

Abstract
Therapeutic targeting of ASCT2, a glutamine transporter that plays a major role in glutamine uptake in cancer cells, is challenging because ASCT2 also has a biological role in normal tissue. In this study, we report our novel finding that ASCT2 is physically associated in a molecular complex with epidermal growth factor receptor (EGFR), which is often overexpressed in human head and neck squamous cell carcinoma (HNSCC). Further, we found that ASCT2 can be co-targeted by cetuximab, an EGFR antibody approved for treating metastatic HNSCC. We demonstrated that cetuximab downregulated ASCT2 in an EGFR expression-dependent manner via cetuximab-mediated EGFR endocytosis. Downregulation of ASCT2 by cetuximab led to decreased intracellular uptake of glutamine and subsequently a decreased glutathione level. Cetuximab thereby sensitized HNSCC cells to reactive oxygen species (ROS)-induced apoptosis and, importantly, it is independent of effective inhibition of EGFR downstream signaling by cetuximab. In contrast, knockdown of EGFR by siRNA or inhibition of EGFR kinase with gefitinib, an EGFR kinase inhibitor, failed to sensitize HNSCC cells to ROS-induced apoptosis. Our findings support a novel therapeutic strategy for EGFR-overexpressing and cetuximab-resistant cancers by combining cetuximab with an oxidative therapy.
Keywords: ASCT2, EGFR, ROS, glutamine, cetuximab, HNSCC
1. Introduction
The solute-linked carrier family A1 member 5 (SLC1A5) encodes a Na+-dependent neutral amino acid transporter, ASCT2. ASCT2 is a glutamine transporter that plays a major role in glutamine uptake in rapidly proliferating cells, including cancer cells [1,2]. Glutamine not only is a building block for protein synthesis and an important nitrogen source for synthesis of purine and pyrimidine bases required for unlimited proliferation of cancer cells, but also participates in several other important biological processes in cells, such as glutaminolysis and glutathione synthesis, both of which processes play key roles in cancer cell metabolism [3,4]. ASCT2, therefore, could be an important target for cancer treatment. However, effective targeting of ASCT2 for cancer therapy is technically challenging because ASCT2 is also expressed in normal tissues and also plays an important role in normal cell metabolism.
The strategy of raising intracellular levels of reactive oxygen species (ROS) to cytotoxic levels selectively in cancer cells, leading to induction of apoptosis as an anticancer therapy, has been extensively investigated for many years [5,6]. One of the agents that has been investigated for this strategy is dichloroacetic acid (DCA), a US Food and Drug Administration (FDA)-approved drug that has been used to treat a rare hereditary lactate metabolism disorder in children for over 30 years [7,8]. Intracellular ROS are generated mainly as a byproduct of oxidative phosphorylation in the mitochondria [9]; DCA has been reported to induce ROS in cancer cells through inhibiting the mitochondrial enzyme pyruvate dehydrogenase (PDH) kinase 1 (PDK1) [10,11]. Inhibition of PDK1, through activation of PDH, redirects the glucose-derived pyruvate for oxidative phosphorylation through the tricarboxylic acid cycle, which can result in overproduction of ROS in the mitochondria, leading to cancer cell death via apoptosis [10]. DCA has been investigated in clinical trials as a PDK1 inhibitor for treatment of cancer [12–15]. However, the intrinsic resistance of cancer cells to ROS-induced apoptosis can significantly affect the antitumor activity of DCA in cancer cells.
Over 90% of head and neck squamous cell carcinomas (HNSCCs) express high levels of epidermal growth factor receptor (EGFR). Cetuximab, an EGFR antibody that blocks ligand-induced EGFR activation in targeted cancer cells, is approved by the FDA for treatment of metastatic HNSCC in combination with conventional chemotherapy or radiation therapy [16]. However, frequent oncogenic mutations of key molecules in EGFR downstream signaling pathways and/or cross-activation of EGFR downstream signaling pathways by receptor tyrosine kinases other than EGFR render many HNSCCs insensitive to cetuximab, which is a major clinical challenge [17,18]. Further investigation to improve the efficacy of cetuximab in HNSCC and other cancers is strongly warranted.
In this paper, we report the novel finding that ASCT2 is physically associated with EGFR in a molecular complex that can be internalized by cetuximab via cetuximab-induced EGFR endocytosis independently of effective inhibition of EGFR downstream cell signaling by cetuximab. Because ASCT2 mediates intracellular uptake of glutamine and of cysteine, and the latter is rate-limiting for biosynthesis of glutathione, a major cellular antioxidant [19,20], such a novel finding could create a unique opportunity to enhance oxidative therapy against cancer by co-targeting ASCT2 using cetuximab. In studies to test this hypothesis, we found that cetuximab could diminish cancer cells’ antioxidant defense via downregulating ASCT2 and thereby sensitize cancer cells to ROS-induced apoptosis. Our findings justify further testing this novel therapeutic strategy in preclinical and clinical studies.
2. Materials and methods
2.1 Cell lines and cell culture
HNSCC cell lines (FaDu, HN5, HN30, MDA1986, UMSCC1, UMSCC2, UMSCC22A, UMSCC22B, and TU167) were maintained in Dulbecco’s modified Eagle’s medium (DMEM)/F12 medium supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin under conditions of 5% CO2 at 37°C in an incubator.
2.2 Reagents
Cetuximab is manufactured by ImClone Systems, an Eli Lilly company. Gefitinib is a product of AstraZeneca. DCA and all other chemicals were purchased from Sigma-Aldrich Co. unless otherwise specified.
2.3 cDNA construct, siRNA duplexes, and transfection
siRNA oligonucleotide duplexes for PDK1 and EGFR were purchased from Sigma-Aldrich Co. The DNA-targeting sequences of siRNAs were as follows: PDK1: sequence 1, GGATGAAATTGCACCTATT; sequence 2, GTCCAGGAGACTGTGTCAT; sequence 3, TGCTAGGCGTCTGTGTGAT. EGFR: sequence 1, GAGGAAATATGTACTACGA; sequence 2, CTATGTGCAGAGGAATTAT. We used ON-TARGETplus SMART pool siRNA oligonucleotide (Dharmacon-Thermo Fisher Scientific) for silencing Rab5 and Rab11.
The cDNA constructs and the siRNA oligonucleotides were transfected into the targeted cells with Lipofectamine 2000 (Life Technologies), according to the manufacturer’s instructions.
2.4 Western blotting and immunoprecipitation
Cell lysates were prepared as we previously reported [21,22]. For Western blotting, whole-cell lysates or cytoplasmic- and membrane-fractionated lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and subjected to Western blotting analysis with primary antibodies and horseradish peroxidase-labeled secondary antibodies. The signals were visualized using the enhanced chemiluminescence detection kit (Amersham Biosciences). For immunoprecipitation studies, equal amounts (800–1000 μg of lysate proteins) of cell lysates were incubated with the complex of primary antibody and protein A-Sepharose beads (Amersham Biosciences) at 4°C for overnight, and the resulting immunoprecipitates were then subjected to immunoblotting analysis with various primary antibodies described in the figure legends.
The antibodies used for Western blotting and immunoprecipitation were all commercially available and purchased from the following vendors: Cell Signal Technology (antibodies against ASCT2 [D7C12], EGFR [D38B1], EGFR-Y1068p, Akt-S473p, total Akt, Ras, PDH, PARP, caspase-3, and cleaved caspase-3), Santa Cruz Biotechnology (antibodies against ASCT2 [H- 52]), Abcam (antibodies against Rab5 and Rab11), Enzo Life Sciences (antibody against PDK1), Novus Biologicals (antibody against S293-phosphorylated PDH), and Sigma-Aldrich Co. (antibodies against EGFR [F4] and β-actin).
2.5 Duolink proximity ligation assay
For Duolink proximity ligation (PLA) assay, cells cultured on coverslips were fixed with cold methanol for 15 min and permeabilized with 0.1% triton-X-100 for 15 min. Then cells grown on the coverslips were incubated with blocking solution provided in the Duolink detection kit for 1 h at room temperature and incubated with EGFR and ASCT2 antibodies, together or individually, at room temperature for 2 h. The proximity ligation reaction and visualization of signals were performed by using the species-specific PLA PLUS and MINUS probe-conjugated secondary antibodies provided in the Duolink detection kit and performing additional steps according to the protocol provided by the manufacturer (Enzo Life Sciences-Axxora). The cells grown on the coverslips were mounted on glass slides with mounting medium containing DAPI (for nuclear counterstain) for observation under a fluorescence microscope [23,24].
2.6 Cytoplasmic and membrane fractionation
Cell pellets were collected and lysed with cold hypotonic lysis buffer containing 25 mM sucrose, 5 mM Tris-HCl, and 0.5 mM MgCl2. After incubation on ice for 20 min, cells were homogenized with 40 strokes in a Dounce homogenizer. Cells were spun at 2,700 rpm for 20 min at 4°C, and the supernatant was collected and subjected to spinning at 14,400 rpm for 20 min at 4°C. The supernatant (cytoplasmic fraction) and pellet (membrane fraction) were separated, and the pellet was resuspended with PBS. The fractionated cytoplasmic and membrane samples were subjected to analysis with immunoblotting [25].
2.7 Glutamine uptake assay
Glutamine uptake was measured in cell suspension in an Eppendorf tube after trypsinization of cells, resuspension of the cells with 0.5 mL of glutamine-deficient medium containing 5 μCi/mL 3H-labeled glutamine (Perkin Elmer), and incubation at 37°C for 5 min. After the incubation, cells were spun down and washed three times with ice-cold PBS, after which cells were lysed with 200 μL of a 0.2% SDS/0.2 N NaOH solution, incubated for 1 h, neutralized with 40 μL of 1 N HCl, and analyzed with a Beckman LS6500 scintillation counter. Relative glutamine uptake in cells of treated groups was expressed as percentage of the glutamine uptake in cells of the untreated group [26,27].
2.8 Intracellular glutathione assay
Intracellular glutathione was measured using a glutathione assay kit purchased from Cayman Chemical [28]. After cells were harvested using a rubber policeman, cell pellets were collected by centrifugation, resuspended in 500 μL of MES buffer (provided in the kit), and sonicated. Following removal of cell debris by centrifugation at 13,000 rpm for 15 min at 4°C, the supernatant was deproteinated by precipitation with an equal volume of 10% metaphosphoric acid and centrifuged again at 5,000 rpm for 5 min. The cleared supernatant was neutralized with 0.2 M triethanolamine. An aliquot of each sample was transferred to a well of a 96-well microplate for detecting total glutathione according to the manufacturer’s recommended protocol, which is based on the reactions catalyzed by glutathione reductase to convert GSSG to GSH and to produce a yellow product that can be quantified at 405 nm using a microplate reader after the reaction of GSH with 5,5′-dithio-bis-2-nitrobenzoic acid (DTNB).
2.9 Quantitative apoptosis ELISA
Induction of apoptosis was quantified by using the Cell Death Detection ELISA (Roche Diagnostics Corp.), which quantitatively measures cytoplasmic histone-associated DNA fragments (mononucleosomes and oligonucleosomes). The ELISA was performed exactly according to the manufacturer’s instructions.
2.10 ROS detection
Intracellular ROS were detected by using the total ROS detection kit (Enzo Life Sciences) according to the manufacturer’s instructions [10]. Briefly, cells were seeded in a 12-well plate at around 40–60% confluency and then treated. At the end of treatment, cells were stained with ROS detection solution at 37°C for 1 h and then observed under a fluorescence microscope.
3. Results
3.1 ASCT2 is physically associated with EGFR in a molecular complex
We first performed co-immunoprecipitation of ASCT2 and EGFR using HN5 cells, which express high levels of both ASCT2 and EGFR. Western blotting with H-52, an ASCT2 antibody that recognizes a peptide sequence near the C-terminus of ASCT2, detected ASCT2 in a complex immunoprecipitated by cetuximab (Figure 1A, left). Conversely, Western blotting detected EGFR in a complex immunoprecipitated by D7C12, an ASCT2 antibody that recognizes a peptide sequence near the N-terminus of ASCT2. To rule out any potential nonspecific activities of H-52 and D7C12, we performed reciprocal experiments by exchanging the ASCT2 antibodies used for immunoprecipitation and Western blotting and using different EGFR antibodies for Western blotting. The results of these reciprocal experiments further confirmed the association between ASCT2 and EGFR (Figure 1B).
Figure 1.
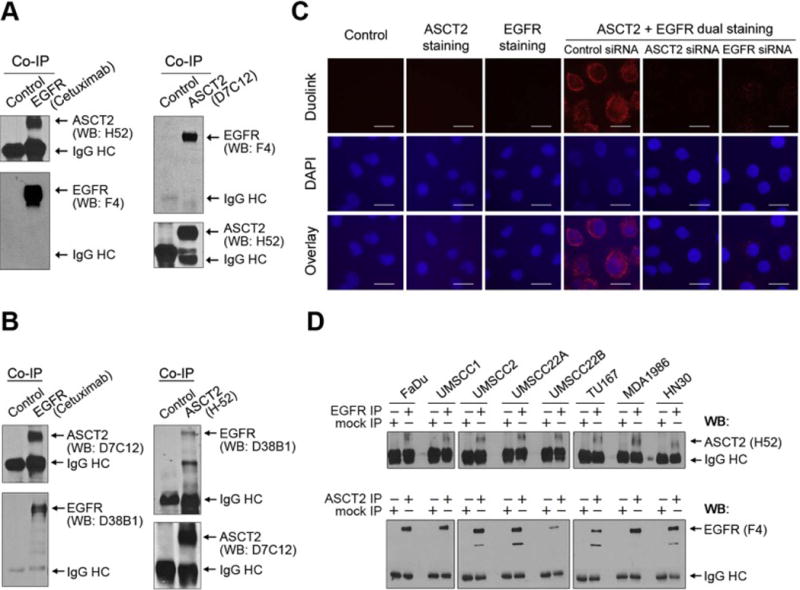
ASCT2 is physically associated with EGFR.
(A) HN5 cell lysates were subjected to EGFR immunoprecipitation (IP) with cetuximab or a control antibody, or subjected to ASCT2 immunoprecipitation with an anti-ASCT2 rabbit monoclonal antibody (D7C12, Cell Signaling) or a control rabbit anti-mouse IgG antibody, followed by Western blotting (WB) of the immunoprecipitates with an anti-ASCT2 rabbit polyclonal antibody (H-52, Santa Cruz Biotechnology) and with an anti-EGFR mouse monoclonal antibody (F4, Sigma-Aldrich), respectively. HC, heavy chain. (B) ASCT2 co-immunoprecipitation by cetuximab was validated by Western blotting using different ASCT2 (D7C12) and EGFR (D38B1, Cell Signaling) antibodies as indicated. (C) HN5 cells were untransfected or transfected with control siRNA, ASCT2 siRNA, or EGFR siRNA for 72 h before being plated on coverslips for overnight. The cells were then incubated with blocking buffer only or with an ASCT2 antibody (H-52) and an EGFR antibody (F4), alone and together, as indicated, and then subjected to Duolink proximity ligation assay as described in Materials and methods. Scale bars, 25 μm. (D) Lysates from the indicated HNSCC cell lines were subjected to EGFR immunoprecipitation with cetuximab or ASCT2 immunoprecipitation with an ASCT2 antibody (H-52), along with a control IgG immunoprecipitation. The immunoprecipitates were then analyzed by Western blotting with the indicated antibodies.
We next examined whether ASCT2 and EGFR are associated in close proximity by using the Duolink proximity ligation assay (PLA), which can detect proteins in a complex less than 40 nm from one another in cells in situ [23,24]. Dual staining of HN5 cells with EGFR and ASCT2 antibodies, but not staining with either antibody alone, resulted in the appearance of fluorescence after addition of PLA probe-conjugated secondary antibodies, indicating ASCT2-EGFR interaction. Knockdown of ASCT2 or EGFR led to a significant decrease in the signal detected by Duolink PLA (Figure 1C). ASCT2-EGFR association was also confirmed by the Duolink PLA in UMSCC1, an HNSCC cell line that expresses much less EGFR than HN5 cells do (Supplementary Figure 1). We next examined the association of ASCT2 and EGFR in an additional eight HNSCC cell lines and confirmed that ASCT2-EGFR association could be detected by co-immunoprecipitation in all the HNSCC cell lines examined (Figure 1D).
As mentioned above, ASCT2 is a glutamine transporter that plays a major role in glutamine uptake, and one of the roles of glutamine after uptake is to participate in the biosynthesis of glutathione, a major component of cellular defenses against oxidative stresses. In a related work that will be reported elsewhere, we found that the levels of ASCT2 expression and glutamine uptake were increased upon exposure to ROS. We therefore examined whether the level of ASCT2-EGFR association was also increased in cells under oxidative stress. As shown in Figure 2, the level of ASCT2-EGFR association was increased in HN5 cells after treatment with H2O2 or with DCA. This interesting finding suggests a role of ASCT2-EGFR association in defending cells against ROS-induced oxidative damage.
Figure 2.
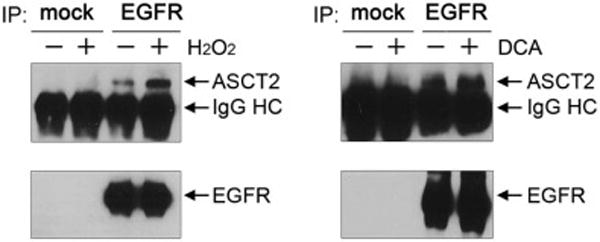
ROS stimulation leads to a higher level of ASCT2-EGFR association.
HN5 cells were untreated or treated with 1 mM H2O2 or 10 mM DCA as indicated for overnight (16 h). Cell lysates were subjected to immunoprecipitation (IP) for EGFR by cetuximab or control IgG, followed by Western blotting with antibodies against ASCT2 (H-52) and EGFR (F4), respectively. HC, heavy chain.
Together, these data strongly indicate that at least some ASCT2 and EGFR proteins are physically associated in a molecular complex that may play a role in cancer response to oxidative stress.
3.2 Cetuximab downregulates ASCT2 via cetuximab-mediated EGFR endocytosis, leading to inhibition of glutamine uptake and decreased intracellular glutathione level
Upon its binding to EGFR, cetuximab can induce EGFR internalization via endocytosis. Because we found that ASCT2 and EGFR are associated in a molecular complex in close proximity, we hypothesized that the EGFR-associated ASCT2 can be co-internalized via cetuximab-induced EGFR endocytosis, leading to decreases in glutamine uptake and glutathione synthesis, and thereby cetuximab can render HNSCC cells susceptible to ROS-induced apoptosis. To test our hypothesis, we analyzed the subcellular location of ASCT2 and EGFR in HN5 cells after treatment with cetuximab for overnight. As shown in Figure 3A, in untreated cells, both EGFR and ASCT2 were located primarily in the membrane fraction (compare lanes 1 and 3); treatment with cetuximab, however, led to detection of EGFR and ASCT2 in the cytoplasmic fraction (compare lanes 3 and 4), indicating that ASCT2 was co-internalized as a result of cetuximab-induced endocytosis of the ASCT2-EGFR complex.
Figure 3.
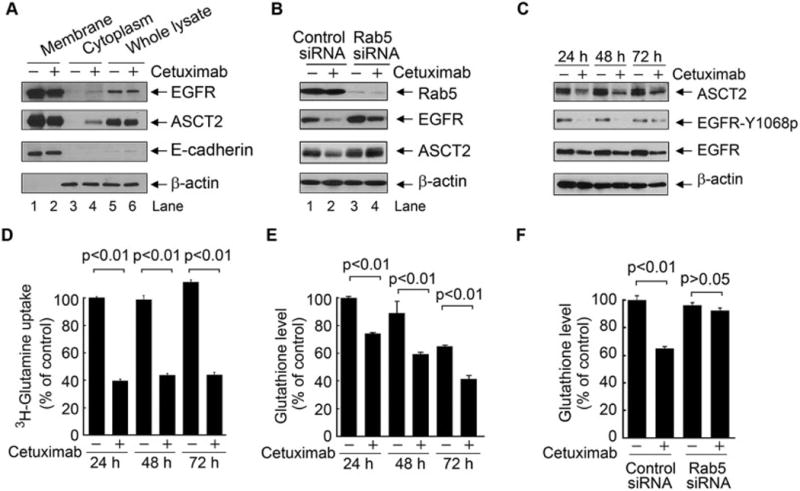
Cetuximab downregulates ASCT2 via cetuximab-mediated EGFR endocytosis, leading to inhibition of glutamine uptake and decreased glutathione synthesis.
(A) HN5 cells were treated with or without 20 nM cetuximab for 10 h. Fractions of cell membrane (indicated by the presence of E-cadherin, a membrane marker) and cytoplasm (indicated by the presence of β-actin, a cytoplasmic marker) were analyzed for Western blotting with the indicated antibodies. (B) HN5 cells were subjected to knockdown of Rab5 for 72 h. Cell lysates were subjected to Western blotting with the indicated antibodies. (C–E) HN5 cells were untreated or treated with 20 nM cetuximab for the indicated times. Cell lysates were subjected to Western blotting with the indicated antibodies (C). 3H-labeled glutamine uptake was determined as described in Materials and methods (D), and glutathione level was measured using the Cayman glutathione assay kit (E). (F) HN5 cells were subjected to knockdown of Rab5 for 72 h as in (B). Glutathione level was measured using the Cayman glutathione assay kit as in (D). All error bars, S.D.
That co-internalization of ASCT2-EGFR complex through cetuximab-induced EGFR endocytosis was further confirmed by our finding that knockdown of Rab5, a regulatory protein needed in biogenesis of early endosomes [29], could inhibit cetuximab-induced ASCT2-EGFR internalization and downregulation (compare lanes 2 and 4 in Figure 3B).
As shown in Figure 3C, the level of ASCT2 was persistently downregulated after extended cetuximab treatment, as were levels of total EGFR (due to EGFR internalization) and Y1068-phosphorylated EGFR (due to inhibition of the EGFR autocrine stimulation loop). As expected, the downregulation of ASCT2 was accompanied by a decreased intracellular uptake of glutamine (Figure 3D) and a decreased intracellular level of glutathione (Figure 3E) in cetuximab-treated cells compared to untreated cells. Knockdown of Rab5 largely abolished cetuximab-induced decrease in glutathione level, which confirmed that ASCT2-EGFR endocytosis was required for the decrease in glutathione level (Figure 3F).
On the basis of the findings in Figures 1 through 3, we conclude that ASCT2 is an EGFR-associated protein that can be downregulated via cetuximab-induced EGFR endocytosis, leading to inhibition of glutamine uptake and decreased intracellular glutathione level.
3.3 Cetuximab sensitizes HNSCC cells to ROS-elevating agents to induce apoptosis in an EGFR-expression-dependent manner
We next explored whether the combination of cetuximab with DCA, which induces ROS via inhibiting PDK1 [10,11], sensitizes cancer cells to DCA-induced apoptosis. As shown in Figure 4A, whereas cetuximab alone and DCA alone failed to induce a significant level of apoptosis in HN5 cells, the combination of cetuximab with DCA did induce apoptosis, indicated by appearance of PARP cleavage, a marker of apoptosis. By contrast, combination of EGFR knockdown with DCA did not induce apoptosis in HN5 cells (Figure 4A). Moreover, knockdown of EGFR expression abolished the apoptosis-inducing effect of cetuximab plus DCA (Figure 4A), indicating that EGFR expression was required for the induction of apoptosis by cetuximab plus DCA. In addition, knockdown of Rab5, but not knockdown of Rab11 (a regulatory protein involved in recycling endosomes), inhibited apoptosis induced by cetuximab plus DCA in both HN5 and FaDu cells (Figure 4B). Together, these data indicate that expression of EGFR on the cell surface is required for induction of apoptosis by cetuximab plus DCA.
Figure 4.
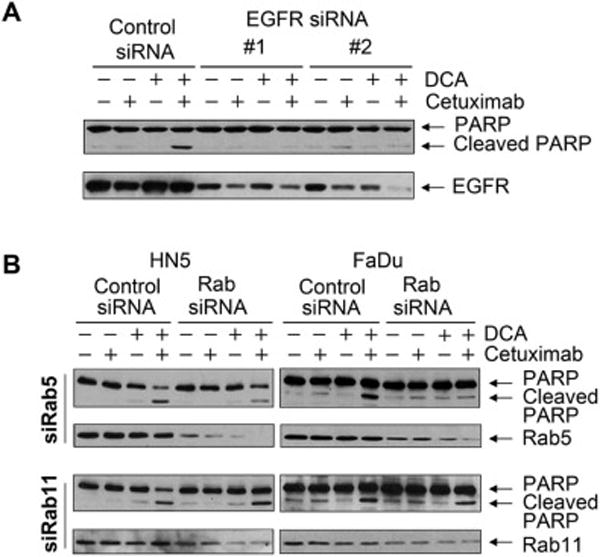
Cetuximab sensitizes cancer cells to ROS-elevating agent-induced apoptosis in an EGFR-expression-dependent manner.
(A) HN5 cells were subjected to knockdown with each of two different EGFR siRNAs or control siRNA for 72 h. During the last 24 h of siRNA transfection, the cells were either untreated or treated with 10 mM DCA, 20 nM cetuximab, or both. Cell lysates were subjected to Western blotting with the indicated antibodies. (B) HN5 and FaDu cells were subjected to knockdown of Rab5 or Rab11 with specific siRNAs or control siRNA for 72 h. During the last 24 h of siRNA transfection, the cells were either untreated or treated with 10 mM DCA, 20 nM cetuximab, or both. Cell lysates were subjected to Western blotting with the indicated antibodies.
3.4 Gefitinib plus DCA fails to induce apoptosis
To confirm that cetuximab-induced EGFR internalization, rather than cetuximab-induced inhibition of EGFR kinase, led to induction of apoptosis by the combination of cetuximab with DCA, we examined whether gefitinib, a small molecule EGFR kinase inhibitor that does not induce EGFR internalization, can induce apoptosis in HN5 cells when used in combination with DCA. As shown in Figure 5A, both cetuximab and gefitinib clearly inhibited EGFR kinase activity, and gefitinib had a greater inhibitory effect at the doses used in the experiment, evidenced by a greater reduction in the level of EGFR Y1068 autophosphorylation. However, whereas cetuximab plus DCA clearly induced apoptosis, gefitinib plus DCA did not, as measured by Western blotting detection of cleaved PARP and caspase 3 (Figure 5A) and by an apoptosis ELISA (Figure 5B).
Figure 5.
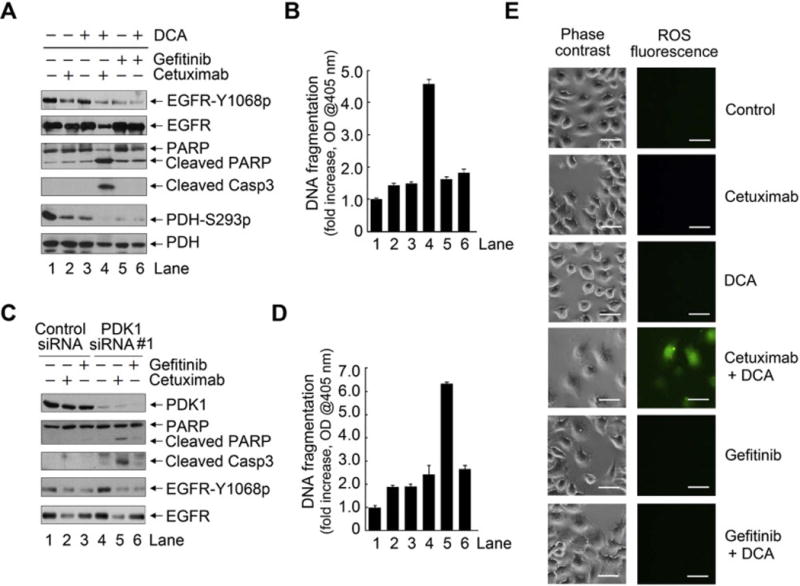
Combination of cetuximab with DCA, but not combination of gefitinib with DCA, induces ROS and apoptosis in HN5 cells.
(A) and (B) HN5 cells were subjected to knockdown of PDK1 followed by treatment with 0.2 μM gefitinib or 20 nM cetuximab for 24 h. (C) and (D) HN5 cells were co-treated with 10 mM DCA plus 0.2 μM gefitinib or 20 nM cetuximab for 24 h. (E) HN5 cells were left untreated or treated with 20 nM cetuximab or 0.2 μM gefitinib, each alone and in combination with 10 mM DCA as indicated. The cells were stained with Enzo’s ROS detection kit and observed under a microscope. Scale bars, 50 μm.
Moreover, as shown in Figure 5A, both cetuximab alone and DCA alone inhibited PDK1 activity, shown by decreased level of PDH phosphorylation on serine 293, and the combination of cetuximab with DCA achieved greater inhibition of PDH phosphorylation. Gefitinib alone and gefitinib plus DCA produced greater inhibition of PDH phosphorylation than cetuximab or cetuximab plus DCA but failed to induce apoptosis. This observation indicates that the apoptosis induced by cetuximab plus DCA was not simply due to inhibition of EGFR or PDK1.
To provide further supportive data, we knocked down the expression of PDK1 in lieu of the effect of DCA treatment. We found that cetuximab plus PDK1 knockdown, but not gefitinib plus PDK1 knockdown, induced apoptosis (Figure 5C and 5D). Further, as shown in Figure 5E, ROS overproduction was detected only with the combination of cetuximab and DCA, not with the combination of gefitinib and DCA, in HN5 cells. Similar results were found in FaDu, another HNSCC cell line (Supplementary Figure 2).
3.5 Cetuximab plus PDK1 knockdown induces apoptosis in cetuximab-resistant HNSCC cells
Last, to further confirm that effective inhibition of EGFR downstream pathways was not required for the induction of apoptosis by cetuximab plus PDK1 inhibition, we transfected a constitutively active Ras mutant (H-RasG12V) into HN5 cells and examined the effect of combination treatment on induction of apoptosis. Overexpression of H-RasG12V conferred strong resistance to cetuximab-induced inhibition of EGFR downstream pathways, as indicated by phosphorylation of Akt and p70S6K (Figure 6A); however, H-RasG12V overexpression had no impact on induction of apoptosis by cetuximab plus PDK1 knockdown with any of three different PDK1 siRNAs as measured by cleavage of PARP and caspase 3 (Figure 6A) and by an apoptosis ELISA (Figure 6B).
Figure 6.
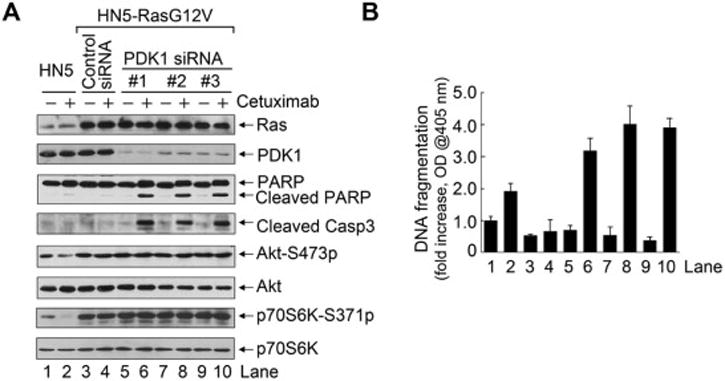
Cetuximab plus DCA induces apoptosis in cetuximab-resistant HNSCC cells.
(A) and (B) HN5 cells and HN5 cells transfected with a constitutively active Ras (H-RasG12V) were subjected to treatment with PDK1 siRNA or control siRNA with or without 20 nM cetuximab as indicated for 24 h. Lysates of HN5 cells treated as indicated were subjected to Western blotting with the indicated antibodies (A) and to ELISA for quantification of apoptosis (B).
4. Discussion
In this paper, we report a novel finding that ASCT2, an important transporter of neutral amino acids, including glutamine, is physically associated with EGFR in close proximity so that ASCT2 can be co-downregulated by cetuximab via cetuximab-induced EGFR endocytosis. This downregulation of ASCT2 by cetuximab leads to decreased uptake of glutamine and a resultant decrease in glutathione synthesis that renders cetuximab-treated HNSCC cells more susceptible than cetuximab-untreated cells to ROS-induced apoptosis. Importantly, this effect is independent of effective inhibition of EGFR downstream cell signaling.
Our findings are of potential clinical significance. First, although glutamine is a nonessential amino acid in normal cells, the demand for glutamine is dramatically increased in cancer cells to support cancer metabolism throughout malignant transformation. Glutamine also plays a role in cell signaling, such as maintaining sustained activation of mTROC1 signaling [30]. Inhibition of ASCT2-mediated glutamine uptake by pharmacological inhibitors of ASCT2 or by shRNA-mediated knockdown of ASCT2 has been shown to successfully inhibit cancer cell growth and proliferation in multiple tumor types, including non-small cell lung cancer, prostate cancer, breast cancer, and melanoma [26,31–34]. However, ASCT2 also plays an important role in normal cell survival and proliferation, which may limit therapeutic approaches to target ASCT2 directly. Our finding that ASCT2 and EGFR are physically associated in a molecular complex provides an alternative approach to co-target ASCT2 with cetuximab in EGFR-overexpressing HNSCC cells.
Second, our findings suggest a new way to improve the efficacy of oxidative therapy for cancer. The strategy of treating cancer by raising intracellular levels of ROS to cytotoxic levels in cancer cells is well justified; however, development of the strategy has been challenging because cancer cells are inherently strong in antioxidant capacity that can maintain a homeostatic balance between formation and removal of ROS [35,36]. Our findings showed that while neither cetuximab-mediated downregulation of ASCT2 nor DCA treatment on its own induced apoptosis, cetuximab-mediated ASCT2 downregulation did sensitize HNSCC cells to induction of apoptosis by DCA through inhibition of PDK1.
Third, our findings suggest a potential strategy to the problem of cetuximab-resistant tumors. Resistance to cetuximab, caused by various mechanisms, is currently a major clinical challenge in the treatment of EGFR-overexpressing tumors. Our finding that the combination of cetuximab with DCA, an ROS-elevating agent that has been approved by the FDA for noncancer diseases, can induce apoptosis independently of effective inhibition of EGFR downstream signaling offers new hope for treating cetuximab-resistant tumors.
Last, cetuximab is currently approved for treating patients with HNSCC in combination with radiation therapy or cisplatin. It has been well documented that both ionizing radiation and cisplatin can induce a high level of ROS, which contribute to the antitumor activity of these therapies [37,38]. It will be interesting, therefore, to determine whether the therapeutic benefits observed in patients treated with the combination of cetuximab with radiation or cisplatin are achieved, in whole or in part, via a mechanism similar to the one we observed with cetuximab plus DCA, i.e., cetuximab downregulates ASCT2 in tumors, thereby sensitizing cancer cells to radiation- or cisplatin-induced ROS. A retrospective study comparing the level of ASCT2 in cetuximab-treated patients with tumors responsive and nonresponsive to cetuximab plus radiation or cisplatin may yield useful information regarding this hypothesis.
A few key biological questions remain to be explored regarding the ASCT2-EGFR association we report in this study. First, our data showed that the extent of ASCT2-EGFR association was increased when cells were under oxidative stress, suggesting that the association has a role in maintaining oxidative homeostasis in cancer cells. However, the biological significance of ASCT2-EGFR association needs to be further explored. Second, our data indicated that ASCT2 is physically associated with EGFR in close proximity and can be co-internalized by cetuximab-induced EGFR endocytosis; however, we cannot rule out the possibility that another protein or proteins are involved in ASCT2-EGFR association. It will be important to investigate how the association between ASCT2 and EGFR is regulated at a molecular level. Third, in the current study, we found that cetuximab sensitized cells to DCA, which induces overproduction of ROS through inhibiting PDK1. It will be interesting to test whether cetuximab can sensitize cancer cells to other agents that elevate ROS via different mechanisms.
In conclusion, we found that ASCT2 is an EGFR-associated protein that can be co-targeted by cetuximab, leading to sensitization of cetuximab-treated cells to ROS-induced apoptosis. Our near future study will focus on determining whether combining cetuximab with DCA is a viable novel approach for improving the therapeutic efficacy of cetuximab, particularly against cetuximab-resistant HNSCCs in xenograft models, and whether combinations of cetuximab with other ROS-elevating agents have any therapeutic applications. Successful completion of such studies will provide strong justification for clinical trials testing this novel combination strategy.
Supplementary Material
Highlights.
ASCT2 is physically associated with EGFR in a molecular complex
Cetuximab downregulates ASCT2 via cetuximab-mediated EGFR endocytosis
Co-targeting ASCT2 by cetuximab sensitizes HNSCC cells to ROS-induced apoptosis
Cetuximab sensitizes ROS-induced apoptosis independently of EGFR kinase inhibition
Acknowledgments
This work was supported in part by US National Institutes of Health (NIH) R01 award (CA179015) and an R21 award (DE021883) to Z. Fan. The work was also supported in part by the NIH through MD Anderson’s Cancer Center Support Grant, CA016672. We thank Stephanie Deming in the Department of Scientific Publications at MD Anderson Cancer Center for editing this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors declare no competing financial interests in relation to the work described.
References
- 1.Kanai Y, Hediger MA. The glutamate/neutral amino acid transporter family SLC1: molecular, physiological and pharmacological aspects. Pflugers Arch. 2004;447:469–479. doi: 10.1007/s00424-003-1146-4. [DOI] [PubMed] [Google Scholar]
- 2.McGivan JD, Bungard CI. The transport of glutamine into mammalian cells. Front Biosci. 2007;12:874–882. doi: 10.2741/2109. [DOI] [PubMed] [Google Scholar]
- 3.DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313–324. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dang CV. Glutaminolysis: supplying carbon or nitrogen or both for cancer cells? Cell Cycle. 2010;9:3884–3886. doi: 10.4161/cc.9.19.13302. [DOI] [PubMed] [Google Scholar]
- 5.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 6.Montero AJ, Jassem J. Cellular redox pathways as a therapeutic target in the treatment of cancer. Drugs. 2011;71:1385–1396. doi: 10.2165/11592590-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 7.Stacpoole PW. The pharmacology of dichloroacetate. Metabolism. 1989;38:1124–1144. doi: 10.1016/0026-0495(89)90051-6. [DOI] [PubMed] [Google Scholar]
- 8.Stacpoole PW, Nagaraja NV, Hutson AD. Efficacy of dichloroacetate as a lactate-lowering drug. J Clin Pharmacol. 2003;43:683–691. [PubMed] [Google Scholar]
- 9.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 11.Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2:31ra34. doi: 10.1126/scitranslmed.3000677. [DOI] [PubMed] [Google Scholar]
- 12.Dunbar EM, Coats BS, Shroads AL, Langaee T, Lew A, Forder JR, Shuster JJ, Wagner DA, Stacpoole PW. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest New Drugs. 2013 doi: 10.1007/s10637-013-0047-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sutendra G, Michelakis ED. Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front Oncol. 2013;3:38. doi: 10.3389/fonc.2013.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strum SB, Adalsteinsson O, Black RR, Segal D, Peress NL, Waldenfels J. Case report: Sodium dichloroacetate (DCA) inhibition of the “Warburg Effect” in a human cancer patient: complete response in non-Hodgkin’s lymphoma after disease progression with rituximab-CHOP. J Bioenerg Biomembr. 2013;45:307–315. doi: 10.1007/s10863-012-9496-2. [DOI] [PubMed] [Google Scholar]
- 15.Garon EB, Christofk HR, Hosmer W, Britten CD, Bahng A, Crabtree MJ, Hong CS, Kamranpour N, Pitts S, Kabbinavar F, Patel C, von EE, Black A, Michelakis ED, Dubinett SM, Slamon DJ. Dichloroacetate should be considered with platinum-based chemotherapy in hypoxic tumors rather than as a single agent in advanced non-small cell lung cancer. J Cancer Res Clin Oncol. 2014 doi: 10.1007/s00432-014-1583-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graham J, Muhsin M, Kirkpatrick P. Cetuximab. Nat Rev Drug Discov. 2004;3:549–550. doi: 10.1038/nrd1445. [DOI] [PubMed] [Google Scholar]
- 17.Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358:1160–1174. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- 18.Wheeler DL, Dunn EF, Harari PM. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat Rev Clin Oncol. 2010;7:493–507. doi: 10.1038/nrclinonc.2010.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mari M, Morales A, Colell A, Garcia-Ruiz C, Fernandez-Checa JC. Mitochondrial glutathione, a key survival antioxidant. Antioxid Redox Signal. 2009;11:2685–2700. doi: 10.1089/ars.2009.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu SC. Glutathione synthesis. Biochim Biophys Acta. 2013;1830:3143–3153. doi: 10.1016/j.bbagen.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu Y, Li X, Liang K, Luwor R, Siddik ZH, Mills GB, Mendelsohn J, Fan Z. Epidermal growth factor receptor (EGFR)ubiquitination as a mechanism of acquired resistance escaping treatment by the anti-EGFR monoclonal antibody cetuximab. Cancer Res. 2007;67:8240–8247. doi: 10.1158/0008-5472.CAN-07-0589. [DOI] [PubMed] [Google Scholar]
- 22.Lu H, Li X, Luo Z, Liu J, Fan Z. Cetuximab reverses the Warburg effect by inhibiting HIF-1-regulated LDH-A. Mol Cancer Ther. 2013;12:2187–2199. doi: 10.1158/1535-7163.MCT-12-1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 24.Thymiakou E, Episkopou V. Detection of signaling effector-complexes downstream of bmp4 using PLA, a proximity ligation assay. J Vis Exp. 2011 doi: 10.3791/2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DiStefano JF, Cotto CA, Lane B, Hagag N. Role of epidermal growth factor in the expression of A431 cancer cell protease and red blood cell cytotoxicity. Cancer Res. 1989;49:179–184. [PubMed] [Google Scholar]
- 26.Hassanein M, Hoeksema MD, Shiota M, Qian J, Harris BK, Chen H, Clark JE, Alborn WE, Eisenberg R, Massion PP. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin Cancer Res. 2013;19:560–570. doi: 10.1158/1078-0432.CCR-12-2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wellen KE, Lu C, Mancuso A, Lemons JM, Ryczko M, Dennis JW, Rabinowitz JD, Coller HA, Thompson CB. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev. 2010;24:2784–2799. doi: 10.1101/gad.1985910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, Lu W, Burger JA, Croce CM, Plunkett W, Keating MJ, Huang P. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol. 2012;14:276–286. doi: 10.1038/ncb2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lanzetti L, Rybin V, Malabarba MG, Christoforidis S, Scita G, Zerial M, Di Fiore PP. The Eps8 protein coordinates EGF receptor signalling through Rac and trafficking through Rab5. Nature. 2000;408:374–377. doi: 10.1038/35042605. [DOI] [PubMed] [Google Scholar]
- 30.Duran RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, Hall MN. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell. 2012;47:349–358. doi: 10.1016/j.molcel.2012.05.043. [DOI] [PubMed] [Google Scholar]
- 31.Hassanein M, Qian J, Hoeksema MD, Wang J, Jacobovitz M, Ji X, Harris FT, Harris BK, Boyd KL, Chen H, Eisenberg R, Massion PP. Targeting SLC1a5-mediated glutamine dependence in non-small cell lung cancer. Int J Cancer. 2015;137:1587–1597. doi: 10.1002/ijc.29535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Q, Hardie RA, Hoy AJ, van GM, Gao D, Fazli L, Sadowski MC, Balaban S, Schreuder M, Nagarajah R, Wong JJ, Metierre C, Pinello N, Otte NJ, Lehman ML, Gleave M, Nelson CC, Bailey CG, Ritchie W, Rasko JE, Holst J. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J Pathol. 2015;236:278–289. doi: 10.1002/path.4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van GM, Wang Q, Nagarajah R, Marshall AD, Thoeng A, Gao D, Ritchie W, Feng Y, Bailey CG, Deng N, Harvey K, Beith JM, Selinger CI, O’Toole SA, Rasko JE, Holst J. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene. 2015 doi: 10.1038/onc.2015.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Q, Beaumont KA, Otte NJ, Font J, Bailey CG, van GM, Sharp DM, Tiffen JC, Ryan RM, Jormakka M, Haass NK, Rasko JE, Holst J. Targeting glutamine transport to suppress melanoma cell growth. Int J Cancer. 2014;135:1060–1071. doi: 10.1002/ijc.28749. [DOI] [PubMed] [Google Scholar]
- 35.Acharya A, Das I, Chandhok D, Saha T. Redox regulation in cancer: a double-edged sword with therapeutic potential. Oxid Med Cell Longev. 2010;3:23–34. doi: 10.4161/oxim.3.1.10095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamamori T, Yasui H, Yamazumi M, Wada Y, Nakamura Y, Nakamura H, Inanami O. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic Biol Med. 2012;53:260–270. doi: 10.1016/j.freeradbiomed.2012.04.033. [DOI] [PubMed] [Google Scholar]
- 38.Ai Z, Lu Y, Qiu S, Fan Z. Overcoming cisplatin resistance of ovarian cancer cells by targeting HIF-1-regulated cancer metabolism. Cancer Lett. 2016;373:36–44. doi: 10.1016/j.canlet.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.