

The binding mode of newly discovered histone demethylase inhibitors could have applications in the design and repurposing of drugs.
Keywords: LSD1, quinazoline, polymyxin, histone demethylation, reversible inhibition
Abstract
Because of its involvement in the progression of several malignant tumors, the histone lysine-specific demethylase 1 (LSD1) has become a prominent drug target in modern medicinal chemistry research. We report on the discovery of two classes of noncovalent inhibitors displaying unique structural features. The antibiotics polymyxins bind at the entrance of the substrate cleft, where their highly charged cyclic moiety interacts with a cluster of positively charged amino acids. The same site is occupied by quinazoline-based compounds, which were found to inhibit the enzyme through a most peculiar mode because they form a pile of five to seven molecules that obstruct access to the active center. These data significantly indicate unpredictable strategies for the development of epigenetic inhibitors.
INTRODUCTION
Lysine-specific demethylase 1 (LSD1/KDM1A) plays fundamental roles in regulating gene expression through the removal of monomethylation and dimethylation marks from Lys4 of histone H3. Its deregulation is frequently observed in human solid cancers and leukemia (1). A large and constantly increasing number of LSD1 inhibition studies are available and use in silico, in vitro, and high-throughput approaches to identify different classes of compounds that could specifically target demethylation activity (2, 3). Various inhibitors have been identified but, nevertheless, the most successful strategy still relies on the sequence and structure similarity of LSD1 to monoamine oxidases A and B, which are well-exploited drug discovery targets (4). In particular, tranylcypromine derivatives, originally developed as covalent inhibitors of monoamine oxidases, remain the best-studied class of specific LSD1 inhibitors and have reached clinical stages. They covalently bind the flavin adenine dinucleotide (FAD) cofactor and, therefore, completely abolish demethylation activity (5). Given the complex biological functions of the enzyme and its involvement in a variety of macromolecular complexes targeting chromatin, the identification of highly specific, reversible LSD1 drug-like inhibitors and druggable sites beyond the region near the flavin at the core of a catalytic site remains an open challenge.
LSD1-CoRESt (RE1-silencing transcription factor co-repressor)features an open cleft that is enriched in negatively charged amino acids and forms the binding site for the H3 N-terminal residues. The histone tail binds in a folded conformation, which enables the establishment of specific interactions with the surrounding enzyme residues and positions methyl-Lys4 in proximity to the flavin (6, 7). Inspired by this knowledge, out of a rather large collection of compounds available in our laboratories (about 2000, including small molecules, natural compounds, and known drugs), we selected a subset of potential ligands with positively charged groups (for example, amines). The compounds were first probed by thermal shift assay using ThermoFAD (8). Promising candidates were then evaluated by established in vitro LSD1 activity assays (9). Such a relatively simple strategy uncovered two new classes of LSD1 inhibitors (Fig. 1), which reveal unexpected binding modes at the same protein surface, establishing specific and common contacts, and highlight novel routes for the development of compounds targeting epigenetic processes.
Fig. 1. Polymyxins and quinazolines bind LSD1-CoREST.
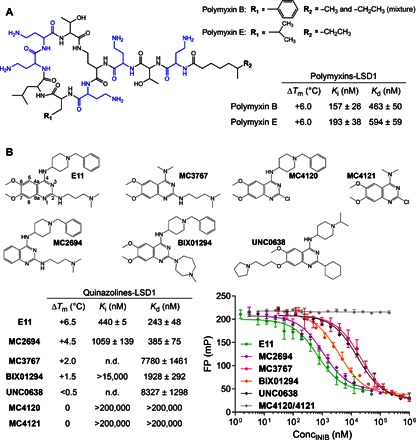
(A) Chemical structures of polymyxin B and polymyxin E (colistin). Highlighted in blue are the positively charged propanamine units. Both antibiotics bind LSD1-CoREST with high affinity, as observed by inhibition and binding assays. (B) Chemical structures of quinazoline-derived compounds binding to LSD1. The atom numbering of the quinazoline core ring is shown for compound E11 as a reference for the compound series. Fluorescence polarization (FP) signal was measured in millipolarization units (mP) and plotted against inhibitor concentration. n.d., not determined.
RESULTS AND DISCUSSION
Polymyxins inhibit LSD1 by binding at the rim of the active-site cleft
The first hits that we identified were polymyxin B and polymyxin E (also known as colistin)—well-known antibiotics used against multidrug-resistant bacteria, such as carbapenemase-producing Enterobacteriaceae or Pseudomonas aeruginosa (10). These compounds are natural cyclic peptides with a linear head group and contain five positively charged propanamine units. Polymyxins B and E differ in terms of hydrophobic side chain (Phe in polymyxin B and Leu in polymyxin E) and an extra methyl in the head group of polymyxin B (Fig. 1A). The magnitude of their binding was immediately evident because they induced a 6°C increase in the melting temperature (Tm) of LSD1-CoREST. Enzymatic assays consistently indicated that both compounds compete against the substrate histone H3 peptide (methylated on Lys4) to inhibit LSD1-CoREST with Ki values of (157 to 193) ± (26 to 38) nM; fluorescence polarization experiments fully confirmed this degree of affinity. These data clearly demonstrated that these antibiotics form a new class of LSD1-CoREST inhibitors. Therefore, we tested polymyxin E (colistin) in cultured leukemia cells (MV4-11) to probe its ability to inhibit LSD1 in a cellular context (11). The inhibitor was used at concentrations comparable to the ones used for antibiotic administration in septic patients (about 1 μM; fig. S1) (12). No remarkable effects on either cell growth or H3-Lys4/H3-Lys9 methylation (either globally or at a specific LSD1 target gene) were observed. It is possible that the compound does not cross the plasma membrane (as is typical of peptides), limiting its cellular efficacy, but we cannot rule out that LSD1 inhibition may, in the long term, contribute to the toxicity observed in polymyxin-treated patients (13).
To elucidate the specific binding modes of the newly discovered inhibitors, we determined the three-dimensional structures of LSD1-CoREST in complex with both compounds (table S1). Over numerous soaking and cocrystallization attempts using either polymyxin B or E, the unbiased electron density maps always displayed large circular peaks (tables S1 and S2 and fig. S2). Inside these maps, we could model the macrocyclic region of the inhibitor, whereas the extended linear aliphatic head group (not visible in the electron density) was assigned zero occupancy during refinement and deposition of the final structural models (Fig. 2A). However, all collected data sets consistently confirmed that these large antibiotics bind with their circular peptide moieties at the entrance of the H3 tail-binding cleft (Fig. 2B). Given the binding affinity for polymyxins and the high concentrations used in the crystallographic experiments, we believe that the electron density genuinely reflects the presence of multiple orientations during binding. The circular nature of the ligands makes it plausible that other “rotated” orientations may exist with lower occupancies. The binding region is characterized by a set of negatively charged residues, which we now find to form a high-affinity binding site for polymyxins (Fig. 2C). In support of this notion, the affinity for these antibiotics is 10-fold decreased (Kd = 4.7 ± 0.7 μM for polymyxin B) in an LSD1 mutant carrying the Glu379Lys substitution (14), which reverses the charge in one of the residues interacting with the inhibitor. In essence, the circular polymyxins form a crown of positive charges that is well suited to interact with the negatively charged entrance to the H3 tail-binding site. The ligands remain relatively distant from the flavin (>5 Å) and do not bind deep in the catalytic region. This binding mode found a remarkable counterpart in the second class of discovered inhibitors.
Fig. 2. Polymyxins B and E bind LSD1 in a similar conformation in the active site.
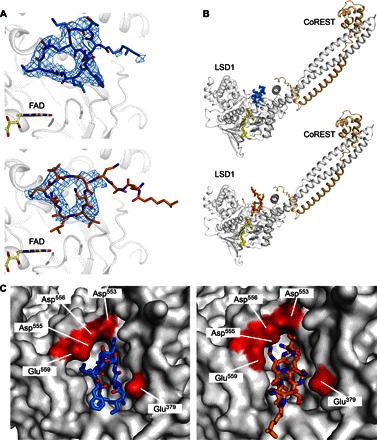
(A and B) When bound, the antibiotics do not establish interactions with FAD (yellow sticks). Polymyxin B is depicted in blue sticks and polymyxin E is depicted in orange sticks. 2Fo − Fc electron density maps (1.2σ level) are calculated before the inclusion of the ligand in the refinement. (C) LSD1-bound polymyxins establish interactions with a patch of negatively charged residues (highlighted in red on the protein surface).
Quinazoline derivatives designed as lysine mimics inhibit LSD1-CoREST
Quinazoline-derived molecules were selected for our screening because they contain a dimethylaminopropanamine moiety that decorates their core ring structure. These compounds were originally synthesized as Lys-mimicking inhibitors of the histone H3K9 methyltransferases G9a and G9a-like protein (11, 15–17). In more detail, the N-(1-benzylpiperidin-4-yl)-6,7-dimethoxy-2-(4-methyl-1,4-diazepan-1-yl)quinazolin-4-amine (BIX01294) was the first well-characterized G9a inhibitor from which compound N4-(1-benzylpiperidin-4-yl)-N2-(3-(dimethylamino)propyl)-6,7-dimethoxyquinazoline-2,4-diamine (E11) and the very potent 2-cyclohexyl-N-(1-isopropylpiperidin-4-yl)-6-methoxy-7-(3-(pyrrolidin-1-yl)propyl)quinazolin-4-amine (UNC0638) were then developed [median inhibitory concentration (IC50) values of 1.7 μM (11), 0.778 μM (15), and <0.03 μM (17), respectively]. We also probed the desmethoxy analog of E11, which is known to be a weaker inhibitor of G9a (the N4-(1-benzylpiperidin-4-yl)-N2-(3-(dimethylamino)propyl)quinazoline-2,4-diamine (MC2694); IC50 = 41.9 μM; see the Supplementary Materials). Thermal shift analysis indicated that these compounds bind to LSD1-CoREST, with MC2694 and E11 yielding large shifts (4° to 6°C) in the Tm of the enzyme. These preliminary findings were then confirmed by enzymatic and fluorescence polarization assays, demonstrating that these quinazolines are high-affinity ligands that competitively inhibit LSD1-CoREST. Compounds MC2694 and E11 tightly bind the demethylase with Kd values in the range of 0.2 to 0.4 μM, whereas BIX01294 and UNC0638 are less potent with a Kd of 2 to 9 μM. In summary, these molecules can be considered as dual demethylase-methyltransferase inhibitors: E11 and BIX01294 show comparable inhibition against both enzymes, whereas MC2694 is >100-fold stronger against LSD1and UNC0638 is >100-fold stronger against G9a.
A unique binding mode
We next determined the three-dimensional structure of LSD1-CoREST in complex with E11, the most potent compound in the quinazoline series (table S1). This study led to a most surprising observation: The inhibitor occupies the same site of polymyxins and binds in a stacked disposition, distant from the FAD cofactor and capping the entrance of the active site with five inhibitor copies (with additional density hinting at a sixth low-occupancy molecule) (Fig. 3). Moving gradually away from the flavin, these multiple ligand copies showed increasing solvent exposure and progressively limited interactions with the LSD1-CoREST heterodimer. We performed several soaking experiments to rule out crystallization artifacts attributable to ligand concentration, which was tested from 0.8 to 5 mM. In all cases, the ligand acquires the same stacking conformation, with the same copy number and highly flexible side chains exhibiting poorer electron density definition. The molecules alternate their orientations, mixing “face-to-face” and “head-to-tail” stacking modes (Fig. 3C). In this way, the molecular stack fully obstructs the active-site cleft, inherently explaining the inhibitory activity exerted by these compounds. The unusual binding stoichiometry was confirmed in solution by isothermal titration calorimetry. Two types of binding sites for E11 were identified: A primary binding site shows high affinity (Kd = 660 ± 260 nM) and 1:1 (N = 0.99 ± 0.07) stoichiometry, whereas a secondary binding site features lower affinity (Kd = 28 ± 11 μM) and ~7:1 (N = 7.1 ± 0.6) stoichiometry. The binding affinities for the primary site measured by calorimetry, fluorescence polarization, and inhibition assays are highly similar (Fig. 1B and fig. S3). Whether the stacks are preformed in solution or assemble upon sequential binding to the protein remains to be seen. Given all data in solution and in crystallo, it is plausible that a first quinazoline molecule binds with high affinity and specificity. The others then rapidly occupy the site in a cooperative fashion, establishing protein-ligand and ligand-ligand interactions.
Fig. 3. Noncovalent quinazoline-derived compound E11 obstructs the LSD1 active site in a unique multiple stacking assembly.
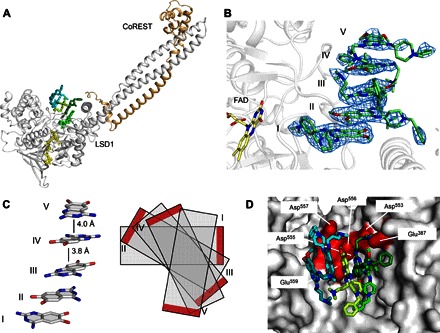
(A) A stack of five inhibitor molecules (green sticks) binding the active site of LSD1-CoREST (white and wheat cartoon, respectively) at >5 Å from FAD (yellow sticks). (B) Side view of the LSD1-CoREST complex with E11 showing the inhibitors at the entrance of the binding site. 2Fo − Fc electron density maps (1.2σ level) are calculated before the inclusion of the ligand in the refinement. I to V indicate each stacking inhibitor, with I being the most proximal to FAD, and V the most distal. (C) Simplified views of inhibitor molecules stacking in alternate flipped orientations at an intermolecular distance of 3.8 to 4.0 Å (side view). Red bars on the right indicate the position of the methoxy groups in each ligand. (D) Surface highlight (red) of negatively charged residues of LSD1 that represent the primary binding region for compound E11.
Given this most unusual mode of binding, we produced and tested other quinazoline analogs. Specifically, we probed the roles of the different substituents on the inhibitor ring to evaluate their roles in ligand stability, inhibition, and stacking binding mode. The removal of the benzylpiperidine group at the quinazoline C-4 position negatively affected inhibitor potency, as demonstrated by reduced affinity for LSD1 [N2-(3-(dimethylamino)propyl)-6,7-dimethoxy-N4,N4-dimethylquinazoline-2,4-diamine (MC3767); Fig. 1B]. However, we were able to solve the three-dimensional structure of the demethylase in complex with this compound, which retained the capability of stacking in multiple copies at the entrance of the LSD1 active site (three visible copies; fig. S4). Removal of the disordered dimethylaminopropylamine branch at the C-2 position yielded more drastic effects, completely abolishing inhibitor binding, as demonstrated by the lack of thermal stabilization and enzyme inhibition [N-(1-benzylpiperidin-4-yl)-2-chloro-6,7-dimethoxyquinazolin-4-amine (MC4120) and 2-chloro-6,7-dimethoxy-N,N-dimethylquinazolin-4-amine (MC4121); Fig. 1B]. In summary, the central quinazoline ring alone is not sufficient to determine efficient inhibition and stable binding. To exert its function, this molecular moiety has to be specifically decorated, in particular by short methoxy groups at the C-6 and C-7 positions (compare E11 with MC2694; Fig. 1B) and by the lysine analog dimethylaminopropanamine at the C-2 position (compare E11 with MC4120).
The two most biochemically active compounds, E11 and MC2694, were then tested in cultured leukemia cells (MV4-11) to probe their ability to inhibit LSD1 in vivo. As a reference, we included benzyl (1-((4-(2-aminocyclopropyl)phenyl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate (MC2580), a potent and well-characterized covalent LSD1 inhibitor (18). The compounds displayed moderate effects on cell growth even after prolonged treatment at a concentration of 1 μM, which is above their measured Kd values (fig. S1A). Likewise, none of the LSD1 inhibitors showed any effect on global H3-Lys4 and H3-Lys9 methylation levels, in agreement with the previous characterization of MC2580 (fig. S1B) (18). Conversely, we did instead observe a more specific effect on LSD1 transcriptional activity. Using quantitative reverse transcription polymerase chain reaction, we analyzed the mRNA levels of Gfi1-B, a gene largely involved in hematopoiesis and previously demonstrated to be a target of LSD1 (19–21). As illustrated in fig. S1C, 24 hours after treatment, the only compound that strongly affected Gfi1-B expression was the reference covalent inhibitor MC2580, but prolonged treatment with compound E11 led to a significant effect. Collectively, these data show that quinazoline compounds, such as E11—though not as potent as the covalent MC2580 inhibitor—are endowed with LSD1 inhibitory activity in the cellular context and can be candidates for developing a potent class of noncovalent LSD1 inhibitors.
The unexpected binding mode of compounds E11 and MC3767 prompted us to perform a thorough research on the Protein Data Bank (PDB) to explore whether such molecular arrangement was reported in other protein-ligand structures. After application of an initial computational filter on the basis of interatomic ligand distances and ligand multiplicity, we identified 599 hits that were visually inspected for possible multicopy stacking interactions, reducing the total list of matching candidates to 13 (fig. S5 and table S3). Among these, only two structures showed a stacking assembly of more than two ligands: the complex between troponin C and the antipsychotic drug trifluoperazine (PDB 1WRL and 1WRK) and the complex formed by the binding of an acetylcholine-binding protein to an isoquinoline derivative (PDB 4BFQ) (22). These crystal structures display average ligand stacking distances of 3.9 ± 0.2 and 3.6 ± 0.2 Å, respectively, which are similar to the distance of 3.9 ± 0.2 Å measured for E11. Intriguingly, there were no hits showing five or more copies of the same molecule adopting a stacked binding mode to inhibit their target, making our case unique.
π-Stacking interactions can be a successful strategy to increase the potency of drugs targeting viral proteins that bind nucleic acids (23, 24). Our data reveal the potential for the use of similar approaches to designing potent inhibitors for nonviral targets as well. In this context, a truly fascinating observation emerges from our studies: Similar or identical inhibitors can have utterly different binding modes, depending on the target. The quinazoline inhibitors do bind to G9a histone methyltransferase as “single molecules” rather than as a multimolecular stack, which is uniquely found in LSD1 (fig. S6) (15). This implies that stacking does not simply reflect an intrinsic property of the quinazoline scaffold but rather arises from the architecture of the binding site that promotes this type of interaction.
An expanded druggable surface for chemically and functionally diverse inhibitors
The rationale for selecting candidate compounds was their potential ability to mimic the dimethyl Lys4 moiety of the H3 tail substrate bound in direct contact with the flavin, in the deepest and narrowest part of the catalytic center (6, 25). Our screening led to the discovery of new inhibitor classes that, though chemically unrelated, both bind at the rim of the active site. This highly charged region was previously indicated as a candidate for the design of nonreversible inhibitors (26). We could now identify a protein surface area for ligand binding larger than that previously exploited in inhibition studies, as observed by comparing the hereby reported inhibitor complexes with all LSD1 structures bound to peptide ligands and covalent inhibitors (fig. S7 and table S4). The outer and more accessible location makes it particularly attractive for inhibitors that not only block Lys4 demethylase activity per se but also impair binding to LSD1-CoREST interactors.
Polymyxins have serious side effects that limit their usage to infections that cannot be otherwise treated. Nevertheless, our findings highlight them as possible drugs to be repurposed for simultaneous targeting epigenetic processes and bacterial infections, for instance, in the context of leukemia. It is fascinating to see how the chemical complexity of natural compounds, such as polymyxins, can exert multiple effects ranging from epigenetic enzyme inhibition to disruption of Gram-negative bacterial membranes. At the same time, our data also demonstrate that a seemingly simple moiety, such as the quinazoline core, can represent a privileged scaffold for developing inhibitors that target epigenetic enzymes. Finely tuned decorations of the core ring structure may provide the appropriate selectivity for the desired target proteins, potentially enabling the design of multitarget compounds that impair the dynamics of demethylation and methylation cycles.
MATERIALS AND METHODS
Chemicals
Polymyxin B and polymyxin E (colistin) were purchased as sulfate salts from Sigma-Aldrich. Syntheses of E11, MC2694, MC3767, MC4120, and MC4121 are reported in the Supplementary Materials and Methods.
Inhibition and binding assays
Protein expression in Escherichia coli and copurification of LSD1Δ124-CoREST1Δ305 (LSD1-CoREST) were performed using previously described procedures (6). Thermal stability, activity on histone H3K4me peptide, and inhibition of human LSD1-CoREST were measured using established protocols (6, 8). Fluorescence polarization experiments were performed on a CLARIOstar plate reader (BMG LABTECH) in a 384-well format using previously described protocols (7). Direct binding of the histone H3 N-terminal tail peptide to LSD1-CoREST was assayed using protein samples (final concentration, 2 μM) with labeled peptides (constant at a final concentration of 1 nM) followed by serial 1:1 dilutions. For competitive experiments, each well contained LSD1-CoREST (constant at a final concentration of 60 nM) and labeled peptides (fixed at a final concentration of 1 nM) to which decreasing concentrations of the competing inhibitors were added: 0 to 200 μM for MC3767, BIX01294, UNC0638, MC4120, and MC4121, and 0 to 50 μM for polymyxins, E11, and MC2694. Given the complexity of the system, including the possibility that the stack does not comprise a fixed number of molecules, we refrained from modifying the standard mathematical models that are used in affinity data analysis and processing (7).
Isothermal titration calorimetry
Experiments were performed at 25°C using a MicroCal iTC200 calorimeter in LSD1-CoREST storage buffer [25 mM KH2PO4 (pH 7.2) and 5% glycerol] with 21.1 μM LSD1-CoREST and 1.5 mM E11. Data were corrected for heat of dilution and fitted with the Origin 7.0 software package (MicroCal).
Crystallization, data collection, and refinement
LSD1-CoREST crystals were prepared at 20°C in 100 mM N-(2-acetamido)iminodiacetic acid (pH 6.5) and 1.2 M Na/K tartrate using the hanging-drop technique. Soaking was performed by incubating crystals with 0.8 to 5 mM compounds for 1 to 48 hours at 20°C; this was followed by washing in a reservoir solution supplemented with 20% glycerol for cryoprotection and immediate freezing in liquid nitrogen. X-ray diffraction data were collected at a wavelength of 1.000 Å on beamlines X06SA and X06DA at the Swiss Light Source (SLS; Villigen, Switzerland) and at a wavelength of 0.976 Å on beamline ID23EH1 at the European Synchrotron Radiation Facility (ESRF; Grenoble, France). Data processing and scaling were carried out using MOSFLM (27), XDS (28), and AIMLESS (29). The structures were solved by molecular replacement using PHASER (30). Structure refinement was performed using REFMAC5 (31) and phenix.refine (32). Topologies for the inhibitors were obtained from the PRODRG server (33). For polymyxins B and E, initial ligand-free electron density maps were subjected to multiple cycles of solvent flattening density modification using PARROT (29) and calculation of feature-enhanced maps using phenix.fem (34), enabling the identification of the macrocycle ring electron density in all data sets (fig. S2). Ligand atoms that could not be identified in the electron density were assigned zero occupancy in the final deposited models. MolProbity (35) and the PDB validation tools (36) were used for structure validation. All structures had less than 0.1% Ramachandran outliers and ~95% of residues in the favorable regions of the Ramachandran plot. Final data collection and refinement statistics are shown in tables S1 and S2. Additional data sets for polymyxins were deposited as open data archive. Structural figures were prepared using PyMOL (The PyMOL Molecular Graphics System; Schrödinger LLC; www.pymol.org).
Supplementary Material
Acknowledgments
We acknowledge the SLS and the ESRF for providing synchrotron radiation facilities and their staff for supervising data collection. We thank C. Binda and P. Roversi for providing technical support with inhibition assays and crystallographic analyses and V. Rybin for performing the isothermal titration calorimetry experiments. Funding: This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC; IG-15208 to A. Mattevi) and Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR) [Progetto Bandiera Epigenomica (EPIGEN to A. Mattevi)]; RF-2010-2318330 (A. Mai), Sapienza Award Project 2014 (D.R.), and IIT-Sapienza Project (A. Mai); AIRC–Fondazione Cariplo TRIDEO Id. 17515 (D.R.); PRIN 2012 (prot. 2012CTAYSY) (D.R.); and FP7 Projects BLUEPRINT/282510 and A-PARADDISE/602080 (A. Mai). F.F. was supported by a career development award from the Armenise-Harvard Foundation and by the Programma Giovani Ricercatori Rita Levi-Montalcini from MIUR. X-ray diffraction experiments were supported by the European Community’s Seventh Framework Programme (FP7/2007-2013) under BioStruct-X (grant agreements 7551 and 10205). Author contributions: V.S., G.C., S.P., B.M., and F.F. purified the proteins and performed biochemical, biophysical, and structural characterizations. D.R. and A. Mai designed the LSD1 inhibitors of the quinazoline series. D.R., M.F., and A.L. performed chemical synthesis and characterization of quinazoline compounds. P.M. carried out cellular assays. S.V. performed the database search for PDB protein-ligand entries. A. Mai and A. Mattevi were responsible for the project’s planning and experimental design. V.S. and A. Mattevi wrote the paper. All authors gave approval for the final version of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Coordinates and structure factors for LSD1-CoREST in complex with E11, MC3767, polymyxin B, and polymyxin E have been deposited in the PDB under accession codes 5L3E, 5LBQ, 5L3F, and 5L3G. Additional data sets for polymyxins (table S2) are available for download in the “movies and data” section of the http://www.unipv.it/biocry website. (Direct link to data set: http://www.unipv.it/biocry/data/Speranzini_et_al_LSD1-polymyxin_data.zip). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/9/e1601017/DC1
Supplementary Materials and Methods
fig. S1. Cellular characterization of LSD1 reversible inhibitors.
fig. S2. Comparison of polymyxin B and polymyxin E conformations from multiple data sets.
fig. S3. Characterization of the binding of E11 to LSD1-CoREST in solution by isothermal titration calorimetry.
fig. S4. Noncovalent quinazoline-derived compound MC3767 binds the LSD1 active site in a multiple stacking assembly as E11.
fig. S5. Workflow of the PDB search for multicopy stacking small-molecule inhibitors.
fig. S6. Comparison of histone methyltransferase G9a-like protein (GLP) and histone demethylase LSD1 in complex with compound E11.
fig. S7. LSD1 druggable space is expanded by the binding of noncovalent compounds.
table S1. Diffraction, data collection, and refinement statistics.
table S2. Diffraction, data collection, and refinement statistics for all data sets of polymyxins B and E.
table S3. List of protein inhibitor structures displaying multiple stacking conformation identified from PDB search.
table S4. List of published LSD1 PDB structures in complex with ligands.
REFERENCES AND NOTES
- 1.Schenk T., Chen W. C., Gӧllner S., Howell L., Jin L., Hebestreit K., Klein H.-U., Popescu A. C., Burnett A., Mills K., Casero R. A. Jr, Marton L., Woster P., Minden M. D., Dugas M., Wang J. C. Y., Dick J. E., Müller-Tidow C., Petrie K., Zelent A., Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 18, 605–611 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stazi G., Zwergel C., Valente S., Mai A., LSD1 inhibitors: A patent review (2010-2015). Expert Opin. Ther. Pat. 26, 565–580 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Zheng Y.-C., Ma J., Wang Z., Li J., Jiang B., Zhou W., Shi X., Wang X., Zhao W., Liu H.-M., A systematic review of histone lysine-specific demethylase 1 and its inhibitors. Med. Res. Rev. 35, 1032–1071 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Zheng Y. C., Yu B., Jiang G. Z., Feng X. J., He P. X., Chu X. Y., Zhao W., Liu H. M., Irreversible LSD1 inhibitors: Application of tranylcypromine and its derivatives in cancer treatment. Curr. Top. Med. Chem. 16, 2179–2188 (2016). [DOI] [PubMed] [Google Scholar]
- 5.McAllister T. E., England K. S., Hopkinson R. J., Brennan P. E., Kawamura A., Schofield C. J., Recent progress in histone demethylase inhibitors. J. Med. Chem. 59, 1308–1329 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Forneris F., Binda C., Adamo A., Battaglioli E., Mattevi A., Structural basis of LSD1-CoREST selectivity in histone H3 recognition. J. Biol. Chem. 282, 20070–20074 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Pilotto S., Speranzini V., Tortorici M., Durand D., Fish A., Valente S., Forneris F., Mai A., Sixma T. K., Vachette P., Mattevi A., Interplay among nucleosomal DNA, histone tails, and corepressor CoREST underlies LSD1-mediated H3 demethylation. Proc. Natl. Acad. Sci. U.S.A. 112, 2752–2757 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forneris F., Orru R., Bonivento D., Chiarelli L. R., Mattevi A., ThermoFAD, a Thermofluor-adapted flavin ad hoc detection system for protein folding and ligand binding. FEBS J. 276, 2833–2840 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Forneris F., Binda C., Vanoni M. A., Mattevi A., Battaglioli E., Histone demethylation catalysed by LSD1 is a flavin-dependent oxidative process. FEBS Lett. 579, 2203–2207 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Falagas M. E., Kasiakou S. K., Toxicity of polymyxins: A systematic review of the evidence from old and recent studies. Crit. Care 10, R27 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kubicek S., O’Sullivan R. J., August E. M., Hickey E. R., Zhang Q., Teodoro M. L., Rea S., Mechtler K., Kowalski J. A., Homon C. A., Kelly T. A., Jenuwein T., Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 25, 473–481 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Grégoire N., Mimoz O., Mégarbane B., Comets E., Chatelier D., Lasocki S., Gauzit R., Balayn D., Gobin P., Marchand S., Couet W., New colistin population pharmacokinetic data in critically ill patients suggesting an alternative loading dose rationale. Antimicrob. Agents Chemother. 58, 7324–7330 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zavascki A. P., Goldani L. Z., Li J., Nation R. L., Polymyxin B for the treatment of multidrug-resistant pathogens: A critical review. J. Antimicrob. Chemother. 60, 1206–1215 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Pilotto S., Speranzini V., Marabelli C., Rusconi F., Toffolo E., Grillo B., Battaglioli E., Mattevi A., LSD1/KDM1A mutations associated to a newly described form of intellectual disability impair demethylase activity and binding to transcription factors. Hum. Mol. Genet. ddw120 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Chang Y., Ganesh T., Horton J. R., Spannhoff A., Liu J., Sun A., Zhang X., Bedford M. T., Shinkai Y., Snyder J. P., Cheng X., Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases. J. Mol. Biol. 400, 1–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang Y., Zhang X., Horton J. R., Upadhyay A. K., Spannhoff A., Liu J., Snyder J. P., Bedford M. T., Cheng X., Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat. Struct. Mol. Biol. 16, 312–317 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vedadi M., Barsyte-Lovejoy D., Liu F., Rival-Gervier S., Allali-Hassani A., Labrie V., Wigle T. J., DiMaggio P. A., Wasney G. A., Siarheyeva A., Dong A., Tempel W., Wang S.-C., Chen X., Chau I., Mangano T. J., Huang X. P., Simpson C. D., Pattenden S. G., Norris J. L., Kireev D. B., Tripathy A., Edwards A., Roth B. L., Janzen W. P., Garcia B. A., Petronis A., Ellis J., Brown P. J., Frye S. V., Arrowsmith C. H., Jin J., A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol. 7, 566–574 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Binda C., Valente S., Romanenghi M., Pilotto S., Cirilli R., Karytinos A., Ciossani G., Botrugno O. A., Forneris F., Tardugno M., Edmondson D. E., Minucci S., Mattevi A., Mai A., Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J. Am. Chem. Soc. 132, 6827–6833 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Saleque S., Kim J., Rooke H. M., Orkin S. H., Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol. Cell 27, 562–572 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Thambyrajah R., Mazan M., Patel R., Moignard V., Stefanska M., Marinopoulou E., Li Y., Lancrin C., Clapes T., Mӧrӧy T., Robin C., Miller C., Cowley S., Gӧttgens B., Kouskoff V., Lacaud G., GFI1 proteins orchestrate the emergence of haematopoietic stem cells through recruitment of LSD1. Nat. Cell Biol. 18, 21–32 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Vianello P., Botrugno O. A., Cappa A., Dal Zuffo R., Dessanti P., Mai A., Marrocco B., Mattevi A., Meroni G., Minucci S., Stazi G., Thaler F., Trifiró P., Valente S., Villa M., Varasi M., Mercurio C., Discovery of a novel inhibitor of histone lysine-specific demethylase 1A (KDM1A/LSD1) as orally active antitumor agent. J. Med. Chem. 59, 1501–1517 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Stornaiuolo M., De Kloe G. E., Rucktooa P., Fish A., van Elk R., Edink E. S., Bertrand D., Smit A. B., de Esch I. J. P., Sixma T. K., Assembly of a π–π stack of ligands in the binding site of an acetylcholine-binding protein. Nat. Commun. 4, 1875 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hare S., Gupta S. S., Valkov E., Engelman A., Cherepanov P., Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 464, 232–236 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lansdon E. B., Liu Q., Leavitt S. A., Balakrishnan M., Perry J. K., Lancaster-Moyer C., Kutty N., Liu X., Squires N. H., Watkins W. J., Kirschberg T. A., Structural and binding analysis of pyrimidinol carboxylic acid and N-hydroxy quinazolinedione HIV-1 RNase H inhibitors. Antimicrob. Agents Chemother. 55, 2905–2915 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baron R., Binda C., Tortorici M., McCammon J. A., Mattevi A., Molecular mimicry and ligand recognition in binding and catalysis by the histone demethylase LSD1-CoREST complex. Structure 19, 212–220 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J., Lu F., Ren Q., Sun H., Xu Z., Lan R., Liu Y., Ward D., Quan J., Ye T., Zhang H., Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer Res. 71, 7238–7249 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leslie A. G. W., Integration of macromolecular diffraction data. Acta Crystallogr. Sect. D Biol. Crystallogr. 55, 1696–1702 (1999). [DOI] [PubMed] [Google Scholar]
- 28.Kabsch W., XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Collaborative Computational Project, Number 4 , The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 50, 760–763 (1994). [DOI] [PubMed] [Google Scholar]
- 30.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murshudov G. N., Vagin A. A., Dodson E. J., Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 53, 240–255 (1997). [DOI] [PubMed] [Google Scholar]
- 32.Headd J. J., Echols N., Afonine P. V., Grosse-Kunstleve R. W., Chen V. B., Moriarty N. W., Richardson D. C., Richardson J. S., Adams P. D., Use of knowledge-based restraints in phenix.refine to improve macromolecular refinement at low resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 68, 381–390 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schüttelkopf A. W., van Aalten D. M. F., PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 1355–1363 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Afonine P. V., Moriarty N. W., Mustyakimov M., Sobolev O. V., Terwilliger T. C., Turk D., Urzhumtsev A., Adams P. D., FEM: Feature-enhanced map. Acta Crystallogr. Sect. D Biol. Crystallogr. 71, 646–666 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen V. B., Arendall W. B. III, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Read R. J., Adams P. D., Arendall W. B. III, Brunger A. T., Emsley P., Joosten R. P., Kleywegt G. J., Krissinel E. B., Lütteke T., Otwinowski Z., Perrakis A., Richardson J. S., Sheffler W. H., Smith J. L., Tickle I. J., Vriend G., Zwart P. H., A new generation of crystallographic validation tools for the protein data bank. Structure 19, 1395–1412 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rotili D., Tarantino D., Marrocco B., Gros C., Masson V., Poughon V., Ausseil F., Chang Y., Labella D., Cosconati S., Di Maro S., Novellino E., Schnekenburger M., Grandjenette C., Bouvy C., Diederich M., Cheng X., Arimondo P. B., Mai A., Properly substituted analogues of BIX-01294 lose inhibition of G9a histone methyltransferase and gain selective anti-DNA methyltransferase 3A activity. PLOS One 9, e96941 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hess H. J., Cronin T. H., Scriabine A., Antihypertensive 2-amino-4(3H)-quinazolinones. J. Med. Chem. 11, 130–136 (1968). [DOI] [PubMed] [Google Scholar]
- 39.Karplus P. A., Diederichs K., Linking crystallographic model and data quality. Science 336, 1030–1033 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rao F. V., Andersen O. A., Vora K. A., Demartino J. A., van Aalten D. M. F., Methylxanthine drugs are chitinase inhibitors: Investigation of inhibition and binding modes. Chem. Biol. 12, 973–980 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Biswal B. K., Wang M., Cherney M. M., Chan L., Yannopoulos C. G., Bilimoria D., Bedard J., James M. N. G., Non-nucleoside inhibitors binding to hepatitis C virus NS5B polymerase reveal a novel mechanism of inhibition. J. Mol. Biol. 361, 33–45 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Lai C., Gum R. J., Daly M., Fry E. H., Hutchins C., Abad-Zapatero C., von Geldern T. W., Benzoxazole benzenesulfonamides as allosteric inhibitors of fructose-1,6-bisphosphatase. Bioorg. Med. Chem. Lett. 16, 1807–1810 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Ekroos M., Sjӧgren T., Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. U.S.A. 103, 13682–13687 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jessop T. C., Tarver J. E., Carlsen M., Xu A., Healy J. P., Heim-Riether A., Fu Q., Taylor J. A., Augeri D. J., Shen M., Stouch T. R., Swanson R. V., Tari L. W., Hunter M., Hoffman I., Keyes P. E., Yu X.-C., Miranda M., Liu Q., Swaffield J. C., Kimball S. D., Nouraldeen A., Wilson A. G. E., Foushee A. M. D., Jhaver K., Finch R., Anderson S., Oravecz T., Carson K. G., Lead optimization and structure-based design of potent and bioavailable deoxycytidine kinase inhibitors. Bioorg. Med. Chem. Lett. 19, 6784–6787 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Chen M., Adeniji A. O., Twenter B. M., Winkler J. D., Christianson D. W., Penning T. M., Crystal structures of AKR1C3 containing an N-(aryl)amino-benzoate inhibitor and a bifunctional AKR1C3 inhibitor and androgen receptor antagonist. Therapeutic leads for castrate resistant prostate cancer. Bioorg. Med. Chem. Lett. 22, 3492–3497 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nomme J., Murphy J. M., Su Y., Sansone N. D., Armijo A. L., Olson S. T., Radu C., Lavie A., Structural characterization of new deoxycytidine kinase inhibitors rationalizes the affinity-determining moieties of the molecules. Acta Crystallogr. Sect. D Biol. Crystallogr. 70, 68–78 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Croci R., Tarantino D., Milani M., Pezzullo M., Rohayem J., Bolognesi M., Mastrangelo E., PPNDS inhibits murine Norovirus RNA-dependent RNA-polymerase mimicking two RNA stacking bases. FEBS Lett. 588, 1720–1725 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Mimasu S., Sengoku T., Fukuzawa S., Umehara T., Yokoyama S., Crystal structure of histone demethylase LSD1 and tranylcypromine at 2.25 Å. Biochem. Biophys. Res. Commun. 366, 15–22 (2008). [DOI] [PubMed] [Google Scholar]
- 49.Yang M., Culhane J. C., Szewczuk L. M., Jalili P., Ball H. L., Machius M., Cole P. A., Yu H., Structural basis for the inhibition of the LSD1 histone demethylase by the antidepressant trans-2-phenylcyclopropylamine. Biochemistry 46, 8058–8065 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Mimasu S., Umezawa N., Sato S., Higuchi T., Umehara T., Yokoyama S., Structurally designed trans-2-phenylcyclopropylamine derivatives potently inhibit histone demethylase LSD1/KDM1. Biochemistry 49, 6494–6503 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Vianello P., Botrugno O. A., Cappa A., Ciossani G., Dessanti P., Mai A., Mattevi A., Meroni G., Minucci S., Thaler F., Tortorici M., Trifiró P., Valente S., Villa M., Varasi M., Mercurio C., Synthesis, biological activity and mechanistic insights of 1-substituted cyclopropylamine derivatives: A novel class of irreversible inhibitors of histone demethylase KDM1A. Eur. J. Med. Chem. 86, 352–363 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Rodriguez V., Valente S., Rovida S., Rotili D., Stazi G., Lucidi A., Ciossani G., Mattevi A., Botrugno O. A., Dessanti P., Mercurio C., Vianello P., Minucci S., Varasi M., Mai A., Pyrrole- and indole-containing tranylcypromine derivatives as novel lysine-specific demethylase 1 inhibitors active on cancer cells. Med. Chem. Commun. 6, 665–670 (2015). [Google Scholar]
- 53.Yang M., Culhane J. C., Szewczuk L. M., Gocke C. B., Brautigam C. A., Tomchick D. R., Machius M., Cole P. A., Yu H., Structural basis of histone demethylation by LSD1 revealed by suicide inactivation. Nat. Struct. Mol. Biol. 14, 535–539 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Tortorici M., Borrello M. T., Tardugno M., Chiarelli L. R., Pilotto S., Ciossani G., Vellore N. A., Bailey S. G., Cowan J., O’Connell M., Crabb S. J., Packham G., Mai A., Baron R., Ganesan A., Mattevi A., Protein recognition by short peptide reversible inhibitors of the chromatin-modifying LSD1/CoREST lysine demethylase. ACS Chem. Biol. 8, 1677–1682 (2013). [DOI] [PubMed] [Google Scholar]
- 55.Hirschi A., Martin W. J., Luka Z., Loukachevitch L. V., Reiter N. J., G-quadruplex RNA binding and recognition by the lysine-specific histone demethylase-1 enzyme. RNA 22, 1250–1260 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/9/e1601017/DC1
Supplementary Materials and Methods
fig. S1. Cellular characterization of LSD1 reversible inhibitors.
fig. S2. Comparison of polymyxin B and polymyxin E conformations from multiple data sets.
fig. S3. Characterization of the binding of E11 to LSD1-CoREST in solution by isothermal titration calorimetry.
fig. S4. Noncovalent quinazoline-derived compound MC3767 binds the LSD1 active site in a multiple stacking assembly as E11.
fig. S5. Workflow of the PDB search for multicopy stacking small-molecule inhibitors.
fig. S6. Comparison of histone methyltransferase G9a-like protein (GLP) and histone demethylase LSD1 in complex with compound E11.
fig. S7. LSD1 druggable space is expanded by the binding of noncovalent compounds.
table S1. Diffraction, data collection, and refinement statistics.
table S2. Diffraction, data collection, and refinement statistics for all data sets of polymyxins B and E.
table S3. List of protein inhibitor structures displaying multiple stacking conformation identified from PDB search.
table S4. List of published LSD1 PDB structures in complex with ligands.