

Abstract
Background: Triple-negative breast cancers (TNBCs) are typically more aggressive and result in poorer outcomes than other breast cancers because treatment options are limited due to lack of hormone receptors or amplified human epidermal growth factor receptor 2 (HER2). Many TNBCs overexpress the epidermal growth factor receptor (EGFR) or manifest amplification of the EGFR gene, supporting EGFR as a therapeutic target. While EGFR-directed small molecule inhibitors have shown limited effectiveness in clinical settings, use of EGFR as a mechanism of delivering enzymatic cytotoxins to TNBC has not been demonstrated.
Methods: Using the single-chain variable fragment (scFv) of the 806 antibody that binds only cells with overexpressed, misfolded, or mutant variants of the EGFR, a recombinant immunotoxin was engineered through gene fusion with Pseudomonas aeruginosa Exotoxin A (806-PE38). The potency of 806-PE38 on reducing TNBC cell growth in vitro and in xenograft models (n ≥ 6) was examined for six TNBC cell lines. All statistical tests were two-sided.
Results: 806-PE38 statistically significantly reduced the viability of all tested TNBC lines, with IC50 values below 10 ng/mL for three of six cell lines, while not affecting cells with wild-type EGFR (IC50 >300 ng/mL). Systemic treatments with 806-PE38 vs vehicle resulted in statistically significantly reduced tumor burdens (806-PE38 mean = 128 mm3 [SD = 46 mm3] vs vehicle mean = 749 mm3 [SD = 395 mm3], P = .001) and increased median survival (806-PE38 median = 82 days vs vehicle median = 50 days, P = .01) in a MDA-MB-468 TNBC mouse xenograft. Deletion of the catalytic residue eliminated both cytotoxic activity in vitro and the reduction in tumor burden and survival (P = .52).
Conclusions: These data support the further development of the 806-PE38 immunotoxin as a therapeutic agent for the treatment of patients with EGFR-positive TNBC. Follow-up experiments with combination therapies will be attempted to achieve full remissions.
Worldwide, breast cancers are one of the most common malignancies of women. While advances in targeted therapies have decreased morbidity and mortality rates for many breast cancer subtypes, triple-negative breast cancers (TNBCs), which account for 10% to 20% of all breast cancers (1), remain a therapeutic challenge. TNBCs disproportionately target younger women and display increased invasiveness, faster times to relapse, and higher mortality rates than other breast cancers (2,3). TNBCs display a wide spectrum of oncogenic mutations (4) but are all defined by the lack of estrogen receptor, progesterone receptor, or amplification of the human epidermal growth factor receptor 2 (HER2) oncogene (5). TNBCs have been identified as the major subtype of BRCA1 mutated cancers (6); approximately 80% display increased expression of epidermal growth factor receptor (EGFR) (7). Currently, TNBCs lack suitable agents for targeted therapy and rely on conventional chemotherapies that often result in drug-resistant relapse (3,8). Development of more effective therapies for TNBC is urgently needed.
Monoclonal antibodies targeting receptors upregulated in an oncogenic setting are a major focus of current therapeutic development (9). One promising molecular target is the EGFR, which is genetically amplified and overexpressed in approximately one-quarter and three-quarters of TNBCs, respectively, with expression levels correlating with poorer prognosis (10,11). Other EGFR family members, especially HER2, have been targeted by directed delivery of cytotoxic therapeutics, with encouraging results (12–15). However, many EGFR-targeted therapies are hampered by targeting of wild-type EGFR. As such, development of a therapeutic agent that can effectively inhibit growth of cells expressing aberrant EGFR while sparing healthy cells is of great priority.
The m806 antibody binds cells expressing misfolded, amplified, or overexpressed EGFR but not to cells expressing wild-type EGFR (16–18). The m806 epitope is not accessible when the receptor is in an inactive monomer or activated dimer but is exposed in the locally misfolded (transitional), or “untethered,” conformation, which preferentially occurs under oncogenic conditions (19,20). Additionally, this epitope is not known to be targeted by current anti-EGFR therapeutics such as cetuximab (21,22). The m806 antibody displayed no targeting of normal tissues and produced minimal toxicities in a phase I clinical trial (23), while demonstrating activity against cancers exhibiting resistance to cetuximab or with EGFR kinase domain mutations (19,24). These properties suggest that m806 might be uniquely suited for delivery of cytotoxins to cancers that overexpress EGFR while sparing healthy cells.
Recombinant immunotoxins (ITs) are fusion proteins consisting of the enzymatic domain of a protein toxin, typified by Pseudomonas aeruginosa Exotoxin A (PE), linked to a single-chain variable antibody fragment (scFv). Targeting to a particular cellular surface receptor is achieved by replacing the toxin's binding domain with the relevant antibody fragment (25). For PE-based ITs, potency is derived from the toxin's ADP ribosylation of cytosolic elongation factor 2 (EF2), resulting in inhibition of protein synthesis and cell death. ITs combine high affinity and selective targeting with potent cell killing, making them well suited for therapeutic use in a cancer setting (26) as demonstrated by promising outcomes in clinical trials (27,28). To determine if m806 could target TNBC cells, a novel IT consisting of the m806 scFv joined to the 38 kDa fragment of PE (806-PE38) was engineered and tested on a panel of TNBC cell lines.
Methods
Cell Culture
Human WI-38 and triple-negative breast cancer cell lines were obtained from the American Type Culture Collection (Manassas, VA): BT20 (ATCC HTB19); MDA-MB-231 (CRM-HTB-26); MDA-MB-468 (HTB-132); HCC70 (CRL-2315). Cells were passaged fewer than 20 times before a freshly thawed vial was obtained. BT-20 cells were cultured in Eagle’s minimal essential medium (ATCC) plus Glutamax (Life Technologies, Grand Island, NY) and 10% fetal bovine serum (FBS). MDA-MB-231, MDA-MB-468, HCC70, HCC1937, and HCC1395 cells were cultured in RPMI 1640 medium (Life Technologies) with Glutamax and 10% heat-inactivated FBS. Cells were maintained humidified at 37 °C and 5% CO2.
SDS-PAGE Analysis and Immunoblotting
Cells were lysed in radioisotope precipitation assay buffer (RIPA; Thermo Scientific; Waltham, MA) containing protease/phosphatase inhibitor cocktail (Thermo Scientific). Postnuclear supernatants were stored at -80 °C until use. An equivalent amount of protein for each cell line was resolved by PAGE using 3% to 8% TrisAcetate NuPage gels (Life Technologies). Proteins were transferred to nitrocellulose membrane and probed with α-EGFR antibody (Pierce, Rockford, IL; MA5-12875; 1:10000) and α-actin (BD Biosciences, San Jose, CA; #612656; 1:30000). Primary antibodies were detected by donkey antimouse HRP-conjugated secondary antibody (Jackson Immunoresearch, West Grove, PA; 1:5000). SuperSignal West Pico (Pierce) was used to develop membrane chemiluminescence.
Flow Cytometry
WI-38, MDA-MB-468, or HCC70 cells were adjusted to 1x106 cells/mL. Cells were washed in FACS buffer (PBS+2%BSA) and incubated at 4 °C with either TGFα-PE40, 806-PE38, or 806-PE38Δ553 (10 µg/mL) in FACS buffer for two hours. Bound toxin was detected by incubation with M40-1 antibody (5 ug/mL), which binds domain II of soluble or immobilized PE (52). α-EGFR (Pierce; MA5-12875) was used as a positive binding control and to determine surface EGFR. Primary antibodies were detected with goat antimouse phycoerythrin-conjugated fluorescent secondary antibody (Jackson Immunoresearch). Cells were assayed on a BD LSRII Flow cytometer (BD Biosciences). Data was analyzed in FlowJo v10 software (FlowJo, LLC, Ashland, OR) and displayed in histogram format.
Cellular Viability
Ten thousand cells per well were plated in 96-well format. After 24 hours, the recombinant immunotoxin was added at the concentration indicated in the figure. After 72 hours, viability was determined with the CellTiter-Glo Viability Assay Kit (Promega, Madison, WI). Data are presented as a percentage of control (untreated) cells. Data are from at least two independent replicates in triplicate.
Protein Synthesis
Ten thousand cells per well were plated in 96-well format. After 24 hours, the recombinant immunotoxin was added for 24 hours. Inhibition of protein synthesis was then measured by the addition of 3H-leucine (1 µCi/mL) in leucine-free RPMI media for 30 minutes. 3H-leucine was measured with a Wallac MicroBeta Trilux plate reader (Perkin Elmer, Waltham, MA). Counts-per-minute data were normalized and presented as a percentage of control. Data are from at least two independent replicates in triplicate.
Triple-Negative Breast Cancer Xenograft Models
All mouse experiments were performed in accordance with National Institutes of Health guidelines and approved by the National Cancer Institute Animal Care and Use Committee. MDA-MB-468 or HCC70 tumors were grown in six- to nine-week-old female nude athymic mice (Charles River Laboratories, Wilmington, MA). 2x106 cells per mouse in serum-free RPMI 1640 were mixed with Matrigel (Corning Life Sciences, Corning, NY) (4 mg/mL) and injected into the second mammary fat pad of mice weighing approximately 25 g. After tumor volume had reached approximately 100 mm3, mice were randomly assigned to groups and treated with vehicle alone (PBS+0.2% human serum albumin), 806-PE38 (5 µg/mouse), or 806-PE38Δ553 (5 µg/mouse). Four injections were performed every other day via tail vein for each mouse. Tumor volume and mouse weight were measured at least three times weekly. Tumor volume was calculated as 0.5*(LxW2). Mice were killed by CO2 inhalation once tumors reached 1200 mm3 or became necrotic. Time to death was displayed on a Kaplan-Meyer plot and statistical significance calculated by log-rank test.
Statistical Analysis
All experimental error bars display standard deviation, with all P values calculated for 95% confidence intervals (CIs). All statistics were performed using Graphpad Prism 6 software. Unpaired two-tailed t tests were performed for each concentration of immunotoxin tested in cell viability assays, comparing 806-PE38 with catalytically inactive 806-PE38Δ553, with P values reported in text. Xenograft tumor volumes were compared at each measurement by unpaired two-tailed t tests, with P values reported in text for comparison between treated and vehicle-treated mice at experimental endpoint. Statistically significant differences in tumor volumes between vehicle-treated and 806-PE38-treated mice were calculated by two-way t test at time points indicated in the figure. Vehicle-treated and Δ553-treated mice were not statistically significantly different at any time point for either xenograft model. A P value of less than .05 was considered statistically significant.
Please see the Supplementary Methods (available online) for additional details.
Results
806-PE38 Immunotoxin Binding to TNBC Cell Lines With Varied Expression of EGFR
To engineer the 806-PE38 immunotoxin, a cDNA encoding the VH and VL domains of m806 was fused with the processing and enzymatic domains of the toxin, termed PE38 (Supplementary Figure 1A, available online). As a control, an identical molecule was produced with a deletion of glutamic acid 553 (Δ553) in the catalytic site, eliminating the toxin’s enzymatic activity. The proteins were produced as a single chain of approximately 60KDa (Supplementary Figure 1B, available online).
To evaluate 806-PE38 as a therapeutic agent for TNBC, EGFR expression and immunotoxin binding to a selection of TNBC cell lines was determined. Individual lines (MDA-MB-468, MDA-MB-231, HCC70, BT-20, HCC1937, and HCC1395) were chosen to reflect the diverse molecular landscape of TNBC (4), but their EGFR expression status was not immediately evident. First, the total quantity of EGFR protein was investigated using immunoblotting (Figure 1). As WI-38 cells (normal fibroblast line) are not known to have EGFR amplification or overexpression, they were chosen as a negative control. All TNBC cell lines had statistically significantly more EGFR than the control cells, with BT-20 and MDA-MB-468 cells displaying the most EGFR, consistent with their amplification of the EGFR gene. The BRCA1-null cell lines HCC1395 and HCC1937 expressed less EGFR than the other TNBC cells but still displayed overexpression relative to WI-38 cells. All lines were judged to be EGFR positive, with differences in expression clearly evident.
Figure 1.

Triple-negative breast cancer cells overexpress epidermal growth factor receptor (EGFR). Cell lysates from triple-negative breast cancer cell lines (BT-20, HCC70, MDA-MB-231, or MDA-MB-468), BRCA1-null (HCC1937, HCC1395) or the control WI-38 fibroblast cell line were probed with α-EGFR antibody to determine levels of EGFR protein. Actin was used as loading control. EGFR = epidermal growth factor receptor.
Flow cytometry was used to establish three features: total surface EGFR, ligand accessible EGFR, and 806-reactive EGFR. Ligand-accessible EGFR was measured by TGFα-PE40 binding (29), indicative of properly folded and surface-accessible EGFR molecules. Surface EGFR was characterized for MDA-MB-468 (EGFR high) and HCC70 (EGFR low). EGFR was detected using a monoclonal Ab to EGFR while bound immunotoxin was detected with a monoclonal antibody against PE domain II. The amount of surface EGFR on MDA-MB-468 cells was approximately five-fold higher than on HCC70 cells (Figure 2, A and B). The 806-immunotoxin bound to MDA-MB-468 cells slightly better than the HCC70 cells, with 5% to 10% of total EGFR accessible to 806-PE38, similar to reports of parental m806 binding (18). 806-PE38Δ553 exhibited similar binding activity as the active immunotoxin. The WI-38 cell line bound TGFα-PE40, confirming the display of properly folded EGFR on these cells, but failed to bind 806-PE38 (Figure 2C). These data confirm that the 806 scFv could potentially be useful for delivering cytotoxic agents to TNBC and other EGFR-aberrant cancers while not interacting with wild-type EGFR.
Figure 2.
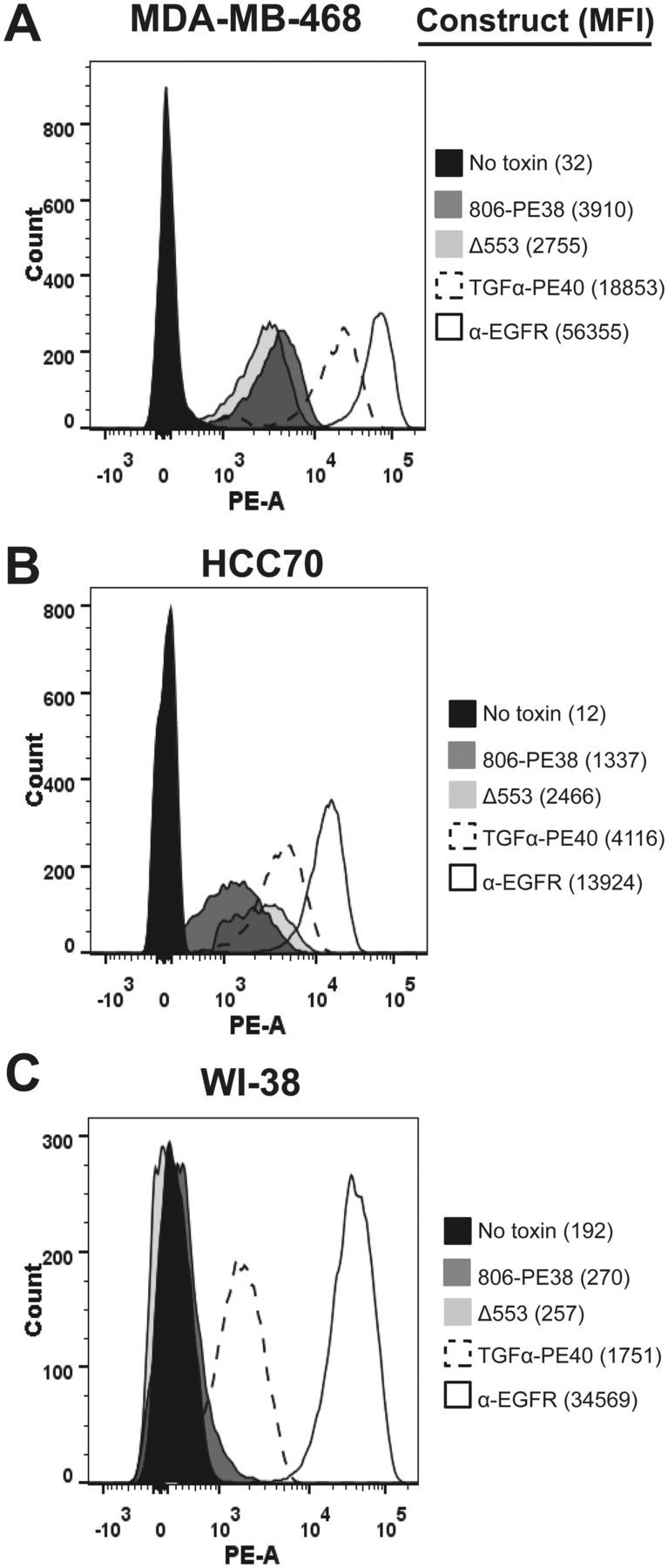
The 806-PE38 immunotoxin binds to triple-negative breast cancer cells but not healthy cells. Assessment of immunotoxin binding was performed by flow cytometry. MDA-MB-468 (A), HCC70 (B), or WI-38 (C) cells were incubated with 10 µg/mL of either 806-PE38 (dark gray), 806-PE38Δ553 (light gray trace), or TGFα-PE40 (dashed trace). Bound immunotoxin was detected by the M40-1 anti-PE antibody. M40-1 in the absence of immunotoxin was used as a negative binding control (black trace). α–epidermal growth factor receptor (EGFR) antibody was used to determine total surface EGFR (empty trace). Data is representative of at least two independent replicates. EGFR = epidermal growth factor receptor.
Effects of Exposure to 806-PE38 on TNBC Cell Viability
PE-based immunotoxins require specific interactions with mammalian cells, including furin processing and KDEL-mediated retrograde trafficking to the ER, leading to the delivery of the toxin's enzymatic domain to the cytosol. To determine if the 806scFv could transport PE38 into this pathway, protein synthesis was measured after a 24-hour exposure to increasing concentrations of the 806-PE38 immunotoxin. Treatment with 806-PE38 resulted in a substantial reduction in 3H-leucine incorporation in all TNBC lines tested, with immunotoxin concentrations at or below 30 ng/mL able to inhibit 50% of protein synthesis (IC50) in six of six cell lines (Table 1; Supplementary Figure 2, available online). The catalytically inactive 806-PE38Δ553 was tested on MDA-MB-468 and HCC70 cells to determine if the antibody component was sufficient to inhibit protein synthesis (Supplementary Figure 3, available online); however, no loss of protein synthesis was noted, even at concentrations of 300 ng/mL (mean % no treatment control ± SD for 806-PE38/Δ553; HCC70: 8.91±2.66/107.94±5.13, P < .001; MDA-MB-468: 10.08±0.98/111.13±8.06, P = .001).
Table 1.
806-PE38 IC50 values: protein synthesis inhibition*
| Cell line | Immunotoxin | IC50 (95% CI), ng/mL |
|---|---|---|
| BT-20 | 806-PE38 | 12.1 (9.7 to 15.9) |
| HCC70 | 806-PE38 | 6.0 (4.4 to 8.5) |
| MDA-MB-231 | 806-PE38 | 31.5 (26.8 to 40.0) |
| MDA-MB-468 | 806-PE38 | 8.2 (6.7 to 9.8) |
| HCC1937 | 806-PE38 | 29.3 (18.7 to 45.4) |
| HCC1395 | 806-PE38 | 7.9 (6.0 to 10.9) |
| WI-38 | 806-PE38 | >300 |
* The concentration of immunotoxin that inhibited 50% of protein synthesis after 24 hours in the indicated cell line is displayed as calculated from best-fit linear regression with the 95% confidence interval. CI = confidence interval.
Having established that the 806scFv was a suitable delivery vehicle, the cytotoxic potential of 806-PE38 was evaluated using cell viability assays. TNBC cells were treated with either catalytically active 806-PE38 or the inactive 806-PE38Δ553 to determine the role of ADP ribosylation in mediating cytotoxicity (Figure 3). Treatment with 806-PE38 resulted in a dose-dependent loss of cell viability in all TNBC cell lines, with IC50 values below 10 ng/mL for three of six cell lines tested (BT-20: 10 ng/mL; HCC70: 4 ng/mL; MDA-MB-468: 7 ng/mL; MDA-MB-231: 90 ng/mL; HCC1395: 30 ng/mL HCC1937: 90 ng/mL). Loss of cell viability trended with inhibition of protein synthesis in all cell lines tested, suggesting that ADP ribosylation of EF2 was the driving factor behind loss of cell viability. In confirmation of the importance of EF2 ADP ribosylation, treatment with Δ553 resulted in no loss in cell viability for any of the TNBC cell lines tested (mean % viability ± SD for 806-PE38/Δ553; BT-20: 20.5±2.12/105.64±3.99, P = .001; HCC70: 1.16±0.47/98.38±1.51, P = .002; MDA-MB-468: 10.10±6.29/92.63±6.89, P = .01; MDA-MB-231: 34.90±13.28/101.85±9.51, P = .02; HCC1395: 29.59±3.52/100.98±3.41, P = .01; HCC1937; 44.82±4.78/96.35±7.72, P = .01) (Figure 3). Confirming the requirement of aberrant EGFR for 806-PE38 binding and toxin delivery, WI-38 cells displayed little or no loss of viability when these cells were treated with the catalytically active immunotoxin, with an IC50 greater than 300 ng/mL (Supplementary Figure 4, available online). However, WI-38 cells were sensitive to TGFα-PE40 (IC50: 3ng/mL, P < .001), confirming that these cells displayed wild-type EGFR on their surface and were not resistant to toxin-mediated killing. These results provide further support for advancing 806-PE38 as a cytotoxic agent to treat cancers expressing aberrant EGFR.
Figure 3.

806-PE38, but not catalytically inactive 806-PE38Δ553, is cytotoxic to triple-negative breast cancer cells. A) BT-20, B) HCC70, C) MDA-MB-231, D) MDA-MB-468, E) HCC1937, F) HCC1395 were incubated with increasing concentrations of either 806-PE38 or 806-PE38Δ553 for 72 hours, at which point cell viability was measured. Data is from at least two independent experiments in triplicate. Error bars display SD value. Two tailed unpaired t tests were performed at each concentration point comparing 806-PE38 with 806-PE38Δ553. *P < .05, †P < .01, ‡P < .001.
806-PE38 Treatment Effects on Tumor Growth in TNBC Mouse Xenograft Models
While 806-PE38 inhibited the viability of TNBC cells in vitro, the microenvironment of solid tumors is often substantially different from cell culture. To determine if 806-PE38 could inhibit the growth of implanted tumors, two xenograft models were established. HCC70 or MDA-MB-468 cells were implanted into the mammary fat pad of female athymic nude mice. Once average tumor volume reached 100 mm3, mice were treated with 806-PE38, 806-PE38Δ553, or vehicle. Mice were killed once they reached the primary endpoint of tumor volume greater than 1200 mm3 per mouse. HCC70 tumors rapidly developed in vehicle-treated and 806-PE38Δ553 treated mice, with average tumor burden of 1094 mm3 and 861 mm3, respectively, when the first mouse reached endpoint (P = .22) (Figure 4A). In contrast, 806-PE38 treated mice showed delayed tumor growth, with an average tumor burden of only 279 mm3 when the first mouse reached endpoint (P = .001). 806-PE38 treatments also statistically significantly extended the time-to-endpoint of mice when compared with vehicle-treated or 806-PE38Δ553-treated mice (P = .001) (Figure 4B). Vehicle or 806-PE38Δ553 -treated mice survived for a median of 27.5 (range = 24–32 days) or 28 (range = 28–36 days) days, respectively. In contrast, 806-PE38-treated HCC70 tumors grew slower, with a median survival of 41 days (range = 30–82 days).
Figure 4.

806-PE38 delays tumor growth and enhances survival time in triple-negative breast cancer xenograft models. A) HCC70 cells were implanted in the mammary fat pad of female athymic nude mice. Mice were treated with 806-PE38 (n = 7), 806-PE38Δ553 (n = 7), or vehicle alone (n = 6) 4 x qod, indicated by arrows. Tumor volume was calculated as (0.5 x LxW2). Tumor volumes were compared by unpaired two-tailed t test at each measurement. †P < .01, ‡P < .001. No statistically significant differences were noted between vehicle-treated and Δ553-treated mice. B) Kaplan-Meyer plot showing time to experimental endpoint for each HCC70 tumor-bearing mouse. Mice were killed once tumor volume was greater than 1200 mm3 or tumors became necrotic. Statistical significance was assessed by log-rank test. †P < .01 Number of mice at risk at weekly intervals is noted below. C) MDA-MB-468 cells were implanted in the mammary fat pad of female athymic nude mice. Mice were treated with 806-PE38 (n = 7), 806-PE38Δ553 (n = 7), or vehicle alone (circles) 4 x qod, indicated by arrows. Tumor volume was calculated as (0.5 x LxW2). Tumor volumes were compared by unpaired two-tailed t test at each measurement. *P < .05, †P < .01, ‡P < .001. No statistically significant differences were noted between vehicle-treated and Δ553-treated mice at any time point. D) Kaplan-Meyer plot showing time to experimental endpoint for each MDA-MB-468 tumor-bearing mouse. Mice were killed once tumor volume was greater than 1200 mm3 or tumors became necrotic. Statistical significance was assessed by log-rank test. ‡P < .001. Number of mice at risk at biweekly intervals is noted below the Kaplan-Meyer plot.
Mice carrying MDA-MB-468 tumors responded similarly, with those treated with 806-PE38 showing statistically significant reduction in tumor burden (Figure 4C) and increased survival time (Figure 4D). Mice had average tumor burdens of 749 mm3 (±SD = 395 mm3, vehicle), 606 mm3 (±304mm3Δ553, P = .52), and 128 mm3 (±46 mm3, 806-PE38, P = .001) when the first vehicle-treated mouse reached endpoint. Mice treated with 806-PE38 had a median survival of 82 (range = 54–>100) days, which was statistically significantly increased compared with vehicle (50 days; range = 42–66, P = .01) or Δ553 treated (52 days, range = 42–68) mice. Notably, two 806-PE38-treated mice had not reached endpoint by 100 days, at which point the experiment was terminated. The number of mice at risk is shown beneath Figure 4, B and D. Treatment regimens were well tolerated, with little or no weight loss observed among mice. Histopathological examination of the tumors showed that treatment with 806-PE38, but not vehicle or Δ553, resulted in focal points of necrotic cell death (Supplementary Figure 5, available online). Further, only in tumors treated with 806-PE38 was there evidence of dead cell clusters, with three to five cells per cluster compared with a few solitary dead cells in the other treatment groups. The lesions showed neither immune-cell infiltrate nor vascular lesions, supporting direct cytoxicity as the mechanism of immunotoxin activity. These results supported the in vitro observations, indicating that the 806-PE38 immunotoxin is cytotoxic for TNBC tumors in vivo and supporting its further clinical development.
Discussion
Cures for TNBC are difficult to achieve because of a lack of targeted therapies, leaving conventional chemotherapies as the standard of care. Increased understanding of the molecular profile of these cancers has suggested that despite the wide variety of oncogenic mutations, a frequent theme is overexpression of the EGFR. Because it was first identified as a family of possible oncogenes, the EGFR receptor family has been an attractive therapeutic target as these receptors are frequently upregulated in many cancers. Development of new HER2-targeted therapies, including antibody-delivered cytotoxins and chimeric antigen-based immune cell repertoires, has shown promise in both research and clinical settings (14,15,30–32). However, while clinical targeting of EGFR function through small molecule inhibitors or antibody inhibition of EGFR signaling has shown some promise, successful long-term outcomes are rare because of compensatory kinase domain mutations or alternate receptor signaling and problems with toxicity to normal tissue (33–36). As ITs target protein synthesis rather than receptor signaling pathways, mutations in the receptor active site or alternate signaling pathways should not affect the activity of this therapeutic.
To augment existing therapeutic approaches, this report demonstrates that the 806 scFv can deliver a potent protein toxin to TNBC cells, using aberrant EGFR as a surface target. Treatment with 806-PE38 killed six of six TNBC cell lines in vitro, with IC50 values at low nanomolar concentrations. Importantly, in two xenograft models there was clear single-agent antitumor activity following systemic treatment with 806-PE38. Deletion of a catalytic residue confirmed mechanistically that antitumor activity was because of the enzymatic activity of the toxin and not merely disruption of receptor signaling (37). The genetic signatures of MDA-MB-468 and HCC70 cells show close similarities to the signatures of clinical basal-like TNBC cases (38), confirming a clinical relevance to our models.
Basal-like TNBCs have been shown to be sensitive to loss of the anti-apoptotic protein MCL-1 (38), which is rapidly turned over in many cell types (39). Immunotoxin-mediated inhibition of protein synthesis results in rapid MCL-1 depletion (40), which suggests that use of an immunotoxin as part of a combination therapy may uniquely sensitize TNBC to other treatments. Follow-up experiments testing a combination of 806-PE38 with other small molecular weight inhibitors may further enhance cell killing, as observed for combination treatments utilizing ITs targeting other cancers (41–43).
While this study shows potential for further development as a therapeutic agent, several limitations are present. Although MDA-MB-468 tumors in two 806-PE38-treated mice were not growing 100 days post-treatment, complete tumor remissions were not observed in either xenograft model. However, the dosing schedule was not optimized for complete tumor killing, with the chosen dose and schedule resulting in slower tumor growth and increased time-to-endpoint with little or no noticeable side effects, allowing for increased dosing in future experiments. Additionally, combination therapies utilizing elements of current TNBC standard of care will be performed. Of potential concern, treatment with protein therapeutics, especially those of nonhuman origin, can result in the generation of neutralizing antibodies. However, co-administration of immunotoxins and selective immunosuppressive agents has resulted in statistically significant reductions in antitoxin antibody formation in mice and humans (27,44). Alternately, identification and removal of major B- and T-cell epitopes from the immunotoxin has proven successful at reducing immune responses (45,46). Another limitation centers on the xenograft model itself. PD-L1 and PD-L2, frequently expressed on the surface of TNBCs (47), would not encounter or influence the functionality of T-cells because tumors are grown in athymic nude mice. Therefore, it remains to be determined whether the antitumor effects of 806-PE38 will be enhanced or diminished when administered to an immunocompetent individual harboring a TNBC tumor.
Finally, it is currently unknown how effective this potential therapeutic will be against other cancers with EGFR overexpression and amplification. The parental m806 antibody was originally raised against the EGFRvIII mutant receptor (16,17), most commonly observed in glioblastomas, which often also display EGFR overexpression. This suggests that 806-PE38 may be effective as a glioblastoma therapeutic. 806-PE38 could also be utilized against non–small cell lung carcinomas (NSCLCs), over half of which overexpress EGFR. EGFR kinase inhibitor therapy is the primary approved treatment for NSCLC; however, secondary kinase domain mutations occur in many cases, and the resulting loss of treatment efficacy confirms that additional treatment options are needed for these cancers (48). Ligand-based toxins with TGFα and EGF have been developed previously (49). Early examples included TGFα-PE40 (29) and DAB389EGF/486EGF (50) while more recent studies have explored dual targeting to EGFR and cytokine receptors (51). While these agents are considered suitable for local administration for treatment of glioblastomas or for intratumoral injections, systemic toxicity precludes wider use, underlining the importance of future development and use of conformation-dependent antibodies, such as 806, to target cancer-expressed EGFRs.
Funding
This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Notes
The funders had no role in design of the study; the collection, analysis, or interpretation of the data; the writing of the manuscript; or the decision to submit the manuscript for publication.
NS conducted experiments, analyzed data, and wrote the paper. AA designed the study, analyzed data, and participated in editing the manuscript. RS constructed the 806-PE38 immunotoxins and produced purified fusion proteins via expression in E. coli and column chromatography. DF designed the study, supervised experimental plans, analyzed data, and cowrote the paper.
The authors thank Stan Lipkowitz for his careful reading of the manuscript. No competing financial interests are reported.
Supplementary Material
References
- 1.Dent R, Trudeau M, Pritchard KI, et al. Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clin Cancer Res. 2007;13(15):4429–4434. [DOI] [PubMed] [Google Scholar]
- 2.Boyle P. Triple-negative breast cancer: epidemiological considerations and recommendations. Ann Oncol. 2012;23:vi7–vi12. [DOI] [PubMed] [Google Scholar]
- 3.Liedtke C, Mazouni C, Hess KR, et al. Response to Neoadjuvant Therapy and Long-Term Survival in Patients With Triple-Negative Breast Cancer. J Clin Oncol. 2008;26(8):1275–1281. [DOI] [PubMed] [Google Scholar]
- 4.Shah SP, Roth A, Goya R, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486(7403):395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abramson VG, Lehmann BD, Ballinger TJ, et al. Subtyping of triple-negative breast cancer: Implications for therapy. Cancer. 2015;121(1):8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tung N, Gaughan E, Hacker MR, et al. Outcome of triple negative breast cancer: comparison of sporadic and BRCA1-associated cancers. Breast Cancer Res Treat. 2014;146(1):175–182. [DOI] [PubMed] [Google Scholar]
- 7.Foulkes WD, Smith IE, Reis-Filho JS. Triple-Negative Breast Cancer. N Engl J Med. 2010;363(20):1938–1948. [DOI] [PubMed] [Google Scholar]
- 8.Cleator S, Heller W, Coombes RC. Triple-negative breast cancer: therapeutic options. Lancet Oncol. 2007;8(3):235–244. [DOI] [PubMed] [Google Scholar]
- 9.Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12(4):278–287. [DOI] [PubMed] [Google Scholar]
- 10.Park HS, Jang MH, Kim EJ, et al. High EGFR gene copy number predicts poor outcome in triple-negative breast cancer. Mod Pathol. 2014;27(9):1212–1222. [DOI] [PubMed] [Google Scholar]
- 11.Liu D, He J, Yuan Z, et al. EGFR expression correlates with decreased disease-free survival in triple-negative breast cancer: a retrospective analysis based on a tissue microarray. Med Oncol. 2012;29(2):401–405. [DOI] [PubMed] [Google Scholar]
- 12.Mazor Y, Noy R, Wels WS, et al. chFRP5-ZZ-PE38, a large IgG-toxin immunoconjugate outperforms the corresponding smaller FRP5(Fv)-ETA immunotoxin in eradicating ErbB2-expressing tumor xenografts. Cancer Lett. 2007;257(1):124–135. [DOI] [PubMed] [Google Scholar]
- 13.Mahmud H, Dalken B, Wels WS. Induction of programmed cell death in ErbB2/HER2-expressing cancer cells by targeted delivery of apoptosis-inducing factor. Mol Cancer Ther. 2009;8(6):1526–1535. [DOI] [PubMed] [Google Scholar]
- 14.Krop IE, LoRusso P, Miller KD, et al. A phase II study of trastuzumab emtansine in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer who were previously treated with trastuzumab, lapatinib, an anthracycline, a taxane, and capecitabine. J Clin Oncol. 2012;30(26):3234–3241. [DOI] [PubMed] [Google Scholar]
- 15.Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367(19):1783–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luwor RB, Johns TG, Murone C, et al. Monoclonal Antibody 806 Inhibits the Growth of Tumor Xenografts Expressing Either the de2–7 or Amplified Epidermal Growth Factor Receptor (EGFR) but not Wild-Type EGFR. Cancer Res. 2001;61(14):5355–5361. [PubMed] [Google Scholar]
- 17.Jungbluth AA, Stockert E, Huang HJ, et al. A monoclonal antibody recognizing human cancers with amplification/overexpression of the human epidermal growth factor receptor. Proc Natl Acad Sci U S A. 2003;100(2):639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johns TG, Stockert E, Ritter G, et al. Novel monoclonal antibody specific for the de2-7 epidermal growth factor receptor (EGFR) that also recognizes the EGFR expressed in cells containing amplification of the EGFR gene. Int J Cancer. 2002;98(3):398–408. [DOI] [PubMed] [Google Scholar]
- 19.Garrett TPJ, Burgess AW, Gan HK, et al. Antibodies specifically targeting a locally misfolded region of tumor associated EGFR. Proc Natl Acad Sci U S A. 2009;106(13):5082–5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johns TG, Adams TE, Cochran JR, et al. Identification of the Epitope for the Epidermal Growth Factor Receptor-specific Monoclonal Antibody 806 Reveals That It Preferentially Recognizes an Untethered Form of the Receptor. J Biol Chem. 2004;279(29):30375–30384. [DOI] [PubMed] [Google Scholar]
- 21.Bardelli A, Janne PA. The road to resistance: EGFR mutation and cetuximab. Nat Med. 2012;18(2):199–200. [DOI] [PubMed] [Google Scholar]
- 22.Li S, Schmitz KR, Jeffrey PD, et al. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7(4):301–311. [DOI] [PubMed] [Google Scholar]
- 23.Scott AM, Lee F-T, Tebbutt N, et al. A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. Proc Natl Acad Sci U S A. 2007;104(10):4071–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li D, Ji H, Zaghlul S, et al. Therapeutic anti-EGFR antibody 806 generates responses in murine de novo EGFR mutant-dependent lung carcinomas. J Clin Invest. 2007;117(2):346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frankel AE, Houston LL, Issell BF, et al. Prospects for immunotoxin therapy in cancer. Annu Rev Med. 1986;37:125–142. [DOI] [PubMed] [Google Scholar]
- 26.Pastan I, Hassan R, Fitzgerald DJ, et al. Immunotoxin therapy of cancer. Nat Rev Cancer. 2006;6(7):559–565. [DOI] [PubMed] [Google Scholar]
- 27.Hassan R, Miller AC, Sharon E, et al. Major cancer regressions in mesothelioma after treatment with an anti-mesothelin immunotoxin and immune suppression. Sci Transl Med. 2013;5(208):208ra147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kreitman RJ, Tallman MS, Robak T, et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol. 2012;30(15):1822–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chaudhary VK, FitzGerald DJ, Adhya S, et al. Activity of a recombinant fusion protein between transforming growth factor type alpha and Pseudomonas toxin. Proc Natl Acad Sci U S A. 1987;84(13):4538–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahmed N, Brawley VS, Hegde M, et al. Human Epidermal Growth Factor Receptor 2 (HER2) –Specific Chimeric Antigen Receptor–Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol. 2015;33(15):1688–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schonfeld K, Sahm C, Zhang C, et al. Selective Inhibition of Tumor Growth by Clonal NK Cells Expressing an ErbB2/HER2-Specific Chimeric Antigen Receptor. Mol Ther. 2015;23(2):330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li B, Meng Y, Zheng L, et al. Bispecific Antibody to ErbB2 Overcomes Trastuzumab Resistance through Comprehensive Blockade of ErbB2 Heterodimerization. Cancer Res. 2013;73(21):6471–6483. [DOI] [PubMed] [Google Scholar]
- 33.Dickler MN, Cobleigh MA, Miller KD, et al. Efficacy and safety of erlotinib in patients with locally advanced or metastatic breast cancer. Breast Cancer Res Treat. 2009;115(1):115–121. [DOI] [PubMed] [Google Scholar]
- 34.Sergina NV, Rausch M, Wang D, et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445(7126):437–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baselga J, Gómez P, Greil R, et al. Randomized Phase II Study of the Anti–Epidermal Growth Factor Receptor Monoclonal Antibody Cetuximab With Cisplatin Versus Cisplatin Alone in Patients With Metastatic Triple-Negative Breast Cancer. J Clin Oncol. 2013;31(20):2586–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee H-J, Zhuang G, Cao Y, et al. Drug Resistance via Feedback Activation of Stat3 in Oncogene-Addicted Cancer Cells. Cancer Cell. 2014;26(2):207–221. [DOI] [PubMed] [Google Scholar]
- 37.Johns TG, Perera RM, Vernes SC, et al. The Efficacy of Epidermal Growth Factor Receptor–Specific Antibodies against Glioma Xenografts Is Influenced by Receptor Levels, Activation Status, and Heterodimerization. Clin Cancer Res. 2007;13(6):1911–1925. [DOI] [PubMed] [Google Scholar]
- 38.Petrocca F, Altschuler G, Tan Shen M, et al. A Genome-wide siRNA Screen Identifies Proteasome Addiction as a Vulnerability of Basal-like Triple-Negative Breast Cancer Cells. Cancer Cell. 2013;24(2):182–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adams KW, Cooper GM. Rapid Turnover of Mcl-1 Couples Translation to Cell Survival and Apoptosis. J Biol Chem. 2007;282(9):6192–6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Du X, Youle RJ, FitzGerald DJ, et al. Pseudomonas Exotoxin A-Mediated Apoptosis Is Bak Dependent and Preceded by the Degradation of Mcl-1. Mol Cell Biol. 2010;30(14):3444–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hassan R, Sharon E, Thomas A, et al. Phase 1 study of the antimesothelin immunotoxin SS1P in combination with pemetrexed and cisplatin for front-line therapy of pleural mesothelioma and correlation of tumor response with serum mesothelin, megakaryocyte potentiating factor, and cancer antigen 125. Cancer. 2014;120(21):3311–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mattoo AR, Pastan I, FitzGerald D. Combination Treatments with the PKC Inhibitor, Enzastaurin, Enhance the Cytotoxicity of the Anti-Mesothelin Immunotoxin, SS1P. PLoS ONE. 2013;8(10):e75576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Traini R, Ben-Josef G, Pastrana DV, et al. ABT-737 overcomes resistance to immunotoxin-mediated apoptosis and enhances the delivery of pseudomonas exotoxin-based proteins to the cell cytosol. Mol Cancer Ther. 2010;9(7):2007–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Onda M, Ghoreschi K, Steward-Tharp S, et al. Tofacitinib suppresses antibody responses to protein therapeutics in murine hosts. J Immunol. 2014;193(1):48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mazor R, Eberle JA, Hu X, et al. Recombinant immunotoxin for cancer treatment with low immunogenicity by identification and silencing of human T-cell epitopes. Proc Natl Acad Sci U S A. 2014;111 (23):8571–8576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu W, Onda M, Lee B, et al. Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human B-cell epitopes. Proc Natl Acad Sci U S A. 2012;109(29):11782–11787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barrett MT, Anderson KS, Lenkiewicz E, et al. Genomic amplification of 9p24.1 targeting JAK2, PD-L1, and PD-L2 is enriched in high-risk triple negative breast cancer. Oncotarget. 2015;6(28):26483–26493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR Mutation and Resistance of Non–Small-Cell Lung Cancer to Gefitinib. N Engl J Med. 2005;352(8):786–792. [DOI] [PubMed] [Google Scholar]
- 49.Berstad MB, Cheung LH, Berg K, et al. Design of an EGFR-targeting toxin for photochemical delivery: in vitro and in vivo selectivity and efficacy. Oncogene. 2015;34(44):5582–5592. [DOI] [PubMed] [Google Scholar]
- 50.Shaw JP, Akiyoshi DE, Arrigo DA, et al. Cytotoxic properties of DAB486EGF and DAB389EGF, epidermal growth factor (EGF) receptor-targeted fusion toxins. J Biol Chem. 1991;266(31):21118–21124. [PubMed] [Google Scholar]
- 51.Oh S, Stish BJ, Sachdev D, et al. A Novel Reduced Immunogenicity Bispecific Targeted Toxin Simultaneously Recognizing Human Epidermal Growth Factor and Interleukin-4 Receptors in a Mouse Model of Metastatic Breast Carcinoma. Clin Cancer Res. 2009;15(19):6137–6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ogata M, Pastan I, FitzGerald D. Analysis of Pseudomonas exotoxin activation and conformational changes by using monoclonal antibodies as probes. Infect Immun. 1991;59(1):407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.