

Abstract
Objective:
To identify the molecular basis of a fatal syndrome of microcephaly, cortical hyperexcitability, and myasthenia.
Methods:
We performed clinical and in vitro microelectrode studies of neuromuscular transmission, examined neuromuscular junctions cytochemically and by electron microscopy (EM), and searched for mutations by Sanger and exome sequencing.
Results:
Neuromuscular transmission was severely compromised by marked depletion of the readily releasable pool of quanta, but the probability of quantal release was normal. Cytochemical and EM studies revealed normal endplate architecture. Exome sequencing identified a homozygous nonsense mutation in the N-terminal domain of MUNC13-1 (UNC13A) truncating the protein after 101 residues.
Conclusions:
Loss of Munc13-1 function predicts that syntaxin 1B is consigned to a nonfunctional closed state; this inhibits cholinergic transmission at the neuromuscular junction and glutamatergic transmission in the brain. Inactivation of syntaxin 1B likely accounts for the patient's cortical hyperexcitability because mutations of syntaxin 1B cause febrile seizures with or without epilepsy, haploinsufficiency of the STX1B is associated with myoclonic astatic epilepsy, and antisense knockdown of stx1b in zebrafish larvae elicits epileptiform discharges. A very recent publication also shows that syntaxin 1B has a separate obligatory role for maintenance of developing and mature neurons and illustrates impaired brain development in syntaxin 1A/1B double knockout mice. We therefore attribute our patient's microcephaly to the truncating homozygous Munc13-1 mutation that consigns syntaxin 1B to a permanently closed nonfunctional state akin to a knockout.
Exocytosis of the synaptic vesicles at central and neuromuscular synapses depends on concerted action of multiple proteins. According to current knowledge, these include the soluble N-ethylmaleimide–sensitive factor attachment protein receptor (SNARE) complex comprising synaptic vesicle–associated synaptobrevin (v-SNARE) and presynaptic membrane–associated SNAP25B and syntaxin 1B (t-SNARES).1 Outside the SNARE complex, Munc18-1 stabilizes a closed conformation of syntaxin 1B in which the N-terminal Habc domain of syntaxin 1B folds over its C-terminal SNARE motif (figure 1).2,3 Munc13-1 (Unc13 homolog A in Caenorhabditis elegans) binds to the N-terminal and perhaps the SNARE motif4 of syntaxin 1B; this shifts the position of Munc18-1 on syntaxin 1B and stabilizes the open conformation of syntaxin 1B. Open syntaxin 1B together with Munc18-1 and Munc13-1 irreversibly associates with SNAP25B and synaptobrevin to form primed SNARE complexes that render the cognate synaptic vesicles ready for exocytosis on Ca2+ entry into the nerve terminal.5–8
Figure 1. Domains of Munc13-1 and syntaxin 1.

MUN domain of Munc13-1 binds to N-terminal region and soluble N-ethylmaleimide–sensitive factor attachment protein receptor motif of syntaxin 1. The Gln102* mutation (arrow) eliminates the syntaxin binding site of Munc13-1. CaMB = calmodulin binding sequence; TMR = transmembrane region.
Munc13-1 is targeted to active zones of cholinergic neuromuscular synapses9 and to nearly all glutamatergic synapses in the brain.10,11 At both sites, Munc13-1 is required for docking12 as well as priming the synaptic vesicles for release.10,13 Our patient's phenotype also indicates that Munc13-1 is essential for normal development and electrical activity of the brain.
METHODS
Standard protocol approvals, registrations, and patient consents.
All human studies were approved by the Institutional Review Board of the Mayo Clinic.
In vitro electrophysiology studies.
An anconeus muscle specimen was obtained from origin to insertion from the patient at age 6.5 months. The miniature endplate (EP) potential (MEPP), and EP potential (EPP) amplitudes and the number of quanta released by nerve impulse (m) were measured as previously reported.14–16 The number of readily releasable quanta (n), estimates of the probability of quantal release (p),17 and the EP acetylcholine receptor (AChR) content were determined as previously described.18
Structural studies.
EPs were localized for electron microscopy (EM) by an established method.19 Peroxidase-labeled α-bungarotoxin was used for the ultrastructural localization of AChR.20 AChR and acetylcholinesterase (AChE) were colocalized in cryosections as described.21
Genetic analysis.
Genomic DNA was isolated from the patient's muscle and from parents' blood by standard methods. Sanger sequencing was performed with PCR primers to sequence exons and flanking noncoding regions. Exome sequencing was performed at the Mayo Medical Genome Facility Sequencing Core. Paired-end libraries were prepared following the manufacturer's protocol (Agilent Technologies, Santa Clara, CA) using genomic DNA. Whole-exome capture was carried out using the protocol for Agilent's SureSelect Human All Exon 71 MB v4 kit. The coverage was above 60× in all samples. The identified putative variants were scrutinized with Ingenuity Variant Analysis software (Qiagen, Redwood City, CA) and focused on variants in genes that affect quantal release from motor nerve terminals.
RESULTS
Clinical data.
A native American girl was born prematurely at 32-week gestation to a gravida 3 and para 1 mother by emergency cesarean section because of premature rupture of the amniotic membranes and fetal distress. She was hypotonic at birth, could not feed or clear her secretions, needed ventilatory support, and required hospitalization for 11 weeks. Because of frequent vomiting and gastroesophageal reflux, Nissen fundoplasty was performed, and she was discharged home with a gastric tube. At home, she had cyanotic episodes and developed aspiration pneumonia.
At age 4 months, she was markedly hypotonic, hyporeflexic, and could barely move. The head circumference was below the fifth percentile, and MRI examination of the head showed a thin corpus callosum (figure 2). She had a high forehead, marked frontotemporal narrowing, a wide nasal bridge, a high-arched palate, a small jaw, thoracic kyphoscoliosis, clinodactyly of the fifth digits, and flexion contractures of the proximal joints and knees. She also had variable eyelid ptosis, no voluntary or tracking eye movements, and an intermittent squint. The EEG showed 2–4 Hz background activity over the posterior head regions, nearly continuous multifocal sharp waves in the central regions, and periodic trains of sharp waves in both hemispheres without overt seizure activity. There was no electric response to photic or auditory stimulation.
Figure 2. Head MRI at age 4 months.
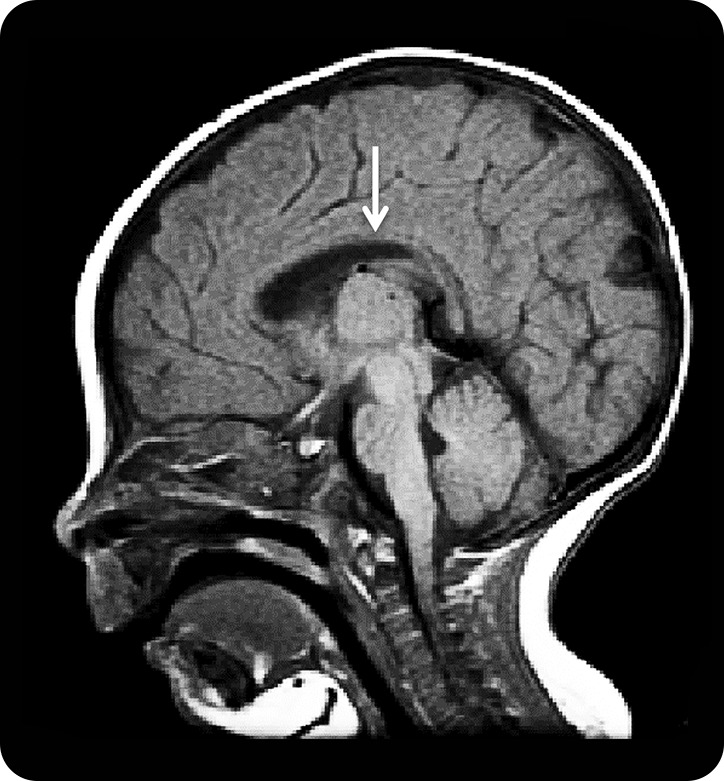
Note thin corpus callosum (arrow).
EMG studies revealed abnormally low-amplitude compound muscle action potentials (CMAPs) at rest, and a 20–40% decrement of the fourth compared with the first evoked CMAP in different muscles. Brief 50-Hz stimulation of the ulnar nerve increased the CMAP amplitude from 0.8 to 4 mV, pointing to a presynaptic defect, as observed in Lambert-Eaton myasthenic syndrome (LEMS). Therapy with pyridostigmine increased the CMAP amplitude to 1.5 mV but had no effect on the patient's strength and caused copious secretions. At age 6 months, additional therapy with 3,4-diaminopyridine increased the ulnar CMAP to 4.1 mV, improved the patient's cough and cry, but increased her limb strength only slightly. Between 10 and 11 months of age, she had bacterial pneumonia associated with a prolonged respiratory arrest and became ventilator dependent. At age 21 months, she was unable to sit up and babbled but could not speak. At age 50 months, she died of respiratory failure.
In vitro analysis of neuromuscular transmission.
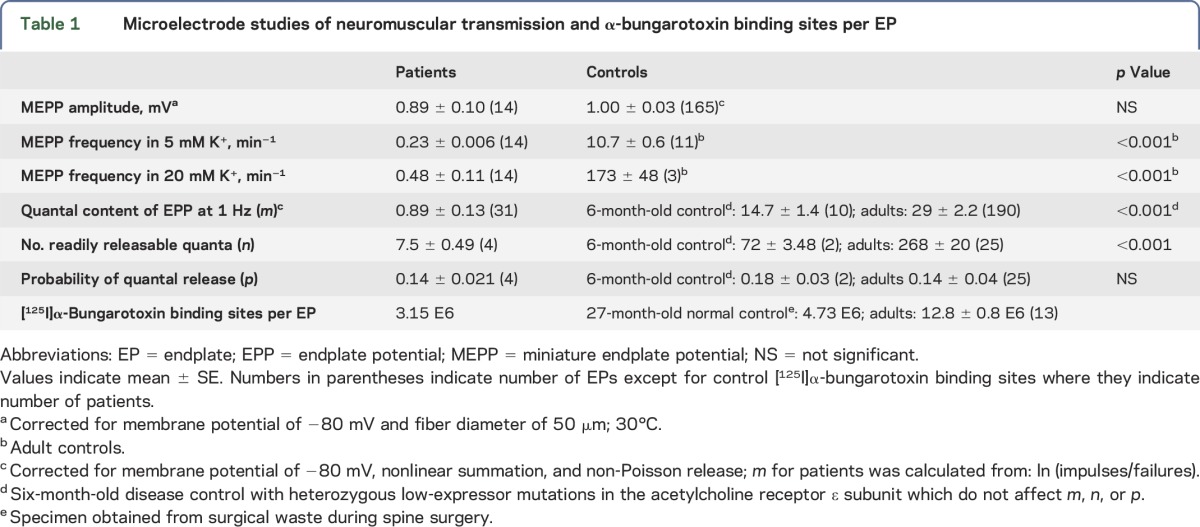
The frequency of the spontaneous MEPPs was reduced to 2% of normal (table 1). Depolarization of the nerve terminals by increasing the external K+ concentration, which enhances Ca2+ entry into the nerve terminal, increased the mean MEPP frequency 2.1-fold at the patient EPs and 16-fold at control EPs. The postsynaptic response to acetylcholine, reflected by the amplitude of the MEPP, fell in the normal range. The quantal content of EPP (m) was only 6%, and the number of readily releasable quanta (n) was only 10% of the corresponding control value, but the probability of quantal release (p) was normal. The number of α-bungarotoxin binding sites per EP was two-thirds of that in a 27-month-old normal control and was likely normal for the patient's age of 6.5 months.
Table 1.
Microelectrode studies of neuromuscular transmission and α-bungarotoxin binding sites per EP
Structural studies.
Conventional histochemical studies of the anconeus muscle revealed marked type 2 fiber atrophy and type 1 fiber hypertrophy (figure 3A). The cholinesterase-reactive synaptic contacts on the muscle fibers had a normal pretzel shape (figure 3B). Paired fluorescence localization of AChR and AChE in cryosections showed strong expression of both proteins at patient EPs (figure 3, C and D). Qualitative inspection of electron micrographs of 61 regions of 37 EPs revealed abundant synaptic vesicles in the nerve terminals. The structural integrity of the junctional folds was preserved (figure 3, E and F). Active zones, identified as thickened segments of the presynaptic membrane with clusters of synaptic vesicles focused on them, were identified at 11 of 13 EPs, and at these sites some vesicles contacted the presynaptic membrane suggesting that they were docked (figure 3E, inset). Ultrastructural localization of AChR with peroxidase-labeled α-bungarotoxin at 24 EPs revealed normal distribution of AChR on the terminal expansions of the junctional folds (figure 3F).
Figure 3. Structural observations.

(A) ATPase reaction after preincubation at pH 9.4 reveals type 2 fiber atrophy and type 1 fiber hypertrophy. (B) Cytochemical reaction for AChE in longitudinally oriented fibers shows pretzel-shaped or linearly arrayed EP regions. (C and D) Colocalization of AChE (green signal) and the AChR (red signal) in cryosections of transversely oriented fibers reveals normal expression of both proteins at the EPs. (E) Electron micrograph depicts a normal EP region. Inset in (E) shows docking of synaptic vesicles at an active zone marked by a thickened segment of the presynaptic membrane. (F) Localization of AChR with peroxidase-labeled α-bungarotoxin. The black reaction product for AChR is normally distributed on terminal expansions of the junctional folds. Bars: (A, B): 50 μm; (C, D): 25 μm; (E, F): 0.5 μm; and inset in (E): 0.1 μm. AChE = acetylcholinesterase; AChR = acetylcholine receptor; AZ = active zone; EP = endplate; JF = junctional fold.
Mutation analysis.
Because the EMG and in vitro electrophysiology studies pointed to a presynaptic defect of neuromuscular transmission resembling that observed in LEMS, we focused on selected genes governing synaptic vesicle exocytosis. Sanger sequencing revealed no mutations in SYN1 (synapsin-1), DNAJC5 (cysteine-string protein), RAB3A (Rab3A), STXBP1 (Munc18-1), CPLX1 and CPLX2 (complexin 1 and 2), SYT1 and SYT2 (synaptotagmin 1 and 2), VAMP1 (synaptobrevin-1), and SNAP25B (synaptosomal-associated protein 25B). Next, we performed exome sequencing using the DNA isolated from the patient's muscle. This revealed a homozygous nonsense mutation in the N-terminal domain of MUNC13-1 (UNC13A) (NM_001080421.2; c.304C>T/p.Gln102*) and was confirmed by Sanger sequencing. Both parents were heterozygous for the same mutation. The identified variant is not listed in the Exome Aggregation Consortium Browser server. The mutant protein is predicted to retain only 101 of the 1,703 residues comprising Munc13-1 (figure 1). No other congenital myasthenic syndrome patient investigated by us has harbored mutations in Munc13-1.
DISCUSSION
The identified homozygous loss-of-function mutation of Munc13-1 adequately accounts for the marked depletion of the readily releasable pool of synaptic vesicles. The decreased number of synaptic vesicles available for release reduces the amplitude of the EPP which compromises the safety margin of neuromuscular transmission. Because Munc13-1 is also required for docking of the synaptic vesicles at the active zones,12 the identified defect in Munc13-1 predicts the absence of docked vesicles from active zones of patient EPs, but EM images of glutaraldehyde-fixed patient EPs display synaptic vesicles docked at active zones (see inset in figure 3E). This apparent contradiction is attributed to a shrinking effect of aldehyde fixation. In a study of Munc13-1 knockout mice, EM of aldehyde-fixed brain slices showed vesicles docked at the active zones, whereas brain slices processed by high-pressure freezing combined with freeze-substitution and then examined by EM tomography revealed no docked vesicles at the active zones.12
EMG studies of our patient showed low-amplitude CMAPs that decremented further on stimulation at 2 Hz but increased by more than 100% on rapid stimulation. A similar facilitation by rapid stimulation of a low-amplitude CMAP is a feature of LEMS22 and synaptotagmin-2 myasthenia,23 but it is not a feature of the presynaptic SNAP25B myasthenia.24 In vitro microelectrode studies on LEMS,22 SNAP25B myasthenia,24 and Munc13-1 myasthenia reveal divergent features. In LEMS, the MEPP frequency and the store of readily releasable vesicles (n) are normal, but the probability of quantal release (p) is markedly reduced22,25 because of depletion of the presynaptic voltage-gated Ca2+ channels.26,27 In SNAP25B myasthenia, the MEPP frequency is one-third of normal and n and p are both reduced, likely because a disrupted coiled-coil configuration of the SNARE complex hinders exocytosis.24 In Munc13-1 myasthenia, p is normal, but the MEPP frequency and n are markedly diminished because of paucity of docked and primed synaptic vesicles (table 2).
Table 2.
In vitro microelectrode studies of Munc13-1 myasthenia, SNAP25B myasthenia, and LEMS

A mouse model of Munc13-1 deficiency had been previously investigated.28 Because these mice were stillborn or died within a few hours after birth, live but totally paralyzed embryos obtained by C-section on day E18.5 were studied. Diaphragms of these mice displayed increased terminal axon branching, enlarged EP areas, abnormally small junctional folds, and a 2.5-fold increase of the MEPP frequency, but stimulation of quantal release by α-latrotoxin or hypertonic sucrose induced much lower MEPP frequencies compared with controls. In contrast to the fetal mice, we detected no abnormal terminal axon branching in frozen sections or on EM of our patient's muscle. In cholinesterase-reacted section (figure 3B), the mean ± SE of the axial length of patient EPs was 16 ± 0.5 μm (n = 40), whereas in a 1-year-old normal control the corresponding value for 32 EPs was 16 ± 0.8 μm (n = 32), which provides little or no evidence for EP enlargement at age 6 months. Finally, the junctional folds of our patient's EPs were well developed (figure 3F). We note but cannot explain the morphologic and physiologic differences between the Munc-13–deficient prenatal mice and our patient.
The homozygous N-terminal stop-codon mutation in Munc13-1 in our patient was associated with microcephaly and abnormal cortical electrical activity. By 2015, no fewer than 11 genomic variants causing primary microcephaly had been reported,29,30 but none implicated a primary defect in a central neurotransmitter. A very recent publication provides an explanation for our patient's microcephaly by documenting that syntaxin 1B has an obligatory role for maintenance of developing and mature neurons and illustrates impaired brain development in syntaxin 1A/1B double knockout mice.31 We therefore attribute our patient's microcephaly to the truncating homozygous Munc13-1 mutation that consigns syntaxin 1B to a permanently closed nonfunctional state akin to a knockout.
Inactivation of syntaxin 1B likely also accounts for the patient's cortical hyperexcitability because mutations of syntaxin 1B are associated with febrile seizures with or without epilepsy,32 and haploinsufficiency of the STX1B is associated with myoclonic astatic epilepsy.33 Moreover, antisense knockdown of stx1b in zebrafish larvae elicits epileptiform discharges.32 Of interest, knockin/knockout mice expressing only open syntaxin 1B also have a decreased pool of release-ready synaptic vesicles but exocytose synaptic vesicle at an enhanced rate and die of generalized seizures at 2–3 months of age.34
GLOSSARY
- AChE
acetylcholinesterase
- AChR
acetylcholine receptor
- EM
electron microscopy
- EP
endplate
- EPP
endplate potential
- LEMS
Lambert-Eaton myasthenic syndrome
- MEPP
miniature endplate potential
- SNARE
soluble N-ethylmaleimide–sensitive factor attachment protein receptor
AUTHOR CONTRIBUTIONS
Andrew G. Engel: study concept and design, data acquisition, analysis and interpretation, and manuscript preparation. Duygu Selcen: data analysis and interpretation and manuscript preparation. Xin-Ming Shen: data acquisition, analysis, and interpretation. Margherita Milone and C. Michel Harper: data acquisition.
STUDY FUNDING
Supported by NIH Grant NS6277 to A.G.E.
DISCLOSURE
Dr. Engel has served on the editorial board of Neuromuscular Disorders; has received an honorarium from Oxford University Press; and has received research support from NIH. Dr. Selcen has served on the editorial boards of Neuromuscular Disorders and the Journal of Neuromuscular Diseases. Dr. Shen reports no disclosures. Dr. Milone has received research support from Mayo Clinic (Department of Neurology and the Center of Individualized Medicine). Dr. Harper reports no disclosures. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Sudhof TC. Neurotransmitter release: the last millisecond in the life of the synaptic vesicle. Neuron 2013;80:675–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dulubova I, Sugita S, Hill S, et al. A conformational switch in syntaxin during exocytosis: role of munc18. EMBO J 1999;18:4372–4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Misura KM, Scheller RH, Weis WI. Self-association of the H3 region of syntaxin 1A. Implications for intermediates in SNARE complex assembly. J Biol Chem 2001;276:13273–13282. [DOI] [PubMed] [Google Scholar]
- 4.Ma C, Li W, Xu Y, Rizo J. Munc13 mediates the transition from the closed syntaxin-Munc18 complex to the SNARE complex. Nat Struct Mol Biol 2011;18:542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Betz A, Thakur P, Junge HJ, et al. Functional interaction of the active zone proteins Munc13-1 and RIM1 in synaptic vesicle priming. Neuron 2001;30:183–196. [DOI] [PubMed] [Google Scholar]
- 6.Ma C, Su L, Seven AB, Yu Y, Rizo J. Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science 2013;339:421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mishima T, Fujiwara T, Sanada M, Kofuji T, Kanai-Azuma M, Akagawa K. Syntaxin 1B, but not syntaxin 1A, is necessary for the regulation of synaptic vesicle exocytosis and of the readily releasable pool at central synapses. PLoS One 2014;9:e90004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rizo J, Xu J. The synaptic vesicle release machinery. Annu Rev Biophys 2015;44:339–367. [DOI] [PubMed] [Google Scholar]
- 9.Betz A, Ashery U, Rickmann M, et al. Munc13-1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron 1998;21:123–1236. [DOI] [PubMed] [Google Scholar]
- 10.Augustin I, Rosenmund C, Sudhof TC, Brose N. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature 1999;400:457–461. [DOI] [PubMed] [Google Scholar]
- 11.Augustin I, Korte S, Rickmann M, et al. The cerebellum-specific Munc13 isoform Munc13-3 regulates cerebellar synaptic transmission and motor learning in mice. J Neurosci 2001;21:10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siksou L, Varoqueaux F, Pascual O, Triller A, Brose N, Marty S. A common molecular basis for membrane docking and functional priming of synaptic vesicles. Eur J Neurosci 2009;30:49–56. [DOI] [PubMed] [Google Scholar]
- 13.Varoqueaux F, Sigler A, Rhee JS, et al. Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc Natl Acad Sci USA 2002;99:9037–9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elmqvist D, Quastel DMJ. A quantitative study of end-plate potentials in isolated human muscle. J Physiol (London) 1965;178:505–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engel AG, Nagel A, Walls TJ, Harper CM, Waisburg HA. Congenital myasthenic syndromes. I. Deficiency and short open-time of the acetylcholine receptor. Muscle Nerve 1993;16:1284–1292. [DOI] [PubMed] [Google Scholar]
- 16.Uchitel O, Engel AG, Walls TJ, Nagel A, Bril V, Trastek VF. Congenital myasthenic syndrome attributed to an abnormal interaction of acetylcholine with its receptor. Ann NY Acad Sci 1993;681:487–495. [DOI] [PubMed] [Google Scholar]
- 17.Kamenskaya MA, Elmqvist D, Thesleff S. Guanidine and neuromuscular transmission 2. Effect on transmitter release in response to repetitive nerve stimulation. Arch Neurol 1975;32:510–518. [DOI] [PubMed] [Google Scholar]
- 18.Engel AG. The investigation of congenital myasthenic syndromes. Ann NY Acad Sci 1993;681:425–434. [DOI] [PubMed] [Google Scholar]
- 19.Engel AG. The muscle biopsy. In: Engel AG, Franzini-Armstrong C, editors. Myology, 3rd ed New York: McGraw-Hill; 2004:681–690. [Google Scholar]
- 20.Engel AG, Lindstrom JM, Lambert EH, Lennon VA. Ultrastructural localization of the acetylcholine receptor in myasthenia gravis and in its experimental autoimmune model. Neurology 1977;27:307–315. [DOI] [PubMed] [Google Scholar]
- 21.Fambrough DM, Engel AG, Rosenberry TL. Acetylcholinesterase of human erythrocytes and neuromuscular junctions: homologies revealed by monoclonal antibodies. Proc Natl Acad Sci U S A 1982;79:1078–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lambert EH, Elmqvist D. Quantal components of end-plate potentials in the myasthenic syndrome. Ann NY Acad Sci 1971;183:183–199. [DOI] [PubMed] [Google Scholar]
- 23.Herrmann DN, Horvath R, Snowden JE, et al. Synaptotagmin 2 mutations cause an autosomal-dominant form of Lambert-Eaton myasthenic syndrome and nonprogressive motor neuropathy. Am J Hum Genet 2014;95:332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen XM, Selcen D, Brengman J, Engel AG. Mutant SNAP25B causes myasthenia, cortical hyperexcitability, ataxia, and intellectual disability. Neurology 2014;83:2247–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Engel AG, Lambert EH, Gomez MR. A new myasthenic syndrome with end-plate acetylcholinesterase deficiency, small nerve terminals, and reduced acetylcholine release. Ann Neurol 1977;1:315–330. [DOI] [PubMed] [Google Scholar]
- 26.Fukunaga H, Engel AG, Osame M, Lambert EH. Paucity and disorganization of presynaptic membrane active zones in the Lambert-Eaton myasthenic syndrome. Muscle Nerve 1982;5:686–697. [Google Scholar]
- 27.Fukunaga H, Engel AG, Lang B, Vincent A. Passive transfer of Lambert-Eaton myasthenic syndrome with IgG from man to mouse depletes the presynaptic membrane active zones. Proc Natl Acad Sci USA 1983;80:7636–7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varoqueaux F, Sons MS, Plomp JJ, Brose N. Aberrant morphology and residual transmitter release at the Munc13-deficient mouse neuromuscular synapse. Mol Cell Biol 2005;25:5973–5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Faheem M, Naseer MI, Rasool M, et al. Molecular genetics of human primary microcephaly: an overview. BMC Med Genomics 2015;8(suppl 1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jamuar SS, Walsh CA. Genomic variants and variations in malformations of cortical development. Pediatr Clin North Am 2015;62:571–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vardar G, Chang S, Arancillo M, Wu YJ, Trimbuch T, Rosenmund C. Distinct functions of syntaxin-1 in neuronal maintenance, synaptic vesicle docking, and fusion in mouse neurons. J Neurosci 2016;36:7911–7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schubert J, Siekierska A, Langlois M, et al. Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat Genet 2014;46:1327–1332. [DOI] [PubMed] [Google Scholar]
- 33.Vlaskamp DR, Rump P, Callenbach PM, et al. Haploinsufficiency of the STX1B gene is associated with myoclonic astatic epilepsy. Eur J Paediatr Neurol 2016;20:489–492. [DOI] [PubMed] [Google Scholar]
- 34.Gerber SH, Rah JC, Min SW, et al. Conformational switch of syntaxin-1 controls synaptic vesicle fusion. Science 2008;321:1507–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]