thrombosis, infection) and mortality (approximately 28% and 7%, respectively) (20).
Figure 1.

Pathophysiologic alterations associated with splenomegaly in myelofibrosis. IFN=interferon; IL-interleukin; JAK=Janus-activated kinase; PDGF=platelet-derived growth factor; TGF=transforming growth factor; TNF=tumor necrosis factor.
Current treatment strategies for myelofibrosis include experimental drug therapy, stem cell transplantation, and conventional drugs that were approved for other indications but not for MF (21). Conventional medications are largely palliative and rarely provide durable benefits, while stem cell transplantation is restricted to a small percentage of patients. These limitations underscore the need to develop more effective disease-targeted therapeutic approaches in patients with MF.
Origins of the JAK2 Hypothesis in Myelofibrosis
The first evidence implicating mutated JAK2 in patients with Ph-negative MPNs came from genetic and functional studies of hematopoietic progenitor cells, which frequently contained a mutation (V617F) of somatic origin (22–26). The acquired mutation in JAK2 implicated in the pathogenesis of Ph-negative MPNs is a point mutation in the negative autoregulatory pseudokinase domain (V617F) that lies adjacent to the tyrosine kinase domain (22); this mutation results in constitutive activation of the kinase and increased sensitivity to cytokine signaling (24). It promotes increased cell survival, cell cycle transition, and differentiation in progenitor cells (Figure 2). Additional strong evidence supporting a role of JAK2 in MF comes from a number of independent studies using mouse models. Transgenic mice that constitutively expressed JAK2V617F in hematopoietic progenitor cells exhibited leukocytosis, erythroblastosis, marked thrombocytosis, and hemoglobin levels consistent with anemia when examined at 1 month of age (27). These mice developed marked splenomegaly with the severity of the phenotype correlated with the level of expression of the mutant transgene. Transgenic mice expressing low levels of JAK2V617F (ie, low mutant allele burden) mimicked ET and those with higher mutant allele burden developed PV (28). Aged mice expressing high levels of JAK2V617F eventually developed MF (27, 29). Although JAK2V617F is expressed by more than 50% of patients with ET, and more than 90% of patients with PV, the role of this mutation in the progression of ET or PV to MF in patients is not well understood. In both PV and ET, the risk of developing MF increases with the duration of disease (eg, in patients with ET progression to MF occurs in 4%, 20%, and 29% at 10, 20, and 30 y, respectively), and the risk is greater in patients who harbor mutated compared with wild-type JAK2 (5, 24, 30).
Figure 2.

Mechanism of action of JAK2 inhibitors. bFGF=basic fibroblast growth factor; ERK=extracellular signal-regulated kinase; JAK=Janus-activated kinase; MEK=MAP/ERK kinase; STAT=signal transducers and activators of transcription; VEGF=vascular endothelial growth factor. Adapted with permission from Levine and colleagues (68).
Mutated JAK2V617F is not the only genetic alteration, however, associated with the development of MF. Some MF patients who are JAK2V617F–negative harbor a point mutation (tryptophan to leucine substitution) in the thrombopoietin receptor (ie, MPLW515L) that confers increased sensitivity to thrombopoietin through activation of wild-type JAK2/signal transducers and activators of transcription (STAT) signaling and induces a spectrum of features of MF when expressed in a mouse model (31). Additional mutations have recently been identified in the MPL receptor including S204P, S505N, K39N, and W515K/L, and in exon 12 (H538-K539delinsL) of JAK2 in patients negative for V617F, suggesting that there are multiple paths that may lead to elevated JAK2 signaling and the development of MPN (32, 33). These results suggest that drugs in development that target JAK2 may be beneficial for MF patients with different molecular/genetic background. JAK2 is a member of the Janus-activated kinase family of cytosolic tyrosine kinases that includes also JAK1, JAK3, and tyrosine kinase 2 (TYK2); for an in-depth review, see Yamaoka et al (34). The ligands for these receptors include erythropoietin, thrombopoietin, granulocyte-macrophage colony-stimulating factor, growth hormone, interferons, interleukins (IL) and many other cytokines. JAKs are used singly or in combination by different cytokine receptor systems to activate specific STATs that regulate gene transcription (35). For example, in response to erythropoietin or thrombopoietin receptor binding, JAK2-mediated activation of STAT3 or STAT5 supports the survival, proliferation, and differentiation of erythroid cells (36) or megakaryocyte-erythroid progenitor cells, respectively (37).
Clinical Relevance of JAK2V617F in Myelofibrosis
Acquisition of the gain-of-function JAK2V617F somatic mutation in patients with Ph-negative MPNs is harbored by approximately 50% of patients with ET and MF, and 95% of patients with PV (38). The presence of JAK2V617F may be a risk factor for morbidity and survival in PMF. Univariate analysis showed that patients with JAK2V617F mutation compared with JAK2-wild type had a significantly increased risk of all-cause mortality and splenomegaly, and the majority of patients who required splenectomy were homozygous for the V617F mutation (39). In a more recent analysis, JAK2V617F allele burden (ratio between JAK2V617 DNA and total JAK2 DNA) was significantly and directly correlated with splenomegaly and constitutional symptoms but inversely correlated with overall survival, and the relationship to survival was manifested in a multivariate analysis (40). A third study revealed a significantly shorter overall survival and leukemia-free survival for PMF patients in the lowest-quartile JAK2V617F allele burden group, but such patients were not at increased risk for splenectomy compared with patients in upper quartiles (41).
JAK2 Inhibitors in Clinical Development
Discovery of JAK2 mutations coupled with the known biology of JAK signaling provided a great deal of enthusiasm for development of JAK2 inhibitors for the treatment of Ph-negative MPNs. Drugs currently in clinical development exhibit differential inhibitory activity against JAK family members (Table 2), and some exhibit effects on other receptor kinases (eg, fibroblast growth factor receptor [FGFR], platelet-derived growth factor receptor [PDGFR], Trk, vascular endothelial growth factor receptor [VEGFR], and FLT3) (42–44).
Table 2.
| Inhibitor | Structure | Relative Inhibitory Activity, nM
|
|||
|---|---|---|---|---|---|
| JAK1 | JAK2 | JAK3 | TYK2 | ||
| CEP701 |
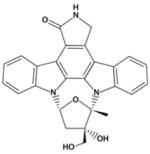
|
NA | 1 | NA | NA |
| INCB018424 |
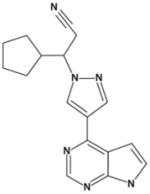
|
3.3 | 2.8 | 428 | 19 |
| TG101348 |
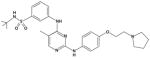
|
105 | 3 | 996 | 405 |
| XL019| | 130 | 2 | 250 | 340 | |
JAK=Janus-activated kinase; NA=not available.
INCB018424
INCB018424 is the first JAK2 inhibitor to enter phase III clinical trials in patients with PMF and post–ET or post–PV MF. It is a potent and selective inhibitor of JAK1 and JAK2 (IC50 of 3.3 and 2.8 nM, respectively), and it demonstrates modest selectivity against TYK2 (~6-fold) and excellent selectivity (≥130-fold) against JAK3 (Table 2) (45). In preclinical studies, INCB018424 inhibited the proliferation of FDCP and BaF/3 cells expressing mutated JAK2 at IC50 values of 100 to 130 nM (46). Selectivity of INCB018424 was further demonstrated by the lack of inhibitory effect in TF-1 cells transformed with BCR-ABL or cell lines expressing activating mutations in c-KIT (45). In a murine model of JAK2V617F–driven malignancy, INCB018424–treated mice exhibited decreased spleen growth and increased survival compared to control mice, outcomes that were accompanied by significant decreases in the circulating levels of IL-6 and tumor necrosis factor-α (TNF-α) (45), proinflammatory cytokines that have been implicated in the pathogenesis of Ph-negative MPNs. Results of a phase I/II clinical study with 153 MF patients show that INCB018424 produces a profound reduction in splenomegaly and improvement of constitutional symptoms regardless of JAK2V617F mutational status (47). Clinical responses have been maintained over the entire duration of treatment, and most patients (115 of 153 patients; 74%) remain on therapy after a median follow-up of 16 months. Thrombocytopenia was identified as the dose-limiting toxicity occurring in approximately 30% of patients at the 25-mg twice daily (BID) dose, which may be related to inhibition of JAK2-dependent thrombopoietin signaling. Treatment resulted in a significant reduction in a number of proinflammatory cytokines (eg, IL-1, TNF-α, and IL-6), and angiogenic and fibrogenic factors, such as VEGF and basic fibroblast growth factor (48). Further optimization of the dosing regimen used baseline platelet count to determine the starting dose (10 or 15 mg BID) and allowed dose titration after 1 and 2 months of therapy; most patients were optimized to 15 or 20 mg BID (47). This approach significantly reduced the incidence of thrombocytopenia (<5%, n=35) while providing equivalent efficacy to higher-dose regimens. With an optimized dosing regimen, spleen volume reduction was evident in as early as 1 month, and durable over 6 months of therapy (33% median decrease after 6 mo, n=23). Based on magnetic resonance imaging, 11 of 23 (48%) of patients achieved a 35% spleen volume reduction (equivalent to International Working Group [IWG] clinical improvement criteria of 50% reduction by palpation) from baseline at month 6. Progression to acute myeloid leukemia [AML] occurred in 3 patients, which is below the expected frequency based on published data (39). Treatment with INCB018424 improved the exercise capacity of MF patients as measured by the 6-minute walk test, with median increase from baseline of 33, 58, and 70 meters after 1, 3, or 6 months of therapy, respectively. Rapid and durable reduction of a total symptom score based on key symptoms (fatigue, abdominal discomfort/pain, bone/muscle pain, night sweats, and pruritus) was noted (51% and 58% of patients achieving 50% reduction in the total score following 1 and 6 mo of treatment, respectively). Improvement in symptoms coincided with a rapid and sustained reduction in proinflammatory cytokines, including IL-1b, IL-1ra, IL-6 and TNF-α, without tachyphylaxis to therapy (47, 49). Although significant clinical improvements have consistently been observed, only modest reductions in the JAK2V617F allele burden have been noted (13% in the marrow and 9% in the peripheral blood) (50). These results suggest that the clinical benefits of INCB018424 treatment may be the result of inhibition of aberrant JAK2 signaling and possibly the inhibition of JAK1 signaling, rather than the elimination of mutant cell clones. Phase III registration studies of INCB018424 recently initiated in the United States and Europe should lead to increased understanding of the therapeutic role for INCB018424 in the clinical management of MF.
CEP-701
CEP-701, also known as lestaurtinib, a staurosporine analog derived from the indolocarbazole K252, is a multikinase inhibitor that targets JAK2 (51) in addition to FLT-3, PDGFR, VEGFR2, and Trk-A with an IC50 in the nM range (Table 2) (42–44, 52, 53). CEP-701 at concentrations of 50 to 100 nM caused nearly complete inhibition of RET autophosphorylation (54). The activity of CEP-701 has been well characterized in preclinical studies. Clinical trials in patients with AML established a recommended CEP-701 oral dose of 80 mg twice daily (55), and this inhibitor is currently being tested in patients with JAK2V617F–positive MPNs (56, 57). In a study of previously treated MF patients (n=22) who were mostly transfusion dependent at study entry, had splenomegaly (median spleen size of 19 cm below costal margin), had a median JAK2V617F allele burden of 53%, and had undergone treatment with CEP-701 for a median duration of 4 months, 6 of 22 (27%) experienced clinical improvement as defined by the IWG for Myelofibrosis Research and Treatment (56, 58). Responses included a reduction in spleen size alone (3 patients), transfusion independence (2 patients), and a reduction in spleen size together with improvement in neutrophil counts and platelets (1 patient). The median time to response was 3 months, and the median duration of response was 14+ months (range, 3–17+). No changes were observed in JAK2V617F allele burden, bone marrow fibrosis, or cytogenetics during therapy. Drug-induced adverse events included anemia (grades 3–4, 14%), thrombocytopenia (grades 3–4, 23%) and diarrhea (all grades, 72%; grades 3–4, 9%).
However, it should be emphasized that results described here with CEP-701 used a liquid formulation because of issues with the solid dosage formulation, A new capsule formulation is currently being tested in a phase I dose-escalation study in MF (59).
XL019
XL019 is a potent and selective inhibitor of JAK2 kinase (IC50 2 nM) that showed a >10-fold selectivity for inhibition of STAT5 phosphorylation following erythropoietin stimulation of erythroid cells (IC50 64 nM) and suppressed STAT phosphorylation by 50% at a dose of 42 mg/kg in a mouse xenograft model.(60) A phase I/II study of XL019 was conducted in patients with MF. Doses of XL019 from 25 to 300 mg (provided daily for 21 days in 28-day cycles) were evaluated in the phase I portion. Therapy caused adverse neurotoxicity in most patients at doses greater than 100 mg (61), and as a result, XL019 doses of 25 to 50 mg once daily or 25 mg every Monday, Wednesday, and Friday were selected for further evaluation. This study enrolled 30 patients, 21 of whom received a dose of ≤50 mg. Treatment resulted in a ≥50% reduction in splenomegaly in 5 of 12 evaluable patients (61). A minority of patients showed improvement in anemia, white blood cell counts, pruritus, and fatigue, and 3 of 4 preleukemic patients showed a reduction in blast counts (circulating and/or bone marrow). Drug-related adverse events associated with XL019 were nonhematologic in nature and included neurotoxicity manifested as formication, peripheral neuropathy, confusional state, balance disorder, and paresthesia. Although reversible in nature, the occurrence of mild neurotoxicity precluded long-term therapy in most patients, and no additional studies of XL019 are planned.
TG101348
TG101348 was synthesized using structure-based drug design to inhibit JAK2 (IC50 3 nM) and demonstrated 300-fold selectivity against JAK3, and 35- and 135-fold selectivity against JAK1 and TYK2, respectively (Table 2) (62). Studies in HEL cells and BaF/3 cells expressing JAK2V617F showed that TG101348 induced dose-dependent apoptosis and inhibited proliferation with an IC50 of approximately 300 nM for either line (63), consistent with the notion that JAK2 activity was required for both proliferation and survival (64). In a mouse model of JAK2V617F–induced PV, TG101348 decreased hematocrit and spleen size and increased overall survival (63). TG101348 is being evaluated in a phase I/II study in patients with MF (65). Patients were administered escalating doses of TG101348 ranging from 30 to 800 mg daily (n=28), or maximum tolerated dose (MTD) of 680 mg daily (n=31). The most frequent nonhematologic toxicities have been grade 1/2 nausea/vomiting and diarrhea, which were seen in the majority of patients and were either self-limiting or easily treated. Other nonhematologic toxicities included grade 1/2 transaminitis (38%), grade 1/2 serum creatinine elevation (38%), and asymptomatic hyperlipasemia (33%). Hematologic toxicities included grade 3/4 thrombocytopenia and neutropenia (30% and 15%, respectively) and grade 3/4 anemia in non–transfusion-dependent patients (50%) (66). The dose-limiting toxicity at 800 mg was asymptomatic amylasemia and lipasemia, and MTD was established at 680 mg/d. Efficacy data in patients who started at ≥680 mg/d showed that a total of 22 patients (67%) have experienced a greater than 50% decrease in spleen size compared with baseline (median spleen size, 18 cm, range, 6–32 cm), including 9 whose spleen became nonpalpable. In addition, 44% of patients who were JAK2V617F–positive had greater than 50% reduction in JAK2V617F allele burden, and patients with leukocytosis at baseline had a marked reduction in their white blood cell counts (67).
Perspective and Future Clinical Direction for JAK2 Inhibitors in Myelofibrosis
The typical clinical features of MF include marked splenomegaly, progressive anemia, and constitutional symptoms (18). Discoveries including the identification of mutations such as JAK2V617F and high circulating cytokine levels in MF have improved our understanding of this complex disease and allowed the development of targeted therapies. Given that the current clinical management of MF patients is largely palliative and minimally effective, significant improvement in 2 of the 3 most important clinical manifestations of MF seen with JAK2 inhibitors is believed to be a major milestone in the development of new therapies for MF.
Before clinical evaluation of JAK2 inhibitors, and based on imatinib experience in chronic myelogenous leukemia and the fact that the BCR-ABL translocation and JAK2V617F mutation both result in kinase activation, a molecular response rendering JAK2V617F alleles undetectable was expected in response to JAK2 inhibitor therapy. However, despite significant clinical benefits observed with JAK2 inhibitors, significant decreases (>50% of baseline) were noted only in a minority of patients. Although it is possible that none of JAK2 inhibitors evaluated clinically have been tested at doses high enough to suppress JAK2 around-the-clock to be able to achieve deeper molecular responses, there may be additional factors that limit such a therapeutic goal. First, because JAK2V617F mutation is not in the adenosine triphosphate binding pocket of the enzyme, this mutation does not lend itself to allow inhibition of mutant JAK2 without impacting wild-type JAK2. Administration of JAK2 inhibitors at high doses will invariably result in significant suppression of wild-type JAK2, which is essential for normal hematopoiesis. Second, the mutation is in the hematopoietic progenitor cells and hence administration of JAK2 inhibitors at high enough doses to eliminate this clone will likely be met with intolerable myelosuppression. The improvement in the quality of life of MF patients observed with INCB018424, which is JAK1 and JAK2 inhibitor, has not been reported to that extent with other investigational agents to date. It is possible that the level of JAK2 inhibition achieved with tolerable doses of CEP-701 and XL019 was lower than needed to demonstrate symptom improvement. Results from ongoing clinical studies with TG101348 and other JAK2 inhibitors, which may be better tolerated, are needed to further understand potential differences between JAK2-selective inhibitors and inhibitors of both JAK1 and JAK2 with regard to symptomatic benefits.
Nonhematologic adverse event profiles also appear to be different among different JAK inhibitors. Although neuropathy was only reported with XL019, gastrointestinal adverse events such as diarrhea and vomiting were observed with CEP-701 and TG101348. In addition, liver and pancreatic enzyme elevations were reported with TG101348 but not other JAK2 inhibitors. Exact mechanisms of nonpharmacologic adverse events are difficult to rationalize with small-molecule drugs and are typically related to the inherent properties of the chemical scaffolds from which they are derived. Although improvement in anemia was reported only in a handful of patients taking JAK2 inhibitors, lack of consistent beneficial effect is likely due to the fact that JAK2 inhibitors, at the doses currently used, interfere with growth factor signaling through wild-type JAK2, an integral component of bone marrow function. Additional studies are needed to determine whether lower doses or alternative schedules of JAK2 inhibitors, with less impact on erythropoietin and/or thrombopoietin signaling, can improve hematologic parameters while maintaining the clinical benefits noted so far. Alternatively, if bone marrow fibrosis is the primary cause of cytopenias in advanced MF, resolution of bone marrow fibrosis will be required to achieve meaningful improvements in cytopenias.
Longer-term results are required to determine the full potential of JAK2 inhibitors in MF and to determine whether they will have an impact on patients’ survival. Ongoing studies will improve understanding of the pathophysiology of MF and the role of JAK inhibitors in clinical management of this chronic life-threatening disorder.
Statement of Translational Relevance.
Primary myelofibrosis and post-polycythemia vera and post-essential thrombocythemia myelofibrosis are chronic and debilitating myeloproliferative neoplasms for which there is no accepted standard of care. Recent studies have implicated a somatic gain-of-function mutation in the Janus-activated kinase 2 (JAK2), namely JAK2V617F, in the pathophysiology and clinical manifestations of myelofibrosis and spurred the development of molecularly targeted agents (JAK2 inhibitors) as therapy for this condition. This review explores the origins of the JAK2 hypothesis, the central role that alterations in signaling via expression of the JAK2V617F mutation play in clinical manifestations of myelofibrosis, and recent clinical progress in targeting JAKs as a therapeutic intervention for patients with this disease. Available clinical evidence provides a strong rationale for targeted modulation of dysregulated JAK signaling as an effective treatment approach in patients with myelofibrosis.
Acknowledgments
Supported in part by P30 CA016672
References
- 1.Mesa RA. Navigating the evolving paradigms in the diagnosis and treatment of myeloproliferative disorders. Hematology Am Soc Hematol Educ Program. 2007;2007:355–62. doi: 10.1182/asheducation-2007.1.355. [DOI] [PubMed] [Google Scholar]
- 2.Campbell PJ, Green AR. The myeloproliferative disorders. N Engl J Med. 2006;355:2452–66. doi: 10.1056/NEJMra063728. [DOI] [PubMed] [Google Scholar]
- 3.Spivak JL, Barosi G, Tognoni G, et al. Chronic myeloproliferative disorders. Hematology Am Soc Hematol Educ Program. 2003:200–24. doi: 10.1182/asheducation-2003.1.200. [DOI] [PubMed] [Google Scholar]
- 4.Tefferi A, Huang J, Schwager S, et al. Validation and comparison of contemporary prognostic models in primary myelofibrosis: analysis based on 334 patients from a single institution. Cancer. 2007;109:2083–8. doi: 10.1002/cncr.22630. [DOI] [PubMed] [Google Scholar]
- 5.Wolanskyj AP, Schwager SM, McClure RF, Larson DR, Tefferi A. Essential thrombocythemia beyond the first decade: life expectancy, long-term complication rates, and prognostic factors. Mayo Clin Proc. 2006;81:159–66. doi: 10.4065/81.2.159. [DOI] [PubMed] [Google Scholar]
- 6.Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med. 2004;117:755–61. doi: 10.1016/j.amjmed.2004.06.032. [DOI] [PubMed] [Google Scholar]
- 7.Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113:2895–901. doi: 10.1182/blood-2008-07-170449. [DOI] [PubMed] [Google Scholar]
- 8.Michiels JJ, Berneman Z, Van Bockstaele D, van der Planken M, De Raeve H, Schroyens W. Clinical and laboratory features, pathobiology of platelet-mediated thrombosis and bleeding complications, and the molecular etiology of essential thrombocythemia and polycythemia vera: therapeutic implications. Semin Thromb Hemost. 2006;32:174–207. doi: 10.1055/s-2006-939431. [DOI] [PubMed] [Google Scholar]
- 9.Pearson TC, Messinezy M, Westwood N, et al. A Polycythemia Vera Updated: Diagnosis, Pathobiology, and Treatment. Hematology Am Soc Hematol Educ Program. 2000:51–68. doi: 10.1182/asheducation-2000.1.51. [DOI] [PubMed] [Google Scholar]
- 10.Schafer AI. Molecular basis of the diagnosis and treatment of polycythemia vera and essential thrombocythemia. Blood. 2006;107:4214–22. doi: 10.1182/blood-2005-08-3526. [DOI] [PubMed] [Google Scholar]
- 11.Haferlach T, Bacher U, Kern W, Schnittger S, Haferlach C. The diagnosis of BCR/ABL-negative chronic myeloproliferative diseases (CMPD): a comprehensive approach based on morphology, cytogenetics, and molecular markers. Ann Hematol. 2008;87:1–10. doi: 10.1007/s00277-007-0403-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dingli D, Schwager SM, Mesa RA, Li CY, Dewald GW, Tefferi A. Presence of unfavorable cytogenetic abnormalities is the strongest predictor of poor survival in secondary myelofibrosis. Cancer. 2006;106:1985–9. doi: 10.1002/cncr.21868. [DOI] [PubMed] [Google Scholar]
- 13.Tefferi A, Thiele J, Orazi A, et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood. 2007;110:1092–7. doi: 10.1182/blood-2007-04-083501. [DOI] [PubMed] [Google Scholar]
- 14.Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer. 2007;109:68–76. doi: 10.1002/cncr.22365. [DOI] [PubMed] [Google Scholar]
- 15.Dupriez B, Morel P, Demory JL, et al. Prognostic factors in agnogenic myeloid metaplasia: a report on 195 cases with a new scoring system. Blood. 1996;88:1013–8. [PubMed] [Google Scholar]
- 16.Cervantes F, Barosi G, Demory JL, et al. Myelofibrosis with myeloid metaplasia in young individuals: disease characteristics, prognostic factors and identification of risk groups. Br J Haematol. 1998;102:684–90. doi: 10.1046/j.1365-2141.1998.00833.x. [DOI] [PubMed] [Google Scholar]
- 17.Tefferi A, Mesa RA, Nagorney DM, Schroeder G, Silverstein MN. Splenectomy in myelofibrosis with myeloid metaplasia: a single-institution experience with 223 patients. Blood. 2000;95:2226–33. [PubMed] [Google Scholar]
- 18.Tefferi A. Myelofibrosis with myeloid metaplasia. N Engl J Med. 2000;342:1255–65. doi: 10.1056/NEJM200004273421706. [DOI] [PubMed] [Google Scholar]
- 19.Panteli KE, Hatzimichael EC, Bouranta PK, et al. Serum interleukin (IL)-1, IL-2, sIL-2Ra, IL-6 and thrombopoietin levels in patients with chronic myeloproliferative diseases. Br J Haematol. 2005;130:709–15. doi: 10.1111/j.1365-2141.2005.05674.x. [DOI] [PubMed] [Google Scholar]
- 20.Mesa RA, Nagorney DS, Schwager S, Allred J, Tefferi A. Palliative goals, patient selection, and perioperative platelet management: outcomes and lessons from 3 decades of splenectomy for myelofibrosis with myeloid metaplasia at the Mayo Clinic. Cancer. 2006;107:361–70. doi: 10.1002/cncr.22021. [DOI] [PubMed] [Google Scholar]
- 21.Tefferi A. Classification, diagnosis and management of myeloproliferative disorders in the JAK2V617F era. Hematology Am Soc Hematol Educ Program. 2006:240–5. doi: 10.1182/asheducation-2006.1.240. [DOI] [PubMed] [Google Scholar]
- 22.Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–61. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 23.James C. The JAK2V617F Mutation in Polycythemia Vera and Other Myeloproliferative Disorders: One Mutation for Three Diseases? Hematology Am Soc Hematol Educ Program. 2008;2008:69–75. doi: 10.1182/asheducation-2008.1.69. [DOI] [PubMed] [Google Scholar]
- 24.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–90. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 25.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–97. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 26.Zhao R, Xing S, Li Z, et al. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem. 2005;280:22788–92. doi: 10.1074/jbc.C500138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shide K, Shimoda HK, Kumano T, et al. Development of ET, primary myelofibrosis and PV in mice expressing JAK2 V617F. Leukemia. 2008;22:87–95. doi: 10.1038/sj.leu.2405043. [DOI] [PubMed] [Google Scholar]
- 28.Tiedt R, Hao-Shen H, Sobas MA, et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood. 2008;111:3931–40. doi: 10.1182/blood-2007-08-107748. [DOI] [PubMed] [Google Scholar]
- 29.Xing S, Wanting TH, Zhao W, et al. Transgenic expression of JAK2V617F causes myeloproliferative disorders in mice. Blood. 2008;111:5109–17. doi: 10.1182/blood-2007-05-091579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood. 2007;110:840–6. doi: 10.1182/blood-2006-12-064287. [DOI] [PubMed] [Google Scholar]
- 31.Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3:e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams DM, Kim AH, Rogers O, Spivak JL, Moliterno AR. Phenotypic variations and new mutations in JAK2 V617F-negative polycythemia vera, erythrocytosis, and idiopathic myelofibrosis. Exp Hematol. 2007;35:1641–6. doi: 10.1016/j.exphem.2007.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356:459–68. doi: 10.1056/NEJMoa065202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamaoka K, Saharinen P, Pesu M, Holt VE, 3rd, Silvennoinen O, O’Shea JJ. The Janus kinases (Jaks) Genome Biol. 2004;5:253–6. doi: 10.1186/gb-2004-5-12-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–9. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- 36.Smithgall TE, Briggs SD, Schreiner S, Lerner EC, Cheng H, Wilson MB. Control of myeloid differentiation and survival by Stats. Oncogene. 2000;19:2612–8. doi: 10.1038/sj.onc.1203477. [DOI] [PubMed] [Google Scholar]
- 37.Kota J, Caceres N, Constantinescu SN. Aberrant signal transduction pathways in myeloproliferative neoplasms. Leukemia. 2008;22:1828–40. doi: 10.1038/leu.2008.236. [DOI] [PubMed] [Google Scholar]
- 38.Levine RL, Wernig G. Role of JAK-STAT signaling in the pathogenesis of myeloproliferative disorders. Hematology Am Soc Hematol Educ Program. 2006:233–9. 510. doi: 10.1182/asheducation-2006.1.233. [DOI] [PubMed] [Google Scholar]
- 39.Barosi G, Bergamaschi G, Marchetti M, et al. JAK2 V617F mutational status predicts progression to large splenomegaly and leukemic transformation in primary myelofibrosis. Blood. 2007;110:4030–6. doi: 10.1182/blood-2007-07-099184. [DOI] [PubMed] [Google Scholar]
- 40.Guglielmelli P, Barosi G, Specchia G, et al. Identification of patients with poorer survival in primary myelofibrosis based on the burden of JAK2V617F mutated allele. Blood. 2009;114:1477–83. doi: 10.1182/blood-2009-04-216044. [DOI] [PubMed] [Google Scholar]
- 41.Tefferi A, Lasho TL, Huang J, et al. Low JAK2V617F allele burden in primary myelofibrosis, compared to either a higher allele burden or unmutated status, is associated with inferior overall and leukemia-free survival. Leukemia. 2008;22:756–61. doi: 10.1038/sj.leu.2405097. [DOI] [PubMed] [Google Scholar]
- 42.Camoratto AM, Jani JP, Angeles TS, et al. CEP-751 inhibits TRK receptor tyrosine kinase activity in vitro exhibits anti-tumor activity. Int J Cancer. 1997;72:673–9. doi: 10.1002/(sici)1097-0215(19970807)72:4<673::aid-ijc20>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 43.Levis M, Stine A, Pham R, et al. Pharmacokinetic and pharmacodynamic studies of lestaurtinib (CEP-701) and PKC-412: cytotoxicity is often dependent on non-FLT30-mediated effects [abstract] Blood. 2005:2463. [Google Scholar]
- 44.George DJ, Dionne CA, Jani J, et al. Sustained in vivo regression of Dunning H rat prostate cancers treated with combinations of androgen ablation and Trk tyrosine kinase inhibitors, CEP-751 (KT-6587) or CEP-701 (KT-5555) Cancer Res. 1999;59:2395–401. [PubMed] [Google Scholar]
- 45.Quintas-Cardama A, Vaddi K, Liu P, et al. Preclinical characterization of the selective AK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2009 doi: 10.1182/blood-2009-04-214957. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fridman J, Nussenzveig R, Liu P, et al. Discovery and preclinical characterization of INCB018424, a selective JAK2 inhibitor for the treatment of myeloproliferative disorders [abstract] Blood. 2007;110:3538. [Google Scholar]
- 47.Verstovsek S, Kantarjian H, Mesa RA, et al. Long-term follow up and optimized dosing regimen of INCB018424 in patients with myelofibrosis: durable clinical, functional and symptomatic responses with improved hematological safety [abstract] Blood. 2009;114:315. [Google Scholar]
- 48.Tefferi A, Kantarjian H, Pardanani A, et al. The clinical phenotype of myelofibrosis encompasses a chronic inflammatory state that is favorably altered by INCB018424, a selective inhibitor of JAK1/2 [Abstract] Blood. 2008;112:2804. [Google Scholar]
- 49.Verstovsek S, Kantarjian H, Pardanani A, et al. The JAK inhibitor, INCB018424, demonstrates durable and marked clinical responses in primary myelofibrosis (PMF) and post-polycythemia/essential thrombocythemia myelofibrosis (Post PV/ETVF) [Abstract] Blood. 2008;112:1762. [Google Scholar]
- 50.Verstovsek S, Kantarjian H, Pardanani A, et al. Characterization of JAK2 V617F allele burden in advanced myelofibrosis (MF) patients: no change in V617F:wt JAK2 ratio in patients with high allele burdens despite profound clinical improvement following treatment with the JAK inhibitor, INCB018424 [abstract] Blood. 2008;112:2802. [Google Scholar]
- 51.Dobrzanski P, Hexner E, Serdikoff C, et al. CEP-701 is a JAK2 inhibitor which attenuates JAK2/STAT5 signaling pathway and the proliferation of primary cells from patients with myeloproliferative disorders [abstract] Blood. 2006;108:3594. doi: 10.1182/blood-2007-04-083402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weisel K, Yildirim S, Xue X, Kanz L, Mohle R. Selective FLT3 inhibition uncovers the role of FL and FLT3 in normal CD34+ hematopoietic stem cells [abstract] Blood. 2006:108. [Google Scholar]
- 53.Levis M, Allebach J, Tse KF, et al. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood. 2002;99:3885–91. doi: 10.1182/blood.v99.11.3885. [DOI] [PubMed] [Google Scholar]
- 54.Strock CJ, Park JI, Rosen M, et al. CEP-701 and CEP-751 inhibit constitutively activated RET tyrosine kinase activity and block medullary thyroid carcinoma cell growth. Cancer Res. 2003;63:5559–63. [PubMed] [Google Scholar]
- 55.Levis M, Smith BD, Beran M, et al. A randomized, open-label study of lestaurtinib (CEP-701), an oral FLT3 inhibitor, administered in sequence with chemotherapy in patients with relapsed AML harboring FLT3 activating mutations: clinical response correlates with successful FLT3 inhibition [abstract] Blood. 2005;106:403. [Google Scholar]
- 56.Santos FP, Kantarjian HM, Jain N, et al. Phase II study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post polycythemia vera/essential thrombocythemia myelofibrosis. Blood. 2009 doi: 10.1182/blood-2009-10-246363. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moliterno A, Roboz G, Carroll M, Luger S, Hexner E, Bensen-Kennedy D. An open-label study of CEP-701 in patients with JAK2 V617F-positive polycythemia vera and essential thrombocythemia [abstract] Blood. 2008;112:99. doi: 10.1111/bjh.12607. [DOI] [PubMed] [Google Scholar]
- 58.Tefferi A, Barosi G, Mesa RA, et al. International Working Group (IWG) consensus criteria for treatment response in myelofibrosis with myeloid metaplasia, for the IWG for Myelofibrosis Research and Treatment (IWG-MRT) Blood. 2006;108:1497–503. doi: 10.1182/blood-2006-03-009746. [DOI] [PubMed] [Google Scholar]
- 59.Hexner E, Goldberg J, Prchal J, et al. A multicenter, open label phase I/II study of CEP701 (Lestaurtinib) in adults with myelofibrosis; report on phase I: a study of the Myeloproliferative Disorders Research Consortium (MPD-RC) [abstract] Blood. 2009;114:314. [Google Scholar]
- 60.Paquette R, Sokol L, Shah N, et al. A phase I study of XL019, a selective JAK2 inhibitor, in patients with polycythemia vera. Blood. 2008;112:2810. [Google Scholar]
- 61.Shah N, Olszynski P, Sokol L, Verstovsek S. A phase I study of XL019, a selective JAK2 inhibitor, in patients with primary myelofibrosis, post-polycythemia vera or post-essential thrombocythemia myelofibrosis [Abstract] Blood. 2008;112:98. [Google Scholar]
- 62.Hood J, Cao J, Chow C, et al. Development of TG101348 for the treatment of JAK2-driven malignancies [Abstract] J Clin Oncol. 2008;26(Suppl):7083. [Google Scholar]
- 63.Wernig G, Kharas MG, Okabe R, et al. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell. 2008;13:311–20. doi: 10.1016/j.ccr.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 64.Ferrajoli A, Faderl S, Van Q, et al. WP1066 disrupts Janus kinase-2 and induces caspase-dependent apoptosis in acute myelogenous leukemia cells. Cancer Res. 2007;67:11291–9. doi: 10.1158/0008-5472.CAN-07-0593. [DOI] [PubMed] [Google Scholar]
- 65.Pardanani A, Gotlib J, Jamieson C, et al. A phase I evaluation of TG101348, a selective JAK2 inhibitor, in myelofibrosis: clinical response is accompanied by significant reduction in JAK2V617F allele burden [abstract] Blood. 2009;114:314. [Google Scholar]
- 66.Verstovsek S. Therapeutic potential of JAK2 inhibitors. Hematology. 2009:636–42. doi: 10.1182/asheducation-2009.1.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pardanani A, Gotlib J, Jamieson C, et al. TG101348, a JAK2-selective inhibitor, is well tolerated in patients with myelofibrosis and shows substantial therpeutic activity accompanied by a reduction in JAK2V617F allele burden [abstract]. 14th Congress of the European Hematology Association; 2009 June 4–9; Berlin, Germany. [Google Scholar]
- 68.Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7:673–83. doi: 10.1038/nrc2210. [DOI] [PubMed] [Google Scholar]