

Abstract
BACKGROUND
The nervous system may include more than 100 residue-specific posttranslational modifications of histones forming the nucleosome core that are often regulated in cell-type-specific manner. On a genome-wide scale, some of the histone posttranslational modification landscapes show significant overlap with the genetic risk architecture for several psychiatric disorders, fueling PsychENCODE and other large-scale efforts to comprehensively map neuronal and nonneuronal epigenomes in hundreds of specimens. However, practical guidelines for efficient generation of histone chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) datasets from postmortem brains are needed.
METHODS
Protocols and quality controls are given for the following: 1) extraction, purification, and NeuN neuronal marker immunotagging of nuclei from adult human cerebral cortex; 2) fluorescence-activated nuclei sorting; 3) preparation of chromatin by micrococcal nuclease digest; 4) ChIP for open chromatin-associated histone methylation and acetylation; and 5) generation and sequencing of ChIP-seq libraries.
RESULTS
We present a ChIP-seq pipeline for epigenome mapping in the neuronal and nonneuronal nuclei from the postmortem brain. This includes a stepwise system of quality controls and user-friendly data presentation platforms.
CONCLUSIONS
Our practical guidelines will be useful for projects aimed at histone posttranslational modification mapping in chromatin extracted from hundreds of postmortem brain samples in cell-type-specific manner.
Keywords: Cell type specific, ChIP-seq, Epigenomics, Postmortem human brain, PsychENCODE, Schizophrenia
The field of neuroepigenomics is shedding light on the mechanisms underlying normal neurodevelopment (1), behavior and learning (2,3), and various neuropsychiatric disorders (4), including schizophrenia (5–8), depression (9), bipolar disorder (7,10), and autism (11). Deeper understanding of the mechanisms governing epigenetic regulation in the brain is necessary for a more complete understanding of complex behaviors and psychopathology, hopefully leading to novel therapeutic and diagnostic approaches in the future. Epigenomic regulation is thought to include a large variety of more than 100 amino acid residue-specific histone posttranslational modifications (histone PTMs). This includes monomethylation, dimethylation, and trimethylation; acetylation; crotonylation; polyADP-ribosylation; and small protein (small ubiquitin-related modifier) modifications of specific lysine residues. Other amino acids are also altered including arginine citrullination, monomethylation and dimethylation, serine phosphorylation, and tyrosine hydroxylation (12). While the large majority of histone PTMs await functional exploration in the nervous system (13), studies conducted in dividing cells and peripheral tissues have shown that many histone PTMs are closely associated with chromatin states, differentiating between active, inactive, or poised promoters and enhancers; gene bodies and exonic or intronic DNA; transcription start sites; and broader regulatory domains including super-enhancers, among others (12,14–16). It has been shown that a significant fraction of the histone PTM landscape shows strong tissue-specific and cell-type-specific regulation in the human brain (17). This observation has important implications for the genetic risk architecture of psychiatric disease. For example, sequences decorated with open chromatin-associated histone methylation and acetylation in fetal brain and in cortical neurons are, on a genome-wide scale, enriched with common polymorphisms conferring heritable risk for schizophrenia, suggesting a potential mechanistic relationship between DNA sequence variation and neural-specific epigenetic modifications (18).
Therefore, comprehensive mapping of histone PTMs in specific regions and cell types of the developing and adult human brain could help elucidate genome organization and function in the context of neuropsychiatric disease. However, with the field still in its infancy, establishing practical guidelines early can lay the foundation for large-scale projects such as PsychENCODE, a National Institutes of Mental Health sponsored initiative. PsychENCODE is aimed to create a public resource of genomic data, including histone PTMs, using tissue-specific and cell-type-specific samples from approximately 1000 human postmortem brains (19). Here, we discuss protocols and develop a set of quality control standards for our chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) pipeline designed to effectively produce and disseminate human brain epigenomic maps and datasets. Using two histone PTMs as examples, H3-trimethyl-lysine 4 (H3K4me3), primarily defined by sharp peaks (~1 kb) and found near transcription start sites-proximal promoters and some enhancers, and H3-acetyl-lysine 27 (H3K27ac), more broadly distributed at various cis-regulatory sequences, we demonstrate that our ChIP-seq pipeline on human postmortem brain is broadly consistent with the Encyclopedia of DNA Elements (ENCODE)-defined criteria for histone PTMs (20) and provides examples for user-friendly data access platforms. These advances are likely to have significant impact because they open the exciting opportunity to study histone PTM landscapes in postmortem brain with quality controls (QC) similar to those applied to peripheral tissues (which are generally much easier to access) or cell lines and to do so in large cohorts involving many hundreds of brain specimens. We expect that the approaches outlined here, when applied to human brain collections across development and disease states, will ultimately lead to deep advancements in our understanding about the neurological functions, particularly of regulatory and noncoding sequences in the human genome, currently left almost completely unexplored.
METHODS AND MATERIALS
The multistep procedure (Figure 1) begins with isolation, immunotagging, and fluorescence-activated cell sorting of nuclei extracted from tissue. Next, chromatin is prepared by enzymatic digestion with micrococcal nuclease (MNase) (N3755; Sigma-Aldrich, St. Louis, MO) to yield primarily mononucleosomes (the elementary unit of chromatin comprised of a histone octamer with 146 base pair [bp] DNA wrapped around it in two and a half loops). DNA enriched for histone modification of interest is obtained using ChIP with a specific antibody. Then, ChIP-seq libraries are prepared and sequenced by Illumina HiSeq 2500 followed by a data quality control pipeline. Our ChIP-seq pipeline includes QC at each step of the protocol (Figure 1). The entire pipeline, including bioinformatical analyses, has so far been completed for more than 100 ChIP-seq samples. Here, we show a representative example of n = 8 ChIP-seq datasets from two prefrontal cortex (PFC) specimens, each processed for Neu-N+H3K4me3, NeuN−H3K4me3, NeuN+H3K27ac, and Neu-N−H3K27ac. A detailed step-by-step protocol, a glossary, and description of postmortem tissue are included in the Supplement.
Figure 1.

Chromatin immunoprecipitation followed by deep sequencing pipeline. Overview of the pipeline flow chart and corresponding quality controls (QC). FACS, fluorescence-activated cell sorting; IGV, Integrative Genomics Viewer; MNase, micrococcal nuclease; N-ChIP, native chromatin immunoprecipitation; NeuN+, neuronal; NeuN−, nonneuronal; qPCR, quantitative polymerase chain reaction.
Nuclei Isolation, Immunotagging, and Sorting
Approximately 300 mg of cortical gray matter, dissected from fresh frozen brain slabs, is homogenized by douncing followed by nuclei extraction via sucrose gradient ultracentrifugation. The nuclei are recovered from the pellet, resuspended, and incubated with mouse monoclonal antibody (clone A60) against neuronal marker NeuN (MAB377X; Millipore). Immunotagging with NeuN antibody conjugated to AlexaFluor 488 allows for sorting of the nuclei into two fractions: NeuN+ (neuronal) and NeuN− (nonneuronal) by fluorescence-activated cell sorting (Figure 2A). Gates are adjusted to 1) select nuclei from debris; 2) select nuclei from cells not undergoing cell division (using DAPI staining); and finally 3) select the NeuN+ and NeuN− populations (Figure 2A). In our hands, cell sorters with gentle sort settings (for example, 100 μm nozzle and 20 psi sheath fluid pressure on a FACSAria [BD Biosciences]) work best. They do not exert too much mechanical force on the nuclei and keep collection volumes reasonable. Under the microscope, sorted cortical gray matter neuronal nuclei can be distinguished from nonneuronal nuclei based on their larger size (Figure 2A). On average, we recover 0.6 to 0.7 million of each NeuN+ and NeuN− nuclei per 100 mg of gray matter, with NeuN+/NeuN− ratio (dorsolateral PFC) close to 1:1, regardless of the amount of the starting material (Figure 2A). For our conventional ChIP protocol with antihistone antibodies, a minimum of 0.4 million nuclei is required as input for each ChIP assay.
Figure 2.

Chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) quality controls (QC). Quality controls include: (A) visual inspection and quantification of nuclei separated into neuronal (NeuN+) and nonneuronal (NeuN−) fraction by fluorescence-activated (cell) sorting of nuclei, including linear correlation of nuclei number with approximate prefrontal cortex gray matter tissue weight, as indicated. (B) DNA agarose gel from native chromatin digested with different amounts of micrococcal nuclease (MNase). The predominant ~150 base pair (bp) band confirms that the bulk of chromatin has been digested into mononucleosomes. (C) Peptide array containing 46 peptides representing 46 different H3 posttranslational modifications, to test specificity of the H3K4me3 antibody by dot blot (see also Supplementary Figure S1). Note no cross-reactivity other than weak activity against the dimethylated form, H3K4me2. (D) Agilent Bioanalyzer QC after ChIP confirms that predominant portion of pulldown was comprised by mononucleosomes as evidenced by sharp peak at ~148 bp. (E) ChIP-quantitative polymerase chain reaction (qPCR) confirms H3K4me3 enrichment in neuronal NeuN+ nuclei fraction (blue curve) and nonneuronal NeuN− nuclei fraction (red curve) for neuronal gene GRIN2B (upper panel) but not for negative control HBB globin sequences (lower panel). Note that the input DNA qPCR signals (dark and light green curves) are similar for these two genes. (F) Agilent Bioanalyzer QC after library preparation, confirming that large majority of DNA molecules locate to 275 bp, representing correct library ligation product (see text). (G) Early bioinformatical analyses include FASTQC, BWA, and other established programs. Note consistent GRIN2B H3K4me3 enrichment observed in the NeuN+ and NeuN− ChIP-seq tracks visualized in Integrative Genomics Viewer (IGV) browser when compared with the corresponding ChIP-qPCR signals above (E) (see text for more details). (H) FASTQC analysis of raw ChIP-seq data, represented here as the sequence quality score (y axis) versus base pair position (x axis), is an important initial step in ChIP-seq data quality control. The graph background colors separate the y axis into very good quality calls (green, score > 28), calls of reasonable quality (orange, score = 20–28), and calls of poor quality (red, score < 20). Note that our representative ChIP-seq data show remarkably high quality throughout the entire sequence, including the sequence toward the end of the read (up to 100 bp).
Chromatin Immunoprecipitation
ChIP assays are the main tools for mapping histone modifications and assaying protein-DNA binding in cells and tissues (21). Antibodies against histone marks or proteins allow for selective enrichment of a chromatin fraction containing the histone modification or protein of interest. The resulting enriched genomic regions are then identified and quantified using, for example, chip over input in ChIP-quantitative polymerase chain reaction (qPCR) or normalized tag counts in ChIP-seq (a tag is the specific DNA sequence read from a single fragment; millions of DNA fragments are sequenced from each ChIP-seq library, see below). ChIP protocols differ primarily in how the starting (input) chromatin is prepared: native ChIP (NChIP) utilizes chromatin fragmented by MNase digestion (22), while cross-linked ChIP uses chromatin treated with formaldehyde (to cross-link proteins to the DNA) followed by fragmentation using sonication (usually 200-bp to 600-bp fragments). NChIP—usually limited to histones and a few other types of protein that are tightly associated with chromatin—has an excellent signal-to-noise ratio, making it a particularly good choice for histone PTM mapping in postmortem tissue (23).
Following sorting and separation by fluorescence-activated cell sorting, neuronal and nonneuronal nuclei are resuspended and MNase is added to digest chromatin primarily into mononucleosomes (Figure 2B). Next, the MNase reaction is stopped, the chromatin is treated with a hypotonic solution and mild detergent, and finally the diluted chromatin is incubated with a site-specific and modification-specific antihistone antibody overnight. We use the following antibodies for chromatin pulldown: anti-H3K4me3 (Cat# 9751BC, lot 7; Cell Signaling) and anti-H3K27ac (Cat# 39133, Lot# 01613007; Active Motif). Histone modification-enriched genomic DNA fragments are recovered the following day using Protein A/G magnetic beads (88803; Thermo Fisher Scientific), which are then washed, eluted, and treated with RNAse A and Proteinase K and further purified using standard phenol-chloroform extraction followed by ethanol precipitation. Before performing NChIP experiments, it is essential to 1) test MNase digestion efficiency; and 2) validate antibody specificity and pulldown efficiency (Figure 2B, C). These two steps are important and should always be performed before processing samples for ChIP-seq.
Testing MNase Digestion Efficiency
Chromatin digestion dictates the resolution of the assay. In NChIP, MNase makes double-stranded cuts between nucleosomes, removes linker DNA, and leaves nucleosomes intact. This allows mapping of histone modifications at nucleosomal resolution (146 bp) provided the chromatin digest was complete (Figure 2B, D). The proper concentration of MNase should be determined for each new lot of the enzyme, as the MNase activity can vary between batches. Efficient digestion is expected to provide mononucleosomal DNA around 146 bp with minimal presence of larger fragments derived from dinucleosomes, trinucleosomes, or polynucleosomes (Figure 2B). Fragments shorter than 146 bp indicate degraded or over-digested DNA.
Testing Antibody Specificity and Efficacy
Antibody specificity and pulldown can vary from lot to lot, which is a challenge for ChIP-seq projects involving hundreds of specimens. For a PTM-specific antihistone antibody, specificity is typically tested using peptide binding assays and immunoblotting of nuclear extracts (20,24). These tests determine that the antibody primarily recognizes the modification of interest and not other related or unrelated modifications or unmodified histones. As an example, Figure 2C shows a dot blot analysis of the anti-H3K4me3 antibody using a commercially available histone H3 peptide array (Cat# 16-667; Millipore). This array contains 100 ng and 10 ng quantities of 46 different H3 peptides, each representing a specific H3 PTM—8 acetylated, 27 methylated, 5 phosphorylated, and 6 unmodified—thus allowing for the comprehensive specificity test (see Supplementary Figure S1 for more details). We find high specificity of the antibody for H3K4me3 and minimal cross-reactivity for H3K4me2. H3K4me1 and other modified or unmodified histones on the array are below the detection threshold. As the H3K4me3 antibody was produced in rabbit, Rabbit IgG serves as a positive control.
In addition to specificity, the antibody should be tested for efficacy. NChIP is performed in a native context, with immunoreactivity for the antibody (which typically is raised against synthetic, unfixed epitopes) largely preserved. However, some antibodies, regardless of vendor advertisements (ChIP-grade), show low efficiency in ChIP assays (24). Therefore, each new lot of antibody should undergo ChIP-qPCR to quantify levels of the histone PTM of interest at sequences known to be enriched (positive control) or void (negative control) for that particular PTM. Using the H3K4me3 antibody, nucleosomal preparations from PFC neurons show the expected enrichment at the proximal promoter of the N-methyl-D-aspartate receptor 2B subunit gene, GRIN2B, but not at the site of a negative control gene, beta globin (HBB) (Figure 2E). The efficacy of ChIP-seq experiments will be confirmed subsequently by sequencing. Ideally, there should be consistency between ChIP-qPCR (Figure 2E) and ChIP-seq (Figure 2G) H3K4me3 enrichments at the corresponding genomic sequences. In general, we recommend ChIP-qPCR pilot studies comparing two or more antibodies recognizing the same histone PTM side by side.
ChIP Quality Controls
Once the basic conditions are established and the experiments are ongoing, three tests provide ongoing information about the quality of each ChIP assay (Figures 2D, E). These tests include 1) Agilent Bioanalyzer of the input DNA (Figure 2D); 2) Qubit fluorometric assay (Thermo Fisher Scientific) concentration measurement of the input DNA and ChIP DNA product; and 3) qPCR of the input DNA and ChIP DNA product (Figure 2E). First, successful chromatin digestion, resulting in an overwhelming share of mononucleosomes, should be confirmed using Agilent Bioanalyzer (the trace will show the major DNA fragment around 150 bp) (Figure 2D). Second, the DNA quantity in the input and in the chromatin immunoprecipitate should be determined. We estimate that the starting material in our protocol (see Supplemental Detailed Protocol) amounts to 2 μg to 4 μg of nucleosomal DNA in the input, of which after pulldown with anti-H3K4me3 and anti-H3K27ac antibodies, up to 0.3% (H3K4me3) and 0.5% (H3K27ac) of the input DNA will be recovered when measured by Qubit fluorometric assay. Note that, potentially, only a small portion of the immunoprecipitated DNA is specifically associated with H3K4me3-tagged or H3K27ac-tagged nucleosomes. Indeed, occasionally the DNA quantity in the immunoprecipitate is low, and therefore ChIP DNA is not detectable by Qubit (concentrations <0.05 ng/μL or <2 ng ChIP DNA). These samples should not be removed from the pipeline provided they pass other QC steps, as they generally yield high-quality H3K4me3 (or H3K27ac) ChIP-seq libraries. Third, an important QC concerns the efficacy and specificity of the ChIP assay; this is confirmed using qPCR for sequences with known enrichment (e.g., the neuronal GRIN2B and DARPP32 promoter regions in neuronal chromatin that was immunoprecipitated with H3K4me3 antibody) and negative controls (e.g., HBB beta-globin locus) (Figure 2E). For example, for the GRIN2B promoter, we expect that neuronal, in comparison with nonneuronal nuclei, should show at a fourfold to eightfold higher enrichment after H3K4me3 and H3K27ac ChIP (Figure 2E, upper panel). In contrast, the HBB signal, since this is not a brain-expressed gene, is expected to be of similar strength as the GRIN2B signal in the input fraction but extremely weak or nondetectable in brain chromatin pulled down with anti-H3K4me3 and anti-H3K27ac antibodies (Figure 2E, lower panel). In our hands, the qPCR-based QCs are very informative. For instance, we regularly see consistent H3K4me3 enrichments when we compare ChIP-qPCR signals (Figure 2E) and the subsequent ChIP-seq tracks (Figure 2G) of the same samples at the analogous genomic sequences. Thus, in our pipeline, PFC NeuN+ and NeuN− samples that fail the ChIP-PCR test would not be further processed for library preparation and sequencing, as they are rarely successful.
Library Preparation
Following ChIP, library preparation is used to prepare DNA (extracted and purified from the chromatin immunoprecipitate) for sequencing. This includes the following steps: end of repair that produces blunt ends on DNA fragments, addition of the 3′ A tails to allow for ligation, adapter ligation, PCR amplification, and size selection. The adapters properly ligated to both ends of DNA are essential as they allow 1) binding of the library to the Illumina flow cell and, hence, sequencing; 2) PCR amplification of the library; and 3) indexing or barcoding of samples so multiple DNA libraries can be multiplexed into a single flow cell lane. The size of a ChIP-seq library depends on DNA fragment size and the adapter length, and in the case of MNase-treated chromatin, adapter-ligated DNA is expected to appear as a sharp peak around 275 bp (150 bp DNA fragment + 125 bp adapters length) (Figure 2F). Details of the library preparation procedure are provided in Supplemental Methods.
Library Preparation QC
The quality of sequencing data is dependent on the quality of library preparation. Two steps are performed to confirm the quality of the next generation sequencing library (Figure 2F): 1) confirmation of the main library product (275 bp) and the absence of the adapter dimer (125 bp) by Agilent Bioanalyzer; and 2) library concentration determination by Qubit. Only libraries that pass Agilent Bioanalyzer quality control and show Qubit measurements >2 ng/μL are further processed for sequencing.
Next Generation (Deep) Sequencing
Our samples are sequenced on HiSeq 2500 Illumina instruments. The following options should be considered when designing a deep sequencing experiment: sequencing depth and multiplexing, read length, and whether to perform single-end or paired-end (PE) sequencing. For detailed discussion on how to make these choices, please see Supplemental Methods. In our study, we aimed for 40 million and 80 million of uniquely mapped 100 bp PE reads for H3K4me3 (more restricted mark) and H3K27ac (broader mark), respectively. To achieve this, samples were sequenced in batches of eight (for H3K4me3) or four (for H3K27ac) samples per flow cell lane.
Sequencing Data Processing and QC
The development of an appropriate analytic pipeline is essential for the efficient processing and QC of the sequencing data. Whenever possible, batch effects should be controlled for at both experimental and computational levels (see Supplemental Methods for detailed discussion). Briefly, the initial processing and QC of the sequencing data includes the following steps: FASTQC analysis, alignment to the reference genome, peak calling, annotation analysis, and data visualization (Figure 2G, H) (25). FASTQC analysis is a fast and simple way to initially assess the quality of raw sequencing data (usually using FASTQ files) before doing any specific data processing (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) (Figure 2H, Supplemental Figure S2). Following the initial quality control, our data processing pipeline (Figure 3) begins with paired FASTQ files aligned to the Human Genome (version HG19) with BWA MEM (version 0.7.8) (26) generating sam files, subsequently converted to indexed and sorted bam files. Picard (version 1.112) MarkDups (http://broadinstitute.github.io/picard) is used to mark duplicates in the bam files and multi-mapped reads and improperly paired reads are filtered out with samtools view-f 2-F 2828-q1′ (version 1.1) (27). Macs2 (28) is used to call peaks from the duplicate marked, filtered bam with a q-value threshold of 0.01. Bigwig files are generated from the bedgraph files output by Macs2 and used for visualization in the Integrative Genomics Viewer (29). See Supplemental Methods for more details regarding data processing.
Figure 3.

Data processing pipeline. (A) Mapped reads—the number of reads mapped to the human genome, after filtering but including duplicate reads. (B) Number of peaks called for H3K4me3 and H3K27ac in two cell types (neuronal and nonneuronal). Distribution of H3K4me3 and H3K27ac around (C) annotated promoters and (D) intergenic genome portions. Encyclopedia of DNA Elements recommended quality metrics: (E) nonredundant fraction (NRF), (F) polymerase chain reaction bottleneck coefficient (PBC), and (G) relative strand correlation (RSC). (H) Bioinformatics pipeline. kb, kilobase.
RESULTS
In this section, we show representative examples from our ChIP-seq pipeline for neuronal and nonneuronal chromatin, assayed in parallel for two histone marks, H3K4me3 and H3K27ac. Data are presented from prefrontal cortex specimens of two subjects with no known history of neurological or psychiatric disease. Supplemental Table S1 provides an overview of the resulting ChIP-seq data from eight samples. As expected, H3K4me3 and H3K27ac ChIP-seq yielded approximately 40 million and 80 million of the aligned PE reads, respectively (Figure 3A). Greater sequencing depth is usually associated with more enrichment peaks (i.e., identified histone modification enrichment regions) (30). For H3K4me3, which is defined by a more narrow genomic distribution, 20 million single-end reads (equivalent to 40 million PE reads) approaches saturation with regard to number of peaks called (~ 90% in humans), with minimal gain achieved by additional sequencing (30). We obtained, on average, 40,014 peaks corresponding to H3K4me3-enriched regions in both the neuronal and nonneuronal fractions (Figure 3B). As expected, a large proportion of H3K4me3 peaks, or 42.1% for NeuN+ and 42.6% for NeuN−, mapped to promoter regions or ≤1 kb from the transcription start sites (Figure 3C), while a lower percentage (19.8% NeuN+ and 22.2% NeuN−) mapped to intergenic regions (Figure 3D). This is consistent with the H3K4me3 mark being primarily associated with active promoter regions (31). The results were different for H3K27ac, for which (in comparison with H3K4me3) a much larger number of peaks were called (Figure 3B), consistent with this histone modification having been associated with both active promoters and enhancers (32,33). This broader mark yielded, on average, 89,664 Macs2-called peaks (Figure 3B), with 17% (NeuN+) and 19.8% (NeuN−) of peaks mapped to promoter regions (Figure 3C) and close to 34.5% (NeuN+) and 23.2% (NeuN−) of peaks mapped to intergenic regions (Figure 3D).
Quality of the Postmortem Brain ChIP-seq Compared With ENCODE Standards
The ENCODE Consortium is an international collaboration of research groups building databases of regulatory elements in the human genome. While this project has not focused on the brain, it has developed extensive quality metrics. Several of ENCODE’s (34) quality metrics are readily applicable to our postmortem brain-derived ChIP-seq data. These include non-redundant fraction and polymerase chain reaction bottleneck coefficient as measures of sequencing library complexity (Figure 3E, F). Complex libraries represent more of the genome, with fewer duplicates (and therefore less noise). The nonredundant fraction is the fraction of reads that remain in a given library after removing duplicates (the ENCODE target is nonredundant fraction ≥0.8) (20). The polymerase chain reaction bottleneck coefficient is the fraction of regions that have been sequenced and contain no duplicates (the most complex library, random DNA, would approach 1.0; based on ENCODE, polymerase chain reaction bottleneck coefficients of 0.8–0.9 and 0.9–1.0 are mild and no bottlenecking, respectively) (34). ENCODE also recommends strand cross-correlation analysis, a measure of enrichment independent of peak calling (20), to assess the degree of immunoprecipitated fragments clustering in ChIP-seq experiments. Specifically, high-quality experiments produce a significant clustering of enriched DNA sequence tags at the locations abundant in the histone modification of interest. The main quality metrics derived from the cross-correlation analysis are 1) normalized strand coefficient; and 2) relative strand correlation (RSC) (Figure 3G). Based on the ENCODE criteria, high-quality ChIP-seq experiments will pass the following metrics: normalized strand coefficient > 1.05 and RSC > 0.8, which holds for 109 of the 127 samples we have processed thus far (Supplemental Table S2). Note that, as expected, our input samples—essentially mononucleosomal preparations from chromatin digested with MNase—do not show any enrichments, resulting in an RSC value approaching 0 [for detailed description and discussion on normalized strand coefficient and RSC, see (20)]. Moreover, sequencing of the input material is considered an important control to correct for potential confounds and bias associated with the ChIP-seq procedure, including amplification and sequencing (35). Taken together, these analyses suggest that our postmortem brain ChIP-seq pipeline generates data of sufficient quality to satisfy some of the common standards in the epigenomics field. Unfortunately, obtaining data for two independent biological replicates is not possible due to tissue limitations and cost when generating population-level data on hundreds of individuals from multiple epigenomic marks, so extensive reproducibility rates calculations cannot be performed.
Resolution of H3K4me3 and H3K27ac Maps in the Brain Using Cell-Specific ChIP-seq Profiling
As discussed above, epigenomic profiles carry a strong cell-type-specific component. Previously, we presented NChIP data on H3K4me3 landscapes in neuronal and nonneuronal nuclei from the prefrontal cortex that were generated with a limited number of 37 samples at lower sequencing depth and shorter read length (36 bp SE) (36–38). Using the current pipeline described here, we are producing ChIP-seq data on 500 samples, which will dramatically extend our earlier studies. To better illustrate the potential benefits of cell-type-specific chromatin assays in the postmortem human brain, we compared H3K4me3 and H3K27ac ChIP-seq data, obtained from neuronal and nonneuronal nuclear fractions of two samples (brain A, brain B, Figure 4), to homogenate H3K27ac ChIP-seq from the same brain region (PFC) derived from the National Institutes of Health Roadmap Epigenomics project (15). Figure 4 depicts the genomic region surrounding the GAD1 gene (encoding the gamma-aminobutyric acid synthesizing enzyme glutamic acid decarboxylase 67 [GAD67]) and the corresponding data for homogenate and sorted cortical tissue. The individual tracks demonstrate that H3K27ac landscapes in particular are cell-type-specific, while the corresponding H3K27ac track from tissue homogenate [downloaded from the National Institutes of Health Roadmap Epigenomics project (15)] appears to represent a combined signal of the H3K27ac track of the NeuN+ and NeuN− fractions. Remarkably, the aforementioned differences between neuronal and nonneuronal H3K27ac (and to a much lesser degree, H3K4me3) profiles are consistently observed across brain A and brain B, suggesting that some cell-type-specific differences in histone PTM landscapes may be more prominent as compared with differences between individuals (Figure 4). Preliminary analyses of the datasets from brain A and brain B suggest that only 50% of peaks, or less, are shared between the NeuN+ and NeuN− fractions. Furthermore, these data argue that cell-type-specific profiling may be critical for the discovery of disease-relevant epigenomic alterations.
Figure 4.

Cell-type-specific epigenomics. Browser tracks for eight chromatin immunoprecipitation followed by deep sequencing libraries. H3K4me3 and H3K27ac chromatin immunoprecipitation followed by deep sequencing tracks from prefrontal cortex (PFC) neuronal and nonneuronal nuclei of brains A and B, as indicated in purple and green, respectively. Tracks show 146 kilobase (kb) centered on GAD1 gamma-aminobutyric acid synthesis gene. Orange track, H3K27ac chromatin immunoprecipitation followed by deep sequencing from Roadmap Epigenomics (PFC tissue homogenate). Note cell-type-specific profiles, particularly for H3K27ac, while there is comparatively little variability between subjects.
DISCUSSION
Dissemination of data to the broader research community is essential for its full use beyond the goals of the original research group. Raw and processed data, analytical results, and code will be made available through the Sage Bionetworks’ (sagebase.org) Synapse system (www.synapse.org). Synapse enables transparency and reproducibility in research by providing easy access to data and metadata, provenance and versioning, and communication (Figure 5). The ChIP-seq data illustrated through this article will be disseminated through the PsychENCODE (www.synapse.org/pec) data release portal in Synapse. Additional complementary data generated on the hospital-based case-control cohort will be released through the PsychENCODE or CommonMind Consortium (www.synapse.org/cmc) data release portals. All data will be made available for users to incorporate into their own research programs and may be analyzed in combination with other data. We are committed to the release of data and results through these portals with the anticipation that results shared in a rapid and transparent manner will speed the pace of research to the benefit of both the PsychENCODE and CommonMind teams and the greater research community. It is our expectation that integrative analyses of transcriptome and epigenome maps generated from the same set of subjects and tissues (and eventually, cell types), in conjunction with other datasets including whole genome sequencing, will exponentially enhance our knowledge about the regulation of the human brain genome in health and disease.
Figure 5.
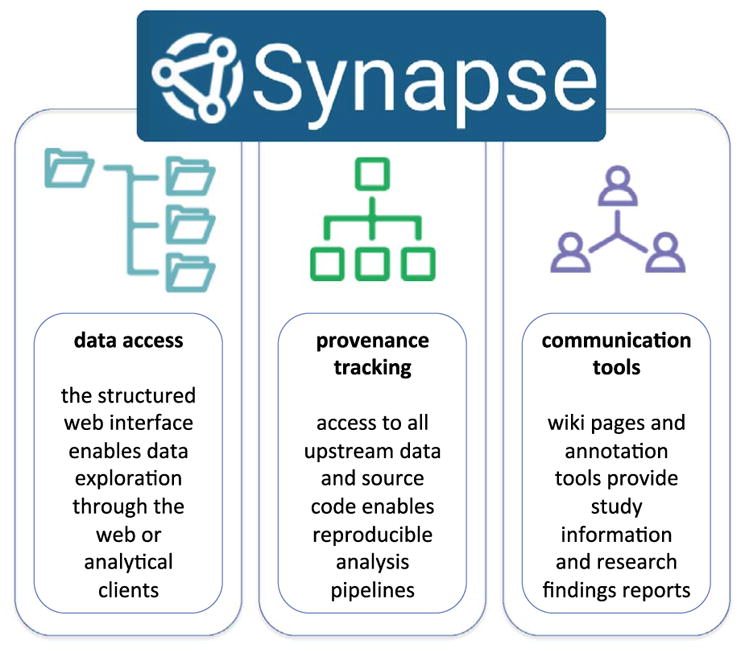
The Sage Bionetworks’ Synapse System. Data generated through this effort will be disseminated to the public through the Synapse system. Synapse enables easy data access, data provenance tracking to enable reproducibility of data processing and analytical output, and tools to communicate study information and research findings.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grant U01 MH103392. MK and YJ were supported, in part, by National Alliance for Research on Schizophrenia and Depression Young Investigator Grants from the Brain & Behavior Research Foundation.
We thank the PsychENCODE consortium for helpful input and discussion.
Footnotes
DISCLOSURES
The authors report no biomedical financial interests or potential conflicts of interest.
References
- 1.Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guo JU, Ma DK, Mo H, Ball MP, Jang MH, Bonaguidi MA, et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci. 2011;14:1345–1351. doi: 10.1038/nn.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sweatt JD. The emerging field of neuroepigenetics. Neuron. 2013;80:624–632. doi: 10.1016/j.neuron.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nestler EJ, Pena CJ, Kundakovic M, Mitchell A, Akbarian S. Epigenetic basis of mental illness [published online ahead of print October 8] Neuroscientist. 2015 doi: 10.1177/1073858415608147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grayson DR, Guidotti A. The dynamics of DNA methylation in schizophrenia and related psychiatric disorders. Neuropsychopharmacology. 2013;38:138–166. doi: 10.1038/npp.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang H-S, Matevossian A, Whittle C, Kim SY, Schumacher A, Baker SP, Akbarian S. Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J Neurosci. 2007;27:11254–11262. doi: 10.1523/JNEUROSCI.3272-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mill J, Tang T, Kaminsky Z, Khare T, Yazdanpanah S, Bouchard L, et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet. 2008;82:696–711. doi: 10.1016/j.ajhg.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morishita H, Kundakovic M, Bicks L, Mitchell A, Akbarian S. Interneuron epigenomes during the critical period of cortical plasticity: Implications for schizophrenia. Neurobiol Learn Mem. 2015;124:104–110. doi: 10.1016/j.nlm.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nestler EJ. Epigenetic mechanisms of depression. JAMA Psychiatry. 2014;71:454–456. doi: 10.1001/jamapsychiatry.2013.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaminsky Z, Tochigi M, Jia P, Pal M, Mill J, Kwan A, et al. A multi-tissue analysis identifies HLA complex group 9 gene methylation differences in bipolar disorder. Mol Psychiatry. 2012;17:728–740. doi: 10.1038/mp.2011.64. [DOI] [PubMed] [Google Scholar]
- 11.Shulha HP, Cheung I, Whittle C, Wang J, Virgil D, Lin CL, et al. Epigenetic signatures of autism: Trimethylated H3K4 landscapes in prefrontal neurons. Arch Gen Psychiatry. 2012;69:314–324. doi: 10.1001/archgenpsychiatry.2011.151. [DOI] [PubMed] [Google Scholar]
- 12.Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brunner AM, Tweedie-Cullen RY, Mansuy IM. Epigenetic modifications of the neuroproteome. Proteomics. 2012;12:2404–2420. doi: 10.1002/pmic.201100672. [DOI] [PubMed] [Google Scholar]
- 14.Houston I, Peter CJ, Mitchell A, Straubhaar J, Rogaev E, Akbarian S. Epigenetics in the human brain. Neuropsychopharmacology. 2013;38:183–197. doi: 10.1038/npp.2012.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roadmap Epigenomics C. Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;16:144–154. doi: 10.1038/nrm3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheung I, Shulha HP, Jiang Y, Matevossian A, Wang J, Weng Z, Akbarian S. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc Natl Acad Sci U S A. 2010;107:8824–8829. doi: 10.1073/pnas.1001702107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roussos P, Mitchell AC, Voloudakis G, Fullard JF, Pothula VM, Tsang J, et al. A role for noncoding variation in schizophrenia. Cell Rep. 2014;9:1417–1429. doi: 10.1016/j.celrep.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akbarian S, Liu C, Knowles JA, Vaccarino FM, Farnham PJ, Crawford GE, et al. The PsychENCODE project. Nat Neurosci. 2015;18:1707–1712. doi: 10.1038/nn.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furey TS. ChIP-seq and beyond: New and improved methodologies to detect and characterize protein-DNA interactions. Nat Rev Genet. 2012;13:840–852. doi: 10.1038/nrg3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Neill LP, Turner BM. Immunoprecipitation of native chromatin: NChIP. Methods. 2003;31:76–82. doi: 10.1016/s1046-2023(03)00090-2. [DOI] [PubMed] [Google Scholar]
- 23.Huang HS, Matevossian A, Jiang Y, Akbarian S. Chromatin immunoprecipitation in postmortem brain. J Neurosci Methods. 2006;156:284–292. doi: 10.1016/j.jneumeth.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 24.Egelhofer TA, Minoda A, Klugman S, Lee K, Kolasinska-Zwierz P, Alekseyenko AA, et al. An assessment of histone-modification antibody quality. Nat Struct Mol Biol. 2011;18:91–93. doi: 10.1038/nsmb.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bailey T, Krajewski P, Ladunga I, Lefebvre C, Li Q, Liu T, et al. Practical guidelines for the comprehensive analysis of ChIP-seq data. PLoS Comput Biol. 2013;9:e1003326. doi: 10.1371/journal.pcbi.1003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jung YL, Luquette LJ, Ho JWK, Ferrari F, Tolstorukov M, Minoda A, et al. Impact of sequencing depth in ChIP-seq experiments. Nucleic Acids Res. 2014;42:e74. doi: 10.1093/nar/gku178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 32.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karlić R, Chung H-R, Lasserre J, Vlahoviček K, Vingron M. Histone modification levels are predictive for gene expression. Proc Natl Acad Sci U S A. 2010;107:2926–2931. doi: 10.1073/pnas.0909344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park PJ. ChIP-seq: Advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheung I, Shulha HP, Jiang Y, Matevossian A, Wang J, Weng Z, Akbarian S. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc Natl Acad Sci U S A. 2010;107:8824–8829. doi: 10.1073/pnas.1001702107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shulha HP, Cheung I, Guo Y, Akbarian S, Weng Z. Coordinated cell type-specific epigenetic remodeling in prefrontal cortex begins before birth and continues into early adulthood. PLoS Genet. 2013;9:e1003433. doi: 10.1371/journal.pgen.1003433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shulha HP, Cheung I, Whittle C, Wang J, Virgil D, Lin CL, et al. Epigenetic signatures of autism: Trimethylated H3K4 landscapes in prefrontal neurons. Arch Gen Psychiatry. 2012;69:314–324. doi: 10.1001/archgenpsychiatry.2011.151. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.