

Abstract
Metabolic reprograming in cancer supports the increased biosynthesis required for unchecked proliferation. Increased glucose utilization is a defining feature of many cancers that is accompanied by altered pyruvate partitioning and mitochondrial metabolism. Cancer cells also require mitochondrial tricarboxylic acid cycle activity and electron transport chain function for biosynthetic competency and proliferation. Recent evidence demonstrates that mitochondrial pyruvate carrier (MPC) function is abnormal in some cancers and that increasing MPC activity may decrease cancer proliferation. Here we examine recent findings on MPC function and cancer metabolism. Special emphasis is placed on the compartmentalization of pyruvate metabolism and the alternative routes of metabolism that maintain the cellular biosynthetic pools required for unrestrained proliferation in cancer.
Keywords: Mitochondrial pyruvate carrier, MPC, cancer, TCA cycle, glycolysis, glutaminolysis
Introduction
Cancer is characterized by aberrant metabolism that decouples biosynthesis and proliferation from normal cell cycle control. As recognized by Otto Warburg decades ago, a common feature of adaptive metabolism in cancer is high utilization of glucose, which supports cancer bioenergetics and biosynthesis in both normoxic and hypoxic environments. Indeed, increased glucose uptake is used as a clinical diagnostic to identify cancer in vivo by 18fluorodeoxyglucose positron emission tomography [1]. However, cancer cells also require intact electron transport chain function and mitochondrial metabolism-dependent biosynthetic pathways [2,3]. High rates of oxidative or reductive glutaminolysis frequently accompany increased glycolysis in cancer as mechanisms for supporting tricarboxylic acid (TCA) cycle-dependent biosynthesis when glycolytic carbon is directed away from mitochondrial metabolism [4]. Numerous mechanisms have been explored and demonstrated to contribute to elevated glycolysis in cancer. The reader is directed to several excellent reviews providing broad coverage of these mechanisms [5-7]. The purpose of this mini review is to highlight emerging research on the recently identified Mitochondrial Pyruvate Carrier (MPC) and the relationship between the critical metabolic decision of mitochondrial pyruvate uptake and overarching adaptive mechanisms in cancer metabolism.
The Mitochondrial Pyruvate Carrier
The Mitochondrial Pyruvate Carrier (MPC) conducts pyruvate from the cytosol into the mitochondrial matrix. The import of pyruvate into mitochondria is a critical metabolic decision because it links glycolysis, which does not require oxygen, with mitochondrial oxidative phosphorylation. The MPC biochemical activity was first measured in 1971 [8]. In 1977, a report on MPC function in cancer found that isolated mitochondria from Ehrlich hyperdiploid ascites tumor cells displayed an MPC activity with a decreased Vmax but unchanged Km relative to rat liver mitochondria [9]. The decreased MPC Vmax in tumor cells was accompanied by a 20-fold decreased rate of pyruvate oxidation. A subsequent study in 1983 observed similar results in Ehrlich ascites tumor cells and also found that MPC activity was decreased in Morris hepatoma tumors [10]. However, mechanistic studies to understand the specific biochemical basis for decreased MPC activity and the potential contribution to aerobic glycolysis were hindered by lack of a molecular identity.
The genes encoding the MPC protein complex were recently identified, which now enables targeted molecular-genetic studies on MPC function [11,12]. The MPC1 and MPC2 genes encode two obligate protein subunits of the MPC, MPC1 and MPC2 respectively. These proteins form a heteroligomeric complex of currently undetermined stoichiometry in mammalian systems [11,13,14]. Both proteins are required for activity because loss of one leads to destabilization and loss of the other and thus loss of the MPC complex [11,12,14].
An important event contributing to the molecular identification of the MPC was the observation of a patient with an in-born error in pyruvate metabolism not explained by decreased pyruvate dehydrogenase or pyruvate carboxylase activity [15]. Fibroblasts from this patient displayed greatly decreased ability to oxidize pyruvate. This phenotype was reversed when the cells were disrupted but not when permeabilized, suggesting an error in mitochondrial pyruvate uptake [15]. The causative mutation was subsequently identified to be in the MPC1 gene, which resulted in a change of a highly conserved arginine residue to tryptophan [11].
Recent molecularly targeted investigations of the MPC have identified important functional contributions to both gluconeogenesis in diabetes [16,17] and proliferation in cancer [18-20], which share the common pathology of excessive biosynthesis. For broad coverage of pyruvate metabolism and historical overviews of scientific advances on understanding of MPC function beyond the intended scope of this work, the reader is referred to several review articles [21-26].
Mechanisms by which decreased MPC activity promotes cancer
Glycolytic support of biosynthesis
High rates of glycolysis enable cancer growth under both normoxic and hypoxic conditions. Glycolysis produces ATP, supports biosynthesis of triglycerides, ceramides, amino acids, and nucleotides for proliferation (Figure 1), and contributes to immune cell evasion and metastasis by acidifying the local tumor environment [6,27-29]. The first glycolytic intermediate, glucose 6-phosphate, may be either channeled for continued glycolytic oxidation or biosynthetic metabolism by the pentose phosphate pathway. Flux of glucose into the pentose phosphate pathway produces ribose for nucleotide synthesis and NADPH for reductive biosynthesis and maintenance of reduced glutathione and thioredoxin pools for defense against reactive oxygen species [30,31]. Further glycolytic breakdown of glucose 6-phosphate to fructose 1,6-bisphosphate followed by aldol cleavage produces dihydroxyacetone phosphate for phospholipid and triacylglycerol biosynthesis and 3-phosphoglycerate for the biosynthesis of amino acids and ceramide [6]. Reduction of dihydroxyacetone phosphate produces glycerol 3-phosphate that is acylated to generate phosphatidic acid upstream of the synthesis of di- and triacylglycerol, membrane lipids, and lipid signaling molecules [32]. Finally, a three-step process reduces, transaminates, and hydrolyzes 3-phosphoglycerate to serine, which supplies the polar head group and amide linkage for the de novo synthesis of ceramide and participates in protein synthesis [33].
Figure 1.
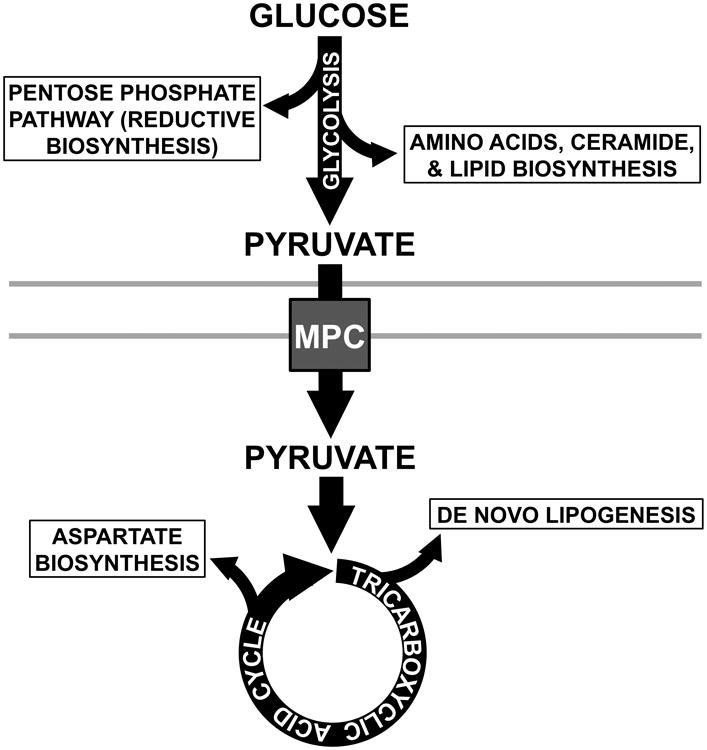
Glycolysis and the TCA cycle are linked by a common molecule, pyruvate, and the activity of the MPC, which conducts pyruvate across the mitochondrial inner membrane. Glycolytic and TCA cycle intermediates support biosynthetic pathways: Glycolysis provides carbon in the form of glucose 6-phosphate to the pentose phosphate pathway for reductive biosynthesis. Dihydroxyacetone supports phospholipid and triacylglycerol biosynthesis. 3-phosphoglycerate supports production of amino acids and ceramide. The TCA cycle generates citrate that supports cytoplasmic production of acetyl-CoA for de novo lipogenesis. Oxaloacetate may be transaminated to aspartate for amino acid biosyntheses.
The terminal step of glycolysis is catalyzed by pyruvate kinase, which converts phosphoenolpyruvate (PEP) to pyruvate. In some cancers the M2 isoform of pyruvate kinase (PKM2), typically a tetramer, is upregulated and adopts a dimeric form [34]. Dimeric PKM2 has decreased affinity for PEP such that at physiological concentrations of PEP the enzyme is essentially inactive (Figure 2A). Loss of PKM2 activity could lead to the accumulation of PEP, however this may be avoided by an alternative glycolytic pathway wherein PEP donates phosphate to the catalytic histidine of phosphoglycerate mutase generating pyruvate [35]. Yet even with this partial outlet for PEP removal, inactive PKM2 could promote the accumulation of other glycolytic intermediates. However, the high biosynthetic rate of rapidly proliferating cancer cells functions as a sink for these intermediates and prevents mass action-driven allosteric inhibition of glycolysis, thus preserving glycolytic flux [36].
Figure 2.
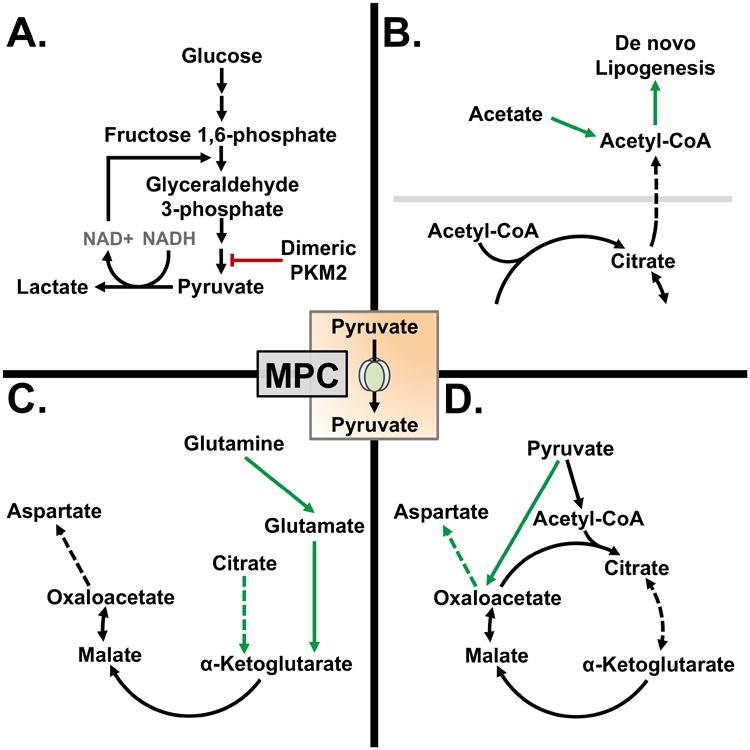
Metabolic reprogramming alters pyruvate metabolism in some cancers: A) Dimeric PKM2 prevents the glycolytic production of pyruvate. Pyruvate is shunted away from mitochondrial metabolism by reduction to lactate. B) Citrate produced by the TCA cycle is converted to acetyl-CoA in the cytoplasm for de novo lipogenesis. Acetyl-CoA may also be generated directly from acetate by acetyl-CoA synthetase 2. C) In the absence of MPC activity in highly oxidative cancers glutaminolysis maintains TCA cycle intermediate pools. When MPC activity is decreased, reductive carboxylation of α-ketoglutarate supports citrate production for de novo lipogenesis. D) Some cancers rely on pyruvate carboxylase to produce oxaloacetate. Oxaloacetate may be transaminated to aspartate to support protein synthesis.
Lactate production enables cancer bioenergetics and tumorigenesis
Complete breakdown of glucose to pyruvate may also promote cancer when coupled to its reduction to lactate. LDHA reduces pyruvate to lactate and simultaneously oxidizes NADH to NAD+, thereby regenerating the NAD+ pool required for the glyceraldehyde phosphate dehydrogenase step of glycolysis and supporting unrestrained flux [37,38] (Figure 2A). A key feature of many solid tumors is their heterogeneous composition. Individual cells are subject to differing physiological conditions, such as oxygen and nutrient availability, depending on location within the tumor. Location within poorly vascularized hypoxic areas of tumors frequently results in induction of a well-studied regulator of pyruvate metabolism HIF-1α, which positively regulates expression of lactate dehydrogenase A (LDHA) and monocarboxylate transporter 4, among other genes [39-42]. Under these conditions, inhibitory mass action effects of lactate buildup are avoided by its transport out of the cell by monocarboxylate transporter proteins (MCTs) 1-4 [43]. The rapid proton-linked export of lactate also functions to acidify the tumor microenvironment, suppress local immune responses, and activate metalloproteases that clear the way for progression to metastasis [27-29,44]. Thus, diversion of glycolytic carbon away from transit through the MPC into mitochondrial metabolism may promote cancer by redox, biosynthetic, and higher order metastatic mechanisms.
Therapeutically, decreasing LDHA activity has been actively investigated to prevent lactate production and NAD+ regeneration in cancer [45-47]. In the absence of LDHA activity, high rates of glycolysis and biosynthetic flux are unsustainable due to limited supply of NAD+ and potential allosteric regulation by pyruvate accumulation. However, LDHA inhibition may not be effective in cancers that do not rely upon this mechanism to sustain biosynthetic flux. Some cancers promote shunting of glycolytic carbon away from mitochondrial oxidation and towards cytosolic biosynthetic pathways by activating pyruvate dehydrogenase kinase, a negative regulator of pyruvate dehydrogenase [48,49]. Therapeutically, dicholoroacetate targets pyruvate dehydrogenase kinase and has been the focus of numerous studies in cell culture and animal models where it has manifested antitumor properties [50,51]. It has been found to decrease cancer cell growth in vivo in animal models and exert synergistic effects in combination with other chemotherapeutics and radiation [52]. These findings in animals have prompted phase I clinical trials [53-55]. In one study with a small cohort of human patients with glioblastomas, dichloroacetate was well-tolerated and decreased tumor-cell proliferation in three of five subjects; however, this study was not designed to determine efficacy [53]. Further investigation is required to fully understand the efficacy of dichloroacetate in the treatment of cancer. However, these studies suggest that an additional mechanism of action for LHDA inhibitors could be to promote oxidative metabolism by increasing MPC-dependent flux of pyruvate into mitochondria, leading to increased TCA cycle activity, electron transport chain activity, and elevated oxidative stress detrimental to cancer cell proliferation and survival [56,57].
Decreased MPC activity and expression in cancer
The relatively low rates of mitochondrial pyruvate oxidation in highly glycolytic cancers suggest that mitochondrial pyruvate metabolism and therefore MPC activity are dispensable. If so, loss of MPC activity could be expected to drive tumorigenic glucose utilization by preventing mitochondrial pyruvate uptake and oxidation. Indeed, several recent molecularly targeted studies indicate decreased MPC activity may contribute to aerobic glycolysis and cancer proliferation. MPC1 expression was found to be decreased in human cancers, which correlated with poor survival rates in lung and colon adenocarcinoma as well as kidney clear cell carcinoma [18]. Overexpression of MPC1 and MPC2 in HCT15 and HT29 colorectal cancer cell lines decreased cell growth in spheroids, decreased tumor size in subcutaneous xenografts, and suppressed markers of stemness including ALDH, Lin28A, LGR5, and NANOG [18]. Decreased growth in spheroids of cells re-expressing MPC1 and MPC2 was reversed by treatment with a specific MPC inhibitor, UK5099. Finally, continued passage of MPC1- and MPC2-overexpressing HCT15 cells in culture prior to xenograft injection resulted in the loss of MPC1 protein, suggesting in vivo selection against MPC1 expression [18]. Similarly, MPC inhibition by UK5099 in prostate cancer cells induced the Oct3/4 and NANOG markers of cell stemness [58].
In addition to decreased MPC expression, cancer may also involve decreased MPC specific activity. Using the recently developed RESPYR system that can detect pyruvate flux through the MPC by bioluminescence resonance energy transfer, MPC activity was found to be decoupled from glycolysis in a cellular lactate export-dependent manner. LS174T colon cancer cells treated with glucose did not manifest significant amounts of MPC flux unless MCT4 was knocked down and the MCT1 and MCT2 inhibitor AR-C155858 was added. In contrast, significant MPC-dependent pyruvate flux was observed in MCT4 knockdown cells incubated with pyruvate in the absence of AR-C155855 [59]. These data suggest that cancer cells preferentially channel glycolytically-derived lactate to MCTs for efflux from the cell, thus decreasing flux of pyruvate through the MPC.
Maintenance of TCA cycle activity when MPC activity is lost
Although glycolysis supplies abundant intermediate molecules and ATP to support biosynthesis, cancer cells also require electron transport chain function and TCA cycle activity for full biosynthetic competency [3,60-62]. In most non-transformed cells pyruvate is a critical substrate for mitochondrially routed biosynthesis. Pyruvate may be utilized to either anaplerotically replenish the TCA cycle by conversion to oxaloacetate, by pyruvate carboxylase, or to directly drive forward TCA cycle flux by conversion to acetyl-CoA, by pyruvate dehydrogenase. Condensation of oxaloacetate with acetyl-CoA generates citrate, a critical intermediate for the de novo synthesis of fatty-acids, which are esterified to glycolytically-derived glycerol 3-phosphate for producing phosphatidic acid upstream of membrane lipid synthesis [63,64] (Figure 2B). Additionally, TCA cycle intermediates help maintain the intracellular redox environment via the malate-aspartate shuttle system and aid in the production of aspartate for protein and purine synthesis [60,62]. If mitochondrial pyruvate metabolism is utilized to support TCA cycle flux and biosynthesis, what sustains these pathways in glycolytically active cancer where MPC activity is low?
Glutaminolysis is a major alternative pathway for sustaining TCA cycle metabolism [65] and is promoted by disruption of MPC activity [16,19,20]. Many cancers, including those of c-Myc and Kras origin, upregulate glutaminolysis to maintain TCA flux [66-69]. Glutaminolysis is the series of reactions by which glutamine enters into the TCA cycle after deamination to glutamate by glutaminase and further deamination to α-ketoglutarate by glutamate dehydrogenase (Figure 2C). α-ketoglutarate may be further oxidized to downstream TCA cycle products such as malate and oxaloacetate. Conversely, α-ketoglutarate may be reductively carboxylated to isocitrate and further isomerized to citrate to support lipid biosynthesis under hypoxic conditions. The enzymatic components required for reductive carboxylation of α-ketoglutarate are found in both the cytoplasm and mitochondria and, indeed, this process has been shown to occur in either compartment [70].
Chemical inhibition of the MPC in SF188 glioblastoma cells modified to express the anti-apoptotic protein Bcl-xL using UK5099 led to increased glutaminolysis, specifically the reductive carboxylation pathway generating M+5 citrate from universally labeled 13C glutamine [20]. Inhibiting glutamate dehydrogenase activity in addition to MPC activity using EGCG and CHC, respectively, decreased cell growth 80% in culture and caused a significant decrease in tumor size in a subcutaneous xenograft model using A549-derived human lung cancer cells. A separate study demonstrated that knockdown of Mpc2 in C2C12 cells increased glutaminolysis and fatty acid oxidation [19]. These results were also observed in differentiated myotubes. Reductive carboxylation of glutamine was not different between Mpc2 knockdown and control cells during culture in hypoxic 1% O2 conditions. Interestingly, significantly more reductive metabolism of glutamine was observed during Complex I inhibition by rotenone compared to MPC inhibition by UK5099. These data may reflect the availability of NADH for the reverse activity of isocitrate dehydrogenase during Complex I inhibition. However, further investigation is required to elucidate the precise mechanisms that result in these unique metabolic programs.
In addition to glutaminolysis, recent studies utilizing Mpc1 and Mpc2 liver-specific knockout mice demonstrated a pyruvate-alanine cycling mechanism to circumvent loss of MPC activity in order to maintain gluconeogenic flux [16,17]. Here, pyruvate is transaminated to alanine in the cytoplasm by alanine aminotransferase 1 (Alt1) and then transported into the mitochondrial matrix by a carrier protein of unknown identity. Once in the matrix, deamination of alanine to pyruvate by alanine aminotransferase 2 (Alt2) produces pyruvate for further metabolism by pyruvate dehydrogenase or pyruvate carboxylase. While this cycle has not been directly investigated in cancer, inhibition of alanine aminotransferases by β-Chloro-alanine was sufficient to impair the growth of malignant fibroblasts and the Lewis lung carcinoma LLC1 cell line [71]. Finally, we note that although not directly investigated in the context of decrease MPC activity, the metabolic reprograming of some cancers allows for the uptake of acetate and its conversion to acetyl-CoA in the cytoplasm by acetyl-CoA synthetase enzyme 2, thus bypassing the TCA cycle for generation of the cytoplasmic acetyl-CoA for lipogenesis [72,73].
Consideration of MPC function in cancers dependent upon oxidative phosphorylation
Although many cancers are characterized by highly glycolytic metabolisms, actively respiring mitochondria are required in some cancers [2,3,74,75]. Xenografts of mitochondrial DNA-depleted, electron transport chain-deficient B16 melonoma and 4T1 breast carcinoma cells displayed decreased tumor growth, yet the cells that established metastatic tumors showed no difference in tumor growth upon re-implantation. Cells isolated from the metastatic tumor were found to contain host mitochondrial DNA and normalized electron transport chain activity, while still being of xenograft origin [3]. In an independent study, inhibition of the electron transport chain at Complex III by antimycin A decreased growth of lung cancer cells, indicating the importance for forward TCA cycle flux and electron transport chain activity [75].
Cancers may employ coordinated adaptations in both glycolytic and glutaminolytic flux to sustain high rates of glycolysis and oxidative phosphorylation, to provide the energy and material required for high rates of biosynthesis. For example, Kras-transformed pancreatic cancers frequently display highly oxidative metabolisms, which utilize both glucose and glutamine [76]. In these cancers, glutaminolytic flux supports forward TCA cycle activity originating from α-ketoglutarate, the production of aspartate, and cytosolic malic enzyme 1 (ME1) activity for the generation of NADPH. Concomitantly, glucose metabolism, in a potentially MPC-dependent manner, supports production of citrate for de novo lipogenesis by providing mitochondrial acetyl-CoA, via pyruvate dehydrogenase activity, to supply the citrate synthase reaction [69] (Figure 2D). Furthermore, some cancers are reliant on pyruvate carboxylase activity, suggesting a dependence on mitochondrial pyruvate metabolism. MDA-MB-231 breast cancer cells displayed high levels of pyruvate carboxylase expression that when knocked down led to decreased migration and invasion in culture [77]. Similarly, human non-small-cell lung carcinoma tumor tissue showed increased pyruvate carboxylase expression relative to adjacent non-transformed tissue [78]. Finally, a recent study demonstrated that pyruvate carboxylase and pyruvate dehydrogenase are each required for tumor growth in a subcutaneous xenograft model using Kras-p53 mutant lung tumor-derived parental cells [79]. Together, these data demonstrate the critical importance of oxidative metabolism in some cancers and suggest a subset may rely on MPC-dependent pyruvate transport to sustain it.
Conclusions
Pyruvate, the product of glycolysis, sits at a central node of cellular metabolism linking glycolysis and the TCA cycle via the activity of the MPC. Evidence supports a model where the biosynthetic needs of cancer require coordination between glycolysis and TCA cycle activity to sustain anabolic metabolite pools and maintain redox balance. In some cases, the loss of MPC expression and activity observed in cancer may be acquired and provide an advantage in cell survival and unchecked proliferation. This phenomenon is still poorly understood and the mechanisms leading to loss of MPC expression and diminished activity in some cancers have yet to be explained in detail. More work is also needed to understand the mechanisms by which glycolytic carbon is preferentially channeled towards cellular efflux by MCTs rather than MPC-dependent mitochondrial metabolism. Conversely, the observation that some cancers require pyruvate carboxylase activity suggests, in these cancers, that MPC-dependent pyruvate metabolism may be important for growth and proliferation. Understanding the divergent regulation of MPC activity in different cancers may help define metabolic events that precede and underlie the unique modes of transformation across different cancers. Further knowledge of the cancer-specific conditions that induce one pathway of pyruvate metabolism versus another in relation to MPC activity may support the development of cancer type-specific therapeutic strategies that target metabolic vulnerabilities elicited by specific alterations in pyruvate metabolism.
Acknowledgments
Research in the Taylor Lab is supported by NIH grants R01 DK104998 (E.B.T.) and R00 AR059190 (E.B.T.). A.J.R. is supported by T32 HL007638 awarded to Michael Welsh. We thank Dr. Lawrence Gray, Dr. Kathleen Markan, and Dr. Douglas Spitz for carefully reading and providing feedback on a draft manuscript.
Footnotes
Conflict of interest: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gallamini A, Zwarthoed C, Borra A. Positron Emission Tomography (PET) in Oncology. Cancers (Basel) 2014;6:1821–1889. doi: 10.3390/cancers6041821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Han YH, Kim SH, Kim SZ, Park WH. Antimycin A as a mitochondrial electron transport inhibitor prevents the growth of human lung cancer A549 cells. Oncol Rep. 2008;20:689–693. [PubMed] [Google Scholar]
- •3.Tan AS, Baty JW, Dong LF, Bezawork-Geleta A, Endaya B, Goodwin J, Bajzikova M, Kovarova J, Peterka M, Yan B, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015;21:81–94. doi: 10.1016/j.cmet.2014.12.003. This investigation used mtDNA depleted cells for xenograft implantation and found tumors had delayed growth. Cells isolated from primary lung metastatic sites formed tumors without lag when re-injected subcutaneosly. It was found primary tumors contained host mtDNA implying horizontal mtDNA or mitochondrial transfer from host to tumor. This resulted in the recovery of respiration and the complete assembly of the respirasome within the tumor. [DOI] [PubMed] [Google Scholar]
- 4.Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest. 2013;123:3685–3692. doi: 10.1172/JCI69741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 7.DeNicola GM, Cantley LC. Cancer's Fuel Choice: New Flavors for a Picky Eater. Mol Cell. 2015;60:514–523. doi: 10.1016/j.molcel.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Papa S, Francavilla A, Paradies G, Meduri B. The transport of pyruvate in rat liver mitochondria. FEBS Lett. 1971;12:285–288. doi: 10.1016/0014-5793(71)80200-4. [DOI] [PubMed] [Google Scholar]
- 9.Eboli ML, Paradies G, Galeotti T, Papa S. Pyruvate transport in tumour-cell mitochondria. Biochim Biophys Acta. 1977;460:183–187. doi: 10.1016/0005-2728(77)90166-9. [DOI] [PubMed] [Google Scholar]
- 10.Paradies G, Capuano F, Palombini G, Galeotti T, Papa S. Transport of pyruvate in mitochondria from different tumor cells. Cancer Res. 1983;43:5068–5071. [PubMed] [Google Scholar]
- 11.Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, Cox JE, Cardon CM, Van Vranken JG, Dephoure N, et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science. 2012;337:96–100. doi: 10.1126/science.1218099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, Kunji ER, Martinou JC. Identification and functional expression of the mitochondrial pyruvate carrier. Science. 2012;337:93–96. doi: 10.1126/science.1218530. [DOI] [PubMed] [Google Scholar]
- 13.Bender T, Pena G, Martinou JC. Regulation of mitochondrial pyruvate uptake by alternative pyruvate carrier complexes. EMBO J. 2015;34:911–924. doi: 10.15252/embj.201490197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colca JR, McDonald WG, Cavey GS, Cole SL, Holewa DD, Brightwell-Conrad AS, Wolfe CL, Wheeler JS, Coulter KR, Kilkuskie PM, et al. Identification of a mitochondrial target of thiazolidinedione insulin sensitizers (mTOT)--relationship to newly identified mitochondrial pyruvate carrier proteins. PLoS One. 2013;8:e61551. doi: 10.1371/journal.pone.0061551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brivet M, Garcia-Cazorla A, Lyonnet S, Dumez Y, Nassogne MC, Slama A, Boutron A, Touati G, Legrand A, Saudubray JM. Impaired mitochondrial pyruvate importation in a patient and a fetus at risk. Mol Genet Metab. 2003;78:186–192. doi: 10.1016/s1096-7192(03)00016-7. [DOI] [PubMed] [Google Scholar]
- 16.Gray LR, Sultana MR, Rauckhorst AJ, Oonthonpan L, Tompkins SC, Sharma A, Fu X, Miao R, Pewa AD, Brown KS, et al. Hepatic Mitochondrial Pyruvate Carrier 1 Is Required for Efficient Regulation of Gluconeogenesis and Whole-Body Glucose Homeostasis. Cell Metab. 2015;22:669–681. doi: 10.1016/j.cmet.2015.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCommis KS, Chen Z, Fu X, McDonald WG, Colca JR, Kletzien RF, Burgess SC, Finck BN. Loss of Mitochondrial Pyruvate Carrier 2 in the Liver Leads to Defects in Gluconeogenesis and Compensation via Pyruvate-Alanine Cycling. Cell Metab. 2015;22:682–694. doi: 10.1016/j.cmet.2015.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••18.Schell JC, Olson KA, Jiang L, Hawkins AJ, Van Vranken JG, Xie J, Egnatchik RA, Earl EG, DeBerardinis RJ, Rutter J. A role for the mitochondrial pyruvate carrier as a repressor of the Warburg effect and colon cancer cell growth. Mol Cell. 2014;56:400–413. doi: 10.1016/j.molcel.2014.09.026. This study was the first investigation of MPC expression in cancer. Bioinformatic analysis showed poor prognosis correlated to decreased MPC1 expression. Using colon cancer cell lines, they established that re-expression of MPC1 and MPC2 was sufficient to decrease cell growth under low-attachment conditions and lessened tumor burden in a xenograft model. Finally, the authors found decreased MPC1 expression lead to increased markers of cell stemness. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••19.Vacanti NM, Divakaruni AS, Green CR, Parker SJ, Henry RR, Ciaraldi TP, Murphy AN, Metallo CM. Regulation of substrate utilization by the mitochondrial pyruvate carrier. Mol Cell. 2014;56:425–435. doi: 10.1016/j.molcel.2014.09.024. Utilizing sophisticated 13C-tracer methodology, this study investigated the changes in mitochondrial metabolism induced by knockdown or phamarcological inhibition of the MPC. They show increases in fatty acid oxidation, glutaminolysis, and branched chain amino acid oxidation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••20.Yang C, Ko B, Hensley CT, Jiang L, Wasti AT, Kim J, Sudderth J, Calvaruso MA, Lumata L, Mitsche M, et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol Cell. 2014;56:414–424. doi: 10.1016/j.molcel.2014.09.025. Glutaminolysis was investigated after siRNA or pharmacological intervention using 13C-tracer methodology in cancer cell lines. They found glutamonlysis was able to mediate TCA cycle anaplerosis in the absence of MPC activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schell JC, Rutter J. The long and winding road to the mitochondrial pyruvate carrier. Cancer Metab. 2013;1:6. doi: 10.1186/2049-3002-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gray LR, Tompkins SC, Taylor EB. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci. 2014;71:2577–2604. doi: 10.1007/s00018-013-1539-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCommis KS, Finck BN. Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem J. 2015;466:443–454. doi: 10.1042/BJ20141171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vanderperre B, Bender T, Kunji ER, Martinou JC. Mitochondrial pyruvate import and its effects on homeostasis. Curr Opin Cell Biol. 2015;33:35–41. doi: 10.1016/j.ceb.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 25.Bender T, Martinou JC. The mitochondrial pyruvate carrier in health and disease: To carry or not to carry? Biochim Biophys Acta. 2016 doi: 10.1016/j.bbamcr.2016.01.017. [DOI] [PubMed] [Google Scholar]
- 26.Olson KA, Schell JC, Rutter J. Pyruvate and Metabolic Flexibility: Illuminating a Path Toward Selective Cancer Therapies. Trends Biochem Sci. 2016;41:219–230. doi: 10.1016/j.tibs.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: a metabolic key player in cancer. Cancer Res. 2011;71:6921–6925. doi: 10.1158/0008-5472.CAN-11-1457. [DOI] [PubMed] [Google Scholar]
- 28.Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, Cova A, Canese R, Jachetti E, Rossetti M, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012;72:2746–2756. doi: 10.1158/0008-5472.CAN-11-1272. [DOI] [PubMed] [Google Scholar]
- 29.Marchiq I, Le Floch R, Roux D, Simon MP, Pouyssegur J. Genetic disruption of lactate/H+ symporters (MCTs) and their subunit CD147/BASIGIN sensitizes glycolytic tumor cells to phenformin. Cancer Res. 2015;75:171–180. doi: 10.1158/0008-5472.CAN-14-2260. [DOI] [PubMed] [Google Scholar]
- 30.Vaughn AE, Deshmukh M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat Cell Biol. 2008;10:1477–1483. doi: 10.1038/ncb1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. 2014;39:347–354. doi: 10.1016/j.tibs.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Athenstaedt K, Daum G. Phosphatidic acid, a key intermediate in lipid metabolism. Eur J Biochem. 1999;266:1–16. doi: 10.1046/j.1432-1327.1999.00822.x. [DOI] [PubMed] [Google Scholar]
- 33.Hanada K. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim Biophys Acta. 2003;1632:16–30. doi: 10.1016/s1388-1981(03)00059-3. [DOI] [PubMed] [Google Scholar]
- 34.Liu VM, Vander Heiden MG. The Role of Pyruvate Kinase M2 in Cancer Metabolism. Brain Pathol. 2015;25:781–783. doi: 10.1111/bpa.12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, Christofk HR, Wagner G, Rabinowitz JD, Asara JM, et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010;329:1492–1499. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong N, Ojo D, Yan J, Tang D. PKM2 contributes to cancer metabolism. Cancer Lett. 2015;356:184–191. doi: 10.1016/j.canlet.2014.01.031. [DOI] [PubMed] [Google Scholar]
- 37.Shi M, Cui J, Du J, Wei D, Jia Z, Zhang J, Zhu Z, Gao Y, Xie K. A novel KLF4/LDHA signaling pathway regulates aerobic glycolysis in and progression of pancreatic cancer. Clin Cancer Res. 2014;20:4370–4380. doi: 10.1158/1078-0432.CCR-14-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 39.Meijer TW, Kaanders JH, Span PN, Bussink J. Targeting hypoxia, HIF-1, and tumor glucose metabolism to improve radiotherapy efficacy. Clin Cancer Res. 2012;18:5585–5594. doi: 10.1158/1078-0432.CCR-12-0858. [DOI] [PubMed] [Google Scholar]
- 40.Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8:705–713. doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- 41.Schodel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood. 2011;117:e207–217. doi: 10.1182/blood-2010-10-314427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao YH, Zhou M, Liu H, Ding Y, Khong HT, Yu D, Fodstad O, Tan M. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene. 2009;28:3689–3701. doi: 10.1038/onc.2009.229. [DOI] [PubMed] [Google Scholar]
- 43.Halestrap AP, Wilson MC. The monocarboxylate transporter family--role and regulation. IUBMB Life. 2012;64:109–119. doi: 10.1002/iub.572. [DOI] [PubMed] [Google Scholar]
- 44.Rofstad EK, Mathiesen B, Kindem K, Galappathi K. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res. 2006;66:6699–6707. doi: 10.1158/0008-5472.CAN-06-0983. [DOI] [PubMed] [Google Scholar]
- 45.Xie H, Hanai J, Ren JG, Kats L, Burgess K, Bhargava P, Signoretti S, Billiard J, Duffy KJ, Grant A, et al. Targeting lactate dehydrogenase--a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab. 2014;19:795–809. doi: 10.1016/j.cmet.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Daniele S, Giacomelli C, Zappelli E, Granchi C, Trincavelli ML, Minutolo F, Martini C. Lactate dehydrogenase-A inhibition induces human glioblastoma multiforme stem cell differentiation and death. Sci Rep. 2015;5:15556. doi: 10.1038/srep15556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qiu H, Jackson AL, Kilgore JE, Zhong Y, Chan LL, Gehrig PA, Zhou C, Bae-Jump VL. JQ1 suppresses tumor growth through downregulating LDHA in ovarian cancer. Oncotarget. 2015;6:6915–6930. doi: 10.18632/oncotarget.3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 49.McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, Wu H, Schell MJ, Tsang TM, Teahan O, et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J Biol Chem. 2008;283:22700–22708. doi: 10.1074/jbc.M801765200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou X, Chen R, Yu Z, Li R, Li J, Zhao X, Song S, Liu J, Huang G. Dichloroacetate restores drug sensitivity in paclitaxel-resistant cells by inducing citric acid accumulation. Mol Cancer. 2015;14:63. doi: 10.1186/s12943-015-0331-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saunier E, Benelli C, Bortoli S. The pyruvate dehydrogenase complex in cancer: An old metabolic gatekeeper regulated by new pathways and pharmacological agents. Int J Cancer. 2015 doi: 10.1002/ijc.29564. [DOI] [PubMed] [Google Scholar]
- 52.Kankotia S, Stacpoole PW. Dichloroacetate and cancer: new home for an orphan drug? Biochim Biophys Acta. 2014;1846:617–629. doi: 10.1016/j.bbcan.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 53.Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2:31ra34. doi: 10.1126/scitranslmed.3000677. [DOI] [PubMed] [Google Scholar]
- 54.Dunbar EM, Coats BS, Shroads AL, Langaee T, Lew A, Forder JR, Shuster JJ, Wagner DA, Stacpoole PW. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest New Drugs. 2014;32:452–464. doi: 10.1007/s10637-013-0047-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garon EB, Christofk HR, Hosmer W, Britten CD, Bahng A, Crabtree MJ, Hong CS, Kamranpour N, Pitts S, Kabbinavar F, et al. Dichloroacetate should be considered with platinum-based chemotherapy in hypoxic tumors rather than as a single agent in advanced non-small cell lung cancer. J Cancer Res Clin Oncol. 2014;140:443–452. doi: 10.1007/s00432-014-1583-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shaw AT, Winslow MM, Magendantz M, Ouyang C, Dowdle J, Subramanian A, Lewis TA, Maglathin RL, Tolliday N, Jacks T. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc Natl Acad Sci U S A. 2011;108:8773–8778. doi: 10.1073/pnas.1105941108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T, et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. 2013;498:109–112. doi: 10.1038/nature12154. [DOI] [PubMed] [Google Scholar]
- 58.Zhong Y, Li X, Yu D, Li X, Li Y, Long Y, Yuan Y, Ji Z, Zhang M, Wen JG, et al. Application of mitochondrial pyruvate carrier blocker UK5099 creates metabolic reprogram and greater stem-like properties in LnCap prostate cancer cells in vitro. Oncotarget. 2015;6:37758–37769. doi: 10.18632/oncotarget.5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••59.Compan V, Pierredon S, Vanderperre B, Krznar P, Marchiq I, Zamboni N, Pouyssegur J, Martinou JC. Monitoring Mitochondrial Pyruvate Carrier Activity in Real Time Using a BRET-Based Biosensor: Investigation of the Warburg Effect. Mol Cell. 2015;59:491–501. doi: 10.1016/j.molcel.2015.06.035. MPC activity was observed in realtime after development of novel BRET technology by tagging MPC1 and MPC2 proteins with a Renilla luciferase variant and venus, respectively. [DOI] [PubMed] [Google Scholar]
- •60.Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell. 2015;162:540–551. doi: 10.1016/j.cell.2015.07.016. This study uncovered a mechanism wherein cytoplasmic aspartate is required for cell survival and proliferation in the absence of electron transport chain function. Cytoplasmic aspartate can be synthesized by the activity of GOT1 from oxaloacetate. Alternatively, mitochondrial produced aspartate can be transported to the cytoplasm via the malate-aspartate shuttle system. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cardaci S, Zheng L, MacKay G, van den Broek NJ, MacKenzie ED, Nixon C, Stevenson D, Tumanov S, Bulusu V, Kamphorst JJ, et al. Pyruvate carboxylation enables growth of SDH-deficient cells by supporting aspartate biosynthesis. Nat Cell Biol. 2015;17:1317–1326. doi: 10.1038/ncb3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell. 2015;162:552–563. doi: 10.1016/j.cell.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin R, Tao R, Gao X, Li T, Zhou X, Guan KL, Xiong Y, Lei QY. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol Cell. 2013;51:506–518. doi: 10.1016/j.molcel.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15:110–121. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Mates JM, DeBerardinis RJ. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci U S A. 2011;108:8674–8679. doi: 10.1073/pnas.1016627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Beuster G, Zarse K, Kaleta C, Thierbach R, Kiehntopf M, Steinberg P, Schuster S, Ristow M. Inhibition of alanine aminotransferase in silico and in vivo promotes mitochondrial metabolism to impair malignant growth. J Biol Chem. 2011;286:22323–22330. doi: 10.1074/jbc.M110.205229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, Walters H, Tantawy MN, Fu A, Manning HC, et al. Acetate dependence of tumors. Cell. 2014;159:1591–1602. doi: 10.1016/j.cell.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C, et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell. 2014;159:1603–1614. doi: 10.1016/j.cell.2014.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Arbini AA, Guerra F, Greco M, Marra E, Gandee L, Xiao G, Lotan Y, Gasparre G, Hsieh JT, Moro L. Mitochondrial DNA depletion sensitizes cancer cells to PARP inhibitors by translational and post-translational repression of BRCA2. Oncogenesis. 2013;2:e82. doi: 10.1038/oncsis.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Han YH, Park WH. Growth inhibition in antimycin A treated-lung cancer Calu-6 cells via inducing a G1 phase arrest and apoptosis. Lung Cancer. 2009;65:150–160. doi: 10.1016/j.lungcan.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 76.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Phannasil P, Thuwajit C, Warnnissorn M, Wallace JC, MacDonald MJ, Jitrapakdee S. Pyruvate Carboxylase Is Up-Regulated in Breast Cancer and Essential to Support Growth and Invasion of MDA-MB-231 Cells. PLoS One. 2015;10:e0129848. doi: 10.1371/journal.pone.0129848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •78.Sellers K, Fox MP, Bousamra M, 2nd, Slone SP, Higashi RM, Miller DM, Wang Y, Yan J, Yuneva MO, Deshpande R, et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J Clin Invest. 2015;125:687–698. doi: 10.1172/JCI72873. Pyruvate carboxylase protein expression and activity was found to be increased in primary non-small cell lung cancer relative to adjacent non-cancerous tissue. Knockdown of pyruvate carboyxlase by shRNA and subsequence decrease in its activity inhibited proliferation, colony formation, and xenograft growth. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O'Brien JP, Pierce KA, et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016 doi: 10.1016/j.cmet.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]