

Abstract
Targeted antifolates with heteroatom replacements of the carbon vicinal to the phenyl ring in 1 by N (4), O (8), or S (9), or with N-substituted formyl (5), acetyl (6), or trifluoroacetyl (7) moieties, were synthesized and tested for selective cellular uptake by folate receptor (FR) α and β or the proton-coupled folate transporter. Results show increased in vitro anti-proliferative activity toward engineered Chinese hamster ovary cells expressing FRs by 4–9 over the CH2 analog 1. Compounds 4–9 inhibited de novo purine biosynthesis and glycinamide ribonucleotide formyltransferase (GARFTase). X-ray crystal structures for 4 with FRα and GARFTase showed that the bound conformations of 4 required flexibility for attachment to both FRα and GARFTase. In mice bearing IGROV1 ovarian tumor xenografts, 4 was highly efficacious. Our results establish that heteroatom substitutions in the 3-atom bridge region of 6-substituted pyrrolo[2,3-d]pyrimidines related to 1 provide targeted antifolates that warrant further evaluation as anticancer agents.
Graphical abstract
INTRODUCTION
Reduced folates are essential cofactors for biosynthesis of purines and pyrimidines.1 Since humans do not synthesize folate, it is necessary to obtain these cofactors from dietary sources. In mammals, three specialized systems exist that mediate membrane transport of folates and antifolates across biological membranes. These include the reduced folate carrier (RFC),2 folate receptor (FRs) α and β,3 and the proton-coupled folate transporter (PCFT).4, 5 RFC and PCFT are facilitative folate transporters, whereas FRs are glycosylphosphatidylinositol-linked proteins that mediate uptake of folates into cells by receptor-mediated endocytosis. RFC is ubiquitously expressed and is the major tissue transporter for folate cofactors.2 FRα and FRβ, as well as PCFT, exhibit narrower patterns of tissue expression and likely serve more specialized physiologic roles.3, 5 For instance, in the proximal tubules of the kidney, FRα contributes to reabsorption of folate from the urine.3 Notably, FRs in normal tissues are either inaccessible to circulating folates (e.g., FRα in renal tubules) or are non-functional (FRβ in thymus).3 PCFT is expressed in the upper gastrointestinal tract where it functions at acidic pH as the major intestinal transporter for absorption of dietary folates.6–8 Although appreciable PCFT can be detected in a number of other normal tissues (e.g., liver, kidney),5 given the requirement for an acidic pH (pH < 7, optimum at pH 5–5.5) for activity,4, 8 PCFT transport should be limited in most normal tissues.
Classical antifolates continue to serve a key role in the therapy of cancer, as well as for other diseases.9–13 Clinically important antifolates include methotrexate (MTX), pemetrexed (PMX), pralatrexate (PDX) and raltitrexed (RTX) (Figure 1). MTX and PDX are both inhibitors of dihydrofolate reductase.10 PMX and RTX inhibit thymidylate synthase as their primary intracellular target, although for PMX, secondary targets have been described, including 5-aminoimidazole-4-carboxamide (AICA) ribonucleotide formyltransferase (AICARFTase) and glycinamide ribonucleotide (GAR) formyltransferase (GARFTase) in de novo purine nucleotide biosynthesis, and dihydrofolate reductase.14–16 Notably, all these compounds are excellent substrates for the RFC and, to varying degrees, are also substrates for FRs α and β and for PCFT.2, 5, 10 Transport by RFC confers limited tumor selectivity, as both tumors and normal tissues express this transporter.2 Interestingly, a substantial cohort of solid tumors (e.g., ovarian, non-small cell lung cancer) also expresses PCFT, often in concert with FRα.17, 18,19 In tumors (unlike normal tissues), FRα is accessible to the circulation, thus facilitating tumor targeting via this mechanism.20, 21 FRβ is expressed in hematologic malignancies such as acute myeloid leukemia3 and in white blood cells of the myeloid lineage,3 including tumor-associated macrophages (TAMs).22 PCFT is active at the acidic pHs typically associated with the tumor microenvironment which confers an additional element of tumor selectivity.5
Figure 1.

Structures of classic antifolate drugs, including methotrexate (MTX), pemetrexed (PMX), pralatrexate (PDX), and raltitrexed (RTX).
In response to the patterns of expression and function of FRs and PCFT in tumors and normal tissues, new cytotoxic agents are being developed for selective tumor targeting by virtue of their specificities for FRs and/or PCFT. FRβ-positive TAMs may play an important role in the tumor microenvironment in relation to tumor metastasis and angiogenesis by releasing proangiogenic factors (e.g., vascular endothelial growth factor, matrix metalloproteinase),23 suggesting that TAMs may constitute an additional potential therapeutic target in cancer for FRβ-targeted agents.22
Folate and pteroate conjugates have been used to deliver cytotoxic agents to FR-expressing tumors including ovarian cancer and non-small cell lung cancer (NSCLC).21, 24, 25 For example, a folic acid-vindesine conjugate (vintafolide) was developed which is internalized by FRs, then cleaved in endosomes to release the cytotoxic vinca alkaloid.24, 25 Based on its success in a randomized Phase II clinical trial with platinum resistant ovarian cancer,26 vintafolide was advanced to a Phase III clinical trial. More recently, a folic acid-tubulysin conjugate,25, 27 was introduced into Phase I clinical trials. N-[4-[2-propyn-1-yl[(6S)-4,6,7,8-tetrahydro-2-(hydroxymethyl)-4-oxo-3H-cyclopenta[g]quinazolin-6-yl]amino]benzoyl]-L-γ-glutamyl-D-glutamic acid (ONX0801) is a small molecule antifolate with selective substrate activity toward FRs over RFC that is an inhibitor of thymidylate synthase.28 This compound is currently being tested in a Phase I clinical trial.
We previously described novel 6-substituted pyrrolo[2,3-d]pyrimidine benzoyl antifolates with selective cellular uptake by FRs and/or PCFT over RFC, resulting in potent inhibitory activity toward human tumor cells.18, 29–35 The most active analog of this series, 1, included a 3 carbon bridge (Figure 2) and showed high level activity toward KB human tumors (IC50 = 1.7 nM)29 expressing FRα and PCFT. Compound 2 has a 4-carbon bridge and showed a better selectivity than 1 for both FRα and PCFT over RFC. An analog of compound 1 with a side-chain thienoyl-for-benzoyl replacement (compound 3) (Figure 2) showed even greater FR- and PCFT-targeted activity.31 Although 1 and 3 were not effective substrates for RFC, an apparent non-mediated uptake process, as reflected in growth inhibition of both RFC and transport-null cells, was detected.29, 31 Compounds 1, 2 and 3 were identified as potent inhibitors of cellular GARFTase and de novo purine nucleotide biosynthesis.29, 31 Notably, these compounds were entirely unique from previously reported GARFTase inhibitors including lometrexol [(6R)5,10-dideazatetrahydrofolate; LMTX],36 (2S)-2-((5-(2-((6R)-2-amino-4-oxo-5,6,7,8-tetrahydro-1H-pyrido[ 2,3-d]pyrimidin-6-yl)ethyl)thiophene-2-carbonyl)amino) pentanedioic acid (LY309887),36 and (2S)-2-((5-(2-((6S)-2-amino-4-oxo-1,6,7,8-tetrahydropyrimido[5,4-b][1,4]thiazin-6-yl)ethyl) thiophene-2-carbonyl)amino) pentanedioic acid (AG2034),37 as all these latter antifolates are excellent substrates for RFC. In Phase I clinical trials with these earlier antifolates, toxicities were dose-limiting,38–40 likely due at least in part to their cellular uptake by RFC and their metabolism to polyglutamates in normal tissues.
Figure 2.

Structures of 6-pyrrolo[2,3-d]pyrimidine antifolates, 1, 2 and 3.
The natural substrate for GARFTase, N10-formyl tetrahydrofolate (10-CHOTHF, Figure 3), is a 6-substituted pteridine with a –CH2-N-two-atom bridge. The N10 is substituted with a formyl moiety which forms a hydroxylated, tetrahedral intermediate prior to transfer of the one-carbon group.41 Thus, replacement of the C10-benzylic CH2 of the 6-substituted pyrrolo[2,3-d]pyrimidine 1 with a NH, N-COCH3, N-CHO, and N-COCF3 [4, 5, 6, and 7, respectively; Figure 4] would afford “mimics” of the natural substrate with a three-atom rather than two-atom bridge. We hypothesized that these 6-substituted three-atom bridged NH and N-substituted analogs would function as potent inhibitors of human GARFTase rather than as substrates. It was further anticipated that these N10-H (4) and N10-substituted compounds (5–7) would provide additional hydrogen bonding with the target enzyme GARFTase that would be absent with the threecarbon bridge analog 1.
Figure 3.

Structure of N10-formyl tetrahydrofolate.
Figure 4.

Structures of 6-substituted pyrrolo[2,-3-d]pyrimidine antifolates with heteroatom bridge substitutions.
We previously reported that in the carbon-bridged analogs the length of the side-chain plays an important role in determining potency and transport selectivity18, 29 such that the most transport selective analogs have a four-carbon bridge (e.g., 2) and the most potent GARFTase and tumor inhibitors have a three carbon bridge (e.g., 1). Thus, it was of interest to compare the effects of replacing the benzylic CH2 of 1 with NH (compound 4), O (compound 8) and S (compound 9) (Figures 4 and 5). The C-S bond length is 1.81 Å, compared to the C-C bond length of 1.54 Å.42 Replacing the benzylic CH2 of the three-atom chain of 1 with larger sulfur would afford a chain length between those for the 3-carbon and 4-carbon bridges of 1 and 2, respectively. In addition to the increased bond length, the C-S-C bond angle is 99° compared to a C-C-C bond angle of 109° (Figure 5).42 This provides a slightly different conformational orientation of the benzene ring, and hence L-glutamate orientation. Similarly, replacement of the benzylic CH2 with NH or an O would afford subtle variations in bond length (C-O = 1.43 Å) and bond angle (C-O-C = 111°, Figure 5). Collectively, these structural alterations could impact transport selectivity, as well as GARFTase inhibition and antitumor efficacy.
Figure 5. Distances and bond angle variations predicted by the nature of the bridge at the benzylic position (X).

Bond angles for X = CH2, O and S obtained from literature.42 Distances and angles for X = NH were measured using energy-minimized conformations of compounds with MOE 2014.09.43
In this study, we report the synthesis and biological activities of analogs of 1 with isosteric heteroatom replacements, including S (9), O (8) and N (4) vicinal to the side chain phenyl ring, along with additional N-substituted analogs including formyl (5), acetyl (6) and trifluoroacetyl (7) moieties.
MOLECULAR MODELING
To provide a rationale for our proposed analogs, molecular modeling studies were initially carried out using our X-ray crystal structure of human GARFTase bound to 3 (PDB: 4ZYW).18 Figure 6A shows the docked pose of the lead compound 1 (green) in the GARFTase active site as a representative example of docking the target molecules. Compound 4 (orange) is also docked in Figure 6A. The pyrrolo[2,3-d]pyrimidine scaffold of 1 binds in the region occupied by the bicyclic scaffold of 3 in the GARFTase crystal structure (PDB: 4ZYW18, 3 not shown for clarity). Hydrogen bonds between the N1 nitrogen of 1 and the backbone of Leu899, 2-NH2 of 1 and the carbonyls of Glu948 and Leu899, N3 of 1 and the amide of Ala947, and 4-oxo of 1 and the amide of Asp951 stabilize the scaffold. Additionally, the N7-nitrogen of 1 forms a hydrogen bond with the carbonyl of Arg897. The pyrrolo[2,3-d]pyrimidine scaffold of 1 forms hydrophobic interactions with Leu892, Ile898, Leu899, and Val904, and the folate binding loop residues 948–951. The amide NH of the L-glutamate of 1 forms a hydrogen bond with the carbonyl of Met896. The L-glutamyl moiety of 1 is oriented similar to 3, with the α-carboxylate interacting with Arg897.
Figure 6. Molecular modeling studies with human GARFTase (PDB: 4ZYW).18.

(Panel A) Superimposition of docked poses of 1 (green) and 4 (orange) (Panel B) Docked pose of 7 (purple). In both panels, GAR is shown in pink.
The docked poses of 4 (Figure 6A, orange) and 7 (Figure 6B) retain the hydrogen bonding and hydrophobic interactions seen in the docked pose of the lead compound 1. In addition, the docked pose of 4 at the N10-H forms hydrogen bonds with the substrate GAR and with Asn913, His915 and Asp951 via a bridging water molecule (Figure 6A). Similar water molecules are seen in other reported X-ray crystal structures of GARFTase.18, 44 This suggests that 4 with the N10-H hydrogen bond would possibly bind better than 1 and 2 with C10-H. Docking scores of 4 (−56.31 kJ/mol) were better than those of 1 (−51.26 kJ/mol) and 2 (−52.57 kJ/mol). Similar interactions are predicted to occur between the heteroatom bridges of 8 (oxygen) and 9 (sulfur) and the conserved water molecule, with the heteroatoms functioning as hydrogen bond acceptors (not shown).
The N10-COCF3 moiety in 7 (Figure 6B) is oriented similar to the N10-H of 4, but binds deeper into the formyl transfer region that is occupied by the formyl group of the natural GARFTase substrate, 10-CHOTHF. The increased bulk at the N10-position of 7 displaces the water molecule involved in H-bonding with the N10-H of 4, and affords direct interaction between the trifluoromethyl of the NCOCF3 moiety of 7 and the terminal amine of substrate GAR. The docking score of 7 in GARFTase was −53.31 kJ/mol. Similar interactions involving the terminal amine of GAR are also observed with the carbonyl of the formyl group of 5 and the carbonyl group of the acetyl of 6 (not shown).
Figure 7 shows the results of molecular modeling studies in the folate binding cleft of FRα(PDB: 4LRH)45 for lead compounds 1 (panel A), 2 (panel B) and 4 (panel C). All three compounds display similar interactions in the pocket, maintaining important protein contacts between the bicyclic scaffolds and benzoyl glutamate tail, as also seen in the crystal structure ligand folic acid.45 In the docked pose of 4 (Figure 7C), the 2-NH2 of 4 interacts with Asp103 (81) (for FR, numbering is that of the full-length gene product with mature protein numbering in parentheses). The 4-oxo moiety forms hydrogen bonds with the side chain hydroxyl of Ser196 (174) and the side chain nitrogens of Arg125 (103) and His157 (135). The pyrrolo[2,3-d]pyrimidine scaffold is stacked between the side chains of Tyr82 (60) and Trp193 (171), similar to that seen with the pteroyl ring of folic acid in its bound conformation.45 The L-glutamate moiety of 4 is oriented similar to the corresponding L-glutamate in folic acid.45 The α-carboxylic acid of 4 forms a network of hydrogen bonds involving the backbone NH of Gly159 (137) and Trp160 (138), while the γ-carboxylic acid of 4 interacts with the amine of Lys158 (136) and the side chain NH of Trp124 (102). The bridge of 4 forms hydrophobic interactions with Tyr82 (60), Phe84 (62), Trp124 (102) and His157 (135).
Figure 7. Molecular modeling studies using the human FR.

αcrystal structure (PDB: 4LRH).45
Panel A, superimposition of the docked pose of 1 (green) with the crystal structure of folic acid (black). Panel B, superimposition of the docked pose of 2 (pink) with crystal structure of folic acid (black) (C) Docked pose of 4 (orange).
Comparing the docked poses of 1 (Figure 7A)and 2 (Figure 7B) with the crystal structure of folic acid in FRα45 indicates that the 3- and 4-atom bridges of 1 and 2 are accommodated within a similar space as the 2-atom bridge in folic acid. Since the bridge NH in folic acid does not form hydrogen bonds with the binding pocket, its replacement with an all-carbon bridge (1 and 2) could, perhaps, better utilize the hydrophobic nature of the pocket for binding interactions. Introduction of heteroatom bridges in the target compounds was not expected to affect FRαtransport capabilities of these compounds, reflected by their highly similar docked pose (e.g., compound 4, Figure 7C) compared to lead 1 and folic acid. The docking score of 4 was −43.87 kJ/mol, similar to the docked scores of 1 (−44.29 kJ/mol) and 2 (−46.99 kJ/mol).
CHEMISTRY
Compounds 10 and 11 (Scheme I) were subjected to Michael addition to afford the β-keto amine 12. Compound 12 was then selectively brominated at the α carbon under acidic conditions using 33% HBr in acetic acid/Br2 to give 13. Protection of the amine in 13 using trifluoroacetic anhydride and subsequent condensation with 2,6-diamino-3H-pyrimidin-4-one in DMF at room temperature for 3 days afforded the 2-amino-4-oxo-6-substituted-pyrrolo[2,3-d]pyrimidine 14. Hydrolysis of 14 afforded the corresponding free acid 15. Subsequent coupling with L-glutamate di-tert-butyl ester using 2-chloro-4,6-dimethoxy-1,3,5-triazine as the activating agent afforded the diester 16. Deprotection of the di-tert-butyl ester afforded the corresponding acid 4. Compound 4 was then reacted with formic acid and acetic anhydride, acetic anhydride or trifluoroacetic anhydride to afford target compounds 5, 6 and 7, respectively.
Scheme I. Reagents and conditions.

(a) Ethanol, reflux, 5 h, 59% ;(b) 33% HBr in CH3COOH, Br2, rt, 2.5 h, 33%; (c) (CF3CO)2O, rt, overnight, 76%; (d) 2,6-diaminopyrimidin-4(3H)-one, DMF, rt, 3 d, 25%; (e) 1N NaOH, rt, 10 h, 88%; (f) N-methylmorpholine, 2-chloro-4,6-methoxy-1,3,5-triazine, L-Glutamate di-tert-butyl, DMF, 12 h, 60%; (g) CF3COOH, CH2Cl2, rt, 2 h, 74%; (h) (CH3CO)2O, rt, 12 h, 47% or (CF3CO)2O, rt, 4 h, 76% or (CH3CO)2O, HCOOH, 1 h, reflux, 35%.
Carboxylic acid 19 (Scheme II) was obtained from β-propiolactone 17 and methyl 4-hydroxybenzoate 18 using NaOH at 50 °C for 30 min. The carboxylic acid 19 was then converted to the acid chloride and immediately reacted with diazomethane, followed by 48% HBr in water, to give the α-bromomethylketone 20. Condensation of 2,6-diamino-3H-pyrimidin-4-one with 20 in DMF at room temperature for 3 days afforded the 2-amino-4-oxo-6-substituted-pyrrolo[ 2,3-d]pyrimidine 21. Hydrolysis of 21 afforded the corresponding free acid 22. Subsequent coupling with the L-glutamate dimethyl ester using 2-chloro-4,6-dimethoxy-1,3,5-triazine as the activating agent afforded the diester 23. Saponification of the diester gave the target compound 8.
Scheme II. Reagents and conditions.

(a) NaOH, H2O, 30 min, 80 °C, 52%; (b) Oxalyl chloride, CH2Cl2, reflux, 1 h, 79%; (c) Diazomethane, (CH3CH2)2O, rt, 1h; (d) 48% HBr in water, 80 °C, 2 h, 94%; (e) 2,6-diaminopyrimidin-4(3H)-one, DMF, rt, 3 d, 31%; (f) 1N NaOH, rt, 12 h, 74%; (g) N-methylmorpholine 2-chloro-4, 6-methoxy-1,3,5-triarzine, L-glutamate dimethyl, DMF, 12 h, 42%; (h) 1N NaOH, rt, 4 h, 88%.
The 6-substituted pyrrolo[2,3-d]pyrimidine 27 (Scheme III) was synthesized by condensation of 2,6-diamino-4-oxopyrimidine 25 with ethyl 4-chloro-3-oxobutanoate 26. Reduction of the ester group of 27 to the alcohol 28 and subsequent mesylation of the alcohol gave 29. Nucleophilic displacement with 4-mercapto-benzoic acid methyl ester under basic conditions gave the thiophene-linked ester 30. Hydrolysis of the ester moiety of 30 to acid 31 and coupling with L-glutamate diethyl ester using 2-chloro-4,6-dimethoxy-1,3,5-triazine as the activating agent afforded the diester 32. Saponication of the diester moieties gave target 9.
Scheme III. Reagents and conditions.

(a) NaOAc, H2O, reflux, 18 h, 54%; (b) LiEt3BH, THF, 0 °C, 30 min, 75%; (c) CH3SO2Cl, DMF, Et3N, 0 °C, 2 h, 80%; (d) 4-mercapto-benzoic acid methyl ester, K2CO3, DMF, rt, 12 h, 37%; (e) 1N NaOH, MeOH, rt, 95–99%; (f) L-glutamic acid diethyl ester hydrochloride, N-methyl morpholine, 2-chloro-4,6-dimethoxy-1,3,5-triazine, DMF, rt, 6 h, 60%.
BIOLOGICAL EVALUATION AND DISCUSSION
Anti-proliferative effects of 6-substituted pyrrolo[2,3-d]pyrimidine benozyl analogs with heteroatom bridge substitutions in relation to mechanisms of folate transport
The goal of this study was to explore the impact of various isosteric heteroatom substitutions including N, O, and S (designated compounds 4, 8, and 9, respectively) at position 10 in the bridge region connecting the bicyclic scaffold and the phenyl ring of 6-pyrrolo[2,3-d]pyrimidine antifolates (Figure 4). Additional N-substituted analogs related to 4 with formyl (5), acetyl (6), and trifluoroacetyl (7) substitutions were also synthesized and tested.
As a primary screen of antiproliferative activities for these novel agents, we initially used a panel of isogenic Chinese hamster ovary (CHO) sublines individually expressing FRα (RT16 cells),29 FRβ (D4),29 RFC (PC43-10),46 or PCFT (R2/PCFT4)47. These engineered CHO cell lines were developed from RFC-, FR- and PCFT-null MTXRIIOuaR2-4 CHO cells48 (hereafter, referred to as R2) by stably transfecting the human transporters and have been documented for their transport characteristics.29, 46, 47, 49 To assess the inhibitory potentials of the 6-substituted pyrrolo[2,3-d]pyrimidine analogs (or classical antifolate inhibitors such as PMX, for comparison), the CHO sublines were cultured in the presence of a range of drug concentrations for up to 96 hours and cell viabilities were measured as a fluorescence read-out. IC50 values, corresponding to the drug concentrations that inhibit growth by 50%, were calculated. Results for the engineered CHO sublines were compared to those for R2 CHO cells.
The 6-substituted pyrrolo[2,3-d]pyrimidine analogs with heteroatom bridge replacements, 4 (N), 8 (O), and 9 (S), all showed potent growth inhibition toward FRα-expressing RT16 cells and toward FRβ-expressing D4 cells with IC50 values equal or less than those previously reported for 129, 49 (Table 1). The impacts of N-substitutions including formyl (5), acetyl (6) or trifluoroacetyl (7) on FR-targeted activity compared to N-unsubstituted 4 were modest. Notably, the N-heteroatom analogs including unsubstituted (4) and N-substituted (5 and 6) forms were significantly more potent toward D4 than toward RT16 cells (<0.003) (Table 1). While 7 was also more active toward D4 cells, the difference from RT16 was not statistically significant. These results suggest an increased FRβ selectivity over FRα for the N heteroatom series of 6- substituted pyrrolo[2,3-d]pyrimidine analogs.
Table 1. IC50s (in nM) for 6-substituted pyrrrolo[2,3-d]pyrimidine benzoyl antifolates with heteroatom replacements, and classical antifolates in RFC-, PCFT- and FR-expressing cell lines.
Proliferation assays were performed for CHO sublines engineered to express human RFC (PC43-10), FRa (RT16), FRβ (D4) or PCFT (R2/PCFT4), and transporter-null (R2) CHO cells,29, 46–48 and KB human tumor cells (express RFC, FRa, and PCFT). For the experiments measuring FR-mediated effects, assays were performed in the presence or absence of 200 nM folic acid (results are shown only for KB cells). Results are presented as IC50 values, corresponding to the concentrations that inhibit growth by 50% relative to cells incubated without drug. The data are mean values from 5–16 experiments (+/− standard errors in parentheses). Some of the data for 1, 2, 3, MTX, PDX, PMX and LMTX have been previously published.29–31, 34, 47, 49 Results are also summarized for KB cells for the protective effects of adenosine (60 μM), thymidine (10 μM), or 5-aminoimidazole-4-carboxamide (320 μM). For compounds 4, 5, 6, 7, 8, and 9, folic acid and nucleoside/AICA protection results are shown in Figure 10. Methods are summarized in the Experimental Section. Undefined abbreviations: Ade, adenosine; AICA, 5-aminoimidazole-4-carboxamide; FA, folic acid; ND, not determined; Thd, thymidine. *Significantly different from compound 4 with RT16 or KB cells, as appropriate (p<0.05); ϕθ¥, significantly different from RT16 with 4 (p=0.0006), 6 (p=0.0026), and 5 (p=0.0001)
| Antifolate | RFC | FRa | FRβ | PCFT | RFC/FRa/PCFT | ||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| PC43-10 | R2 | RT16 | D4 | R2/PCFT4 | R2 | KB | KB (+FA) | KB + Ade/Thd/AICA | |
|
| |||||||||
| 1 | 649(38) | >1000 | 4.1(1.6) | 5.6(1.2) | 23.0(3.3) | >1000 | 1.7(0.4) | >1000 | Ade/AICA |
| 2 | >1000 | >1000 | 6.3(1.6) | 10(2) | 213(28) | >1000 | 1.7(0.4) | >1000 | Ade/AICA |
| 3 | 101.0 (16.6) | 273.5 (49.1) | 0.31 (0.14) | 0.17 (0.03) | 3.34(0.26) | 288(12) | 0.26(0.03) | 101(7) | Ade/AICA |
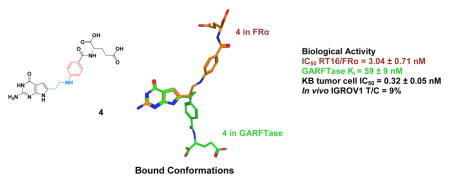
| 4 | 510(90) | >1000 | 3.04 (0.71) | 0.62 (0.20)ϕ | 87.4(9.9) | >1000 | 0.32(0.05) | 666(46) | Ade/AICA |
| 5 | 642(239) | >1000 | 3.01 (0.57) | 0.53 (0.11)¥ | 88.5(13.4) | >1000 | 2.09(0.72)* | >1000 | Ade/AICA |
| 6 | 808(124) | >1000 | 4.87 (0.81) | 1.57 (0.53)θ | 81.7(91) | >1000 | 2.57(0.60)* | >1000 | Ade/AICA |
| 7 | 783(109) | >1000 | 1.08 (0.69) | 0.45 (0.11) | 109 (26) | >1000 | 0.56(0.05)* | 924(40) | Ade/AICA |
| 8 | 641(140) | 940(60) | 1.89 (0.85) | 2.59 (1.17) | 57.5(5.8) | >1000 | 0.34(0.05) | 704(73) | Ade/AICA |
| 9 | >1000 | >1000 | 0.51 (0.10)* | 0.34 (0.02) | 267(19) | >1000 | 1.66(0.26)* | >1000 | Ade/AICA |
| MTX | 12(1.1) | 114(31) | 114 (31) | 106 (11) | 121 (17) | >1000 | 6.00(0.60) | 20(2.4) | Thd/Ade |
| PMX | 138(13) | 42(9) | 42 (9) | 60 (8) | 13.2(2.4) | 974 (18) | 68(12) | 327(103) | Thd/Ade |
| RTX | 6.3(1.3) | 15(5) | 15 (5) | 22 (10) | 99.5(11.4) | >1000 | 5.90(2.20) | 22(5) | Thd |
| LMTX | 12(2.3) | 12(8) | 12 (8) | 2.6 (1.0) | 38.0(5.3) | >1000 | 1.20(0.60) | 31(7) | Ade/AICA |
With PCFT-expressing R2/PCFT4 CHO cells, anti-proliferative activities were reduced for 4 (N), 8 (O) and 9 (S), compared to that for compound 1 (Table 1). N-substitution of 4 (i.e., 5, 6, and 7) resulted in no significant increase in growth inhibition of R2/PCFT4 cells compared to the non-substituted compound (4).
Toward RFC-expressing PC43-10 cells, very minor inhibitory effects were detected for all the heteroatom compounds, with IC50s which were far in excess of the inhibition recorded for FRα-, FRβ- or PCFT-expressing CHO cells (Table 1). The modest inhibitions of the heteroatom analogs were likewise less than those previously reported toward PC43-10 cells for 129 or 331. Thus, the selectivity for this series (as reflected in the relative IC50s for FRα or FRβ versus RFC-expressing CHO cells) is greater than previously reported for compounds 1, 2 or 3.29, 31
The results with the engineered CHO sublines were generally recapitulated in KB human nasopharengeal carcinoma cells, which express FRα and PCFT along with RFC18, although there were some differences, with 4 and 8 showing potencies essentially equivalent to 3 in this tumor model (Table 1). In KB tumor cells, 4 was significantly more active than 5, 6, 7, and 9 (p<0.05), with the antiproliferative effects for the entire series substantially reversed in the presence of 200 nM folic acid. This is consistent with a FR-mediated cellular uptake process, although non-FR uptake (i.e., PCFT) manifested (as reflected by a lack of folic acid protection18) at higher concentrations of 4, 6, 7 and 8 (Figure 10 shows results for KB cells).
Figure 10. Growth inhibition of KB human tumor cells by 6-substituted pyrrolo[2,3-d]pyrimidine antifolates 1, 4, 5, 6, 7, 8 and 9, and protective effects of excess folic acid, nucleosides, or 5- aminoimidazole-4-carboxamide (AICA).

KB cells were plated (4000 cells/well) in folate-free RPMI 1640 medium with 10% dialyzed FBS, antibiotics, L-glutamine, and 2 nM LCV with a range of drug concentrations, in presence of folic acid (200 nM), adenosine (60 μM), thymidine (10 μM), or AICA (320 μM). Cell proliferation was assayed with a fluorescence-based assay. Data are representative of at least triplicate experiments. These results are summarized in Table 1. The methods are described in the Experimental Section. Abbreviation: Cpd, compound.
The inhibitory potencies measured for the 6-substituted pyrrolo[2,3-d]pyrimidine heteroatom compounds 4, 5, 6, 7, 8, and 9 toward the engineered CHO and human tumor cell lines exceeded those for standard antifolates including MTX, PMX, RTX and LMTX (Table 1). Further, there was limited selectivity for FR and PCFT over RFC for the standard agents.
Transport characteristics of 6-substituted pyrrolo[2,3-d]pyrimidine benozyl analogs with heteroatom bridge substitutions
The growth inhibition data with the engineered CHO cell lines in Table 1 strongly imply that the heteroatom analogs 4–9 are internalized by FRα and FRβ, as well as by PCFT.
The binding affinity for folic acid, as reflected in the dissociation constant (Kd) value measured by isothermal calorimetry, was greater for FRα (Kd=0.01 nM) than for FRβ (Kd=2.7 nM).50 To determine relative binding affinities individually for FRα and FRβ compared to folic acid for this series, we incubated RT16 and D4 cells with [3H]folic acid at 4 °C in the presence of a range of concentrations of the unlabeled pyrrolo[2,3-d]pyrimidine antifolates 1 and 4–9 (0–1000 nM).
Controls for these experiments included a comparable concentration range of non-radioactive folic acid (positive control) and MTX (negative control). Relative binding affinities are reflected in the extents to which the analogs compete with [3H]folic acid for binding to FR, with FR-bound [3H]folic acid normalized to total cell proteins. Relative FR binding affinities were expressed as inverse molar ratios of the unlabeled compounds required to reduce the level of FR-bound [3H]folic acid by 50%, with the folic acid affinity assigned a value of 1.
In this assay, with both FRα- and FRβ-expressing cells, relative binding profiles for 4, 8, and 9 were similar and ~60–110% of the affinity for folic acid (Figure 8). These results indicate that heteroatom substitution for the benzylic CH2 in 1 preserves binding affinity for both FRα (RT16) and FRβ (D4). For FRα, the N-formyl-substituted 5 and the N-acetyl- and N-trifluoroacetyl-substituted 6 and 7, respectively, all showed binding that was substantially reduced (<15% of that for folic acid and for 4). A similar pattern was detected with FRβ-expressing D4 cells and 4, 5, 6, and 7, in spite of increased in vitro drug efficacies toward D4 over RT16 cells (Table 1). Thus, relative FR binding affinities were only partly reflected in the differences in in vitro drug efficacies toward RT16 or D4 cells (Table 1), suggesting that factors unrelated to FR binding are necessary to explain the differential drug sensitivities at low nanomolar or subnanomolar concentrations of these novel antifolates.
Figure 8. FRα and FRβ binding affinities for compounds 4.

–9, compared to folic acid, MTX and compound 1.
Results are shown for the relative binding affinities of the (anti)folates with FRα-expressing RT16 and FRβ-expressing D4 CHO cells. Relative binding affinities were determined over a range of (anti)folate concentrations and were calculated as the inverse molar ratios of the unlabeled ligands required to inhibit [3H]folic acid binding by 50%. By this definition, the relative affinity of folic acid is 1. Results are shown as mean values plus/minus standard errors from 3–4 experiments. Detailed experimental methods are provided in the Experimental Section. Abbreviation: FA, folic acid.
Since the 6-substituted pyrrolo[2,3-d]pyrimidine heteroatom compounds were transported by PCFT in R2/PCFT4 CHO cells (based on patterns of growth inhibition; Table 1), we determined relative binding affinities for this series compared to PMX in order to assess a possible basis for differences in relative PCFT-targeted activities. We compared the effects of the heteroatom compounds (at 1 or 10 μM) on PCFT-mediated transport of [3H]MTX (0.5 μM) over 5 min at pH 5.5 (the pH optimum of PCFT) and at pH 6.8 (approximating the pH of tumor microenvironment) 5 (Figure 9). Results were compared to those for PMX, among the best PCFT substrates,4, 5 and for PT523 (N(alpha)-(4-amino-4-deoxypteroyl)-N(delta)-hemiphthaloyl-L-ornithine), 30 an antifolate substrate for RFC with no significant transport by PCFT. At pH 5.5, PMX potently inhibited [3H]MTX uptake (≥90% at both 1 and 10 μM), whereas 4, 5, 6, 7, 8, and 9, like 1 were somewhat less inhibitory (80–90% inhibition at 10 μM and 30–60% inhibition at 1 μM). At pH 6.8, inhibitions for all compounds were decreased, although appreciable transport inhibitions (~40–60%) compared to controls were detected at 10 μM for compounds 1, 4, 7 and 8. Ki values were calculated from the extent of [3H]MTX transport inhibition at pH 5.5 over a range of drug concentrations by Dixon analysis (Table, Figure 9). Ki values ranged from ~7-fold (for 8) to ~18-fold (for 6) higher than for PMX. Thus, the patterns of growth inhibition toward PCFT-expressing R2/PCFT4 cells are only partially reflected in analog binding to PCFT.
Figure 9. Transport competition by 6-substituted pyrrolo[2,3-d]pyrimidine analogs for PCFT in R2/PCFT4.

Panels A and B: Data are shown for the inhibitory effects of the unlabeled ligands (1 or 10 nM) on [3H]MTX uptake (5 min) with human PCFT-expressing R2/PCFT4 CHO cells at pH 5.5 and pH 6.8. The extent of inhibition in this assay is a reflection of relative PCFT binding affinities for assorted antifolate substrates. Results are presented as average values plus/minus ranges for 2–4 experiments. Results are compared to those for normalized rates in the absence of any additions (“No additions”) and to PCFT-null R2 cells without additions. Panel C: In the table are shown Ki values for inhibition of [3H]MTX uptake at pH 5.5, as measured by Dixon analysis for 3–8 separate experiments. Experimental details are provided in the Experimental Section. Cpd, compound.
Identification of GARFTase as the intracellular enzyme target of 6-substituted pyrrolo [2,3-d]pyrimidine antifolates with heteroatom bridge substitutions
To identify the targeted pathway, we determined the effects of nucleoside additions on the growth inhibitory effects of compounds 4–9 toward KB tumor cells. The results are shown in Figure 10 and are summarized in Table 1. The antiproliferative effects of all compounds were abolished by adenosine (60 μM) but not by thymidine (10 μM), identifying de novo purine nucleotide rather than thymidylate biosynthesis as the targeted pathway, analogous to 1.29 Since AICA (320 μM), a precursor of AICA ribonucleotide (ZMP) which circumvents the step catalyzed by GARFTase (by providing downstream substrate for AICARFTase, the 2nd folate-dependent reaction) also protected (Figure 10), the likely enzyme target is GARFTase.
We used both in situ cell-based (in KB tumor cells) and in vitro GARFTase assays with recombinant GARFTase to confirm GARFTase as the enzyme target for our analogs. The former measures the generation of [14C]formyl GAR from [14C]glycine (and GAR) by GARFTase in KB cells treated with azaserine. In these experiments, we incubated KB cells with a range of concentrations of compounds 4–9 and [14C]glycine for 16 h, under conditions approximating those in our proliferation experiments (Table 1). Our results demonstrated dramatically decreased [14C]formyl GAR in cells in drug-treated cells, with nanomolar IC50 values approximating or slightly exceeding those for inhibition of cell proliferation (Figure 11).
Figure 11. In situ GARFTase assay in KB tumor cells treated with 4, 5, 6, 7, 8, and 9.

Inhibition of cellular GARFTase activity by the pyrrolo[2,3-d]pyrimidine antifolates was assayed in KB tumor cells. KB cells were treated with drug and [14C]glycine for 16 h, then [14C]formyl GAR was extracted and isolated by anion-exchange chromatography for quantitation. Relative [14C]formy GAR was expressed as percent of the untreated control as a function of drug concentrations. IC50s were as follows: 2.64+/−0.24 nM, 4; 2.11+/−0.37 nM, 5; 5.58+/−0.40 nM, 6; 4.95+/−0.44 nM, 7; 6.62+/−1.63 nM, 8; and 5.25+/−0.60 nM, 9. For comparison, the IC50 for GARFTase inhibitions in KB cells by compound 1, PMX and LMTX were 1.92 (+/−0.40) nM (not shown), 30 nM47 and 14 nM,47 respectively. The methods are described in the Experimental Section. Abbreviation: Cpd, compound.
We used an in vitro spectrophotometric enzyme assay with recombinant formyltransferase domain of human GARFTase to measure inhibition by monoglutamyl 6-pyrrolo[2,3-d]pyrimidine antifolates, 1, 4, 5, 6, 7, 8, and 9, along with 3 and PMX from our previous study.18 Absorbance was measured at 295 nm accompanying one-carbon transfer from 10-formyl-5,8-dideazafolic acid to GAR to form 5,8-dideazafolic acid and formyl GAR. Assays were performed in the presence and absence of the inhibitors, and the initial rates were plotted versus inhibitor concentrations and fit to a hyperbola (Figure 1S, Supporting Information) for calculating Ki values (Table 2). Nanomolar Ki values were calculated that varied over a ~3–4-fold range, with the most potent GARFTase inhibition by compounds 4, 6, and 8.
Table 2. Ki values for inhibition of human GARFTase by 6-pyrrolo[2,3-d]pyrimidine heteroatom analogs.
GARFTase activity was determined by measuring formation of 5,8-dideazafolate from 10-formyl-5,8-dideazafolic acid using a spectrophotometric assay (295 nm) in the presence of the indicated antifolate. Detailed methods are described in the Experimental Section. Results are shown as mean values +/− standard errors from three replicate assays. The results for compound 3 and PMX were previously reported.18 Abbreviation; Cpd, compound
| Antifolate | Ki (nM) |
|---|---|
| PMX | 1000 +/− 160 |
| Cpd 1 | 160 ± 17 |
| Cpd 3 | 68 +/− 11 |
| Cpd 4 | 59 ± 9 |
| Cpd 5 | 99 ± 15 |
| Cpd 6 | 62 ± 7 |
| Cpd 7 | 122 ± 14 |
| Cpd 8 | 61 ± 4 |
| Cpd 9 | 201 ± 36 |
Collectively, our results identify GARFTase, the first folate-dependent step in de novo purine biosynthesis, as the principal intracellular target for the heteroatom series. The data in Table 2 suggest that as the monoglutamate forms, heteroatom substitution with N (4) or O (8) affords an approximately 3-fold improvement in GARFTase inhibitory potency over C (1). In addition, N-acetyl substitution (6) maintains this inhibitory potency. The formyl (5) and trifluoroacetyl (7) N-substitutions decrease potency somewhat against isolated GARFTase. Further, all of the heteroatom analogs, 4–9, as well as the carbon analogs 1 and 3, are 5- to 17-fold better GARFTase inhibitors than PMX.
X-Ray crystal structure of 6-pyrrolo[2,3-d]pyrimidine heteroatom antifolates with FRα and human GARFTase
Since compound 4 is selectively transported via FRα over RFC, we determined the x-ray crystal structure of FRα bound to 4 using our published methods.45 We expressed human FRα in HEK293 cells and purified FRα by affinity and size-exclusion chromatography. To confirm functionality of the purified soluble FRα, we measured competition for binding to purified FRα by folic acid or 4 with [3H]folic acid. Compound 4 showed an IC50 only slightly higher than that of folic acid in this assay (not shown), consistent with the results of our FRα binding experiments in RT16 CHO cells (Figure 8). For structural determinations, FRα was deglycosylated, stripped of bound folate, then incubated overnight with a molar excess of 4. The complex was purified and used to set up crystallization screens. The FRα/compound 4 complex formed crystals that permitted the determination of its structure at a resolution of 3.6 Å.
Compound 4 assumes a position in the pocket very similar to that of folic acid (Figures 12A and 12B) with interactions with the same 12 key residues [Tyr82 (60), Asp103 (81), Tyr107 (85), Trp124 (102), Arg125 (103), Arg128 (106), His157 (135), Lys158 (136), Trp160 (138), Trp162 (140), Ser196 (174), and Tyr197 (175); again, numbering for FRα is that of the full-length gene product with mature protein numbering in parentheses]. When compound 4 was computationally docked into the FRα pocket (Figure 7C), the position of the docked 4 was very close (RMSD 0.37 Å) to that of 4 in the experimentally determined FRα/4 co-crystal (Figure 12).
Figure 12. Crystal structures of FRα/folic acid (Panel A) andFRα/compound 4 (Panel B).

Superimposition of the crystal structure of folic acid with FRα onto the crystal structure of 4 indicates that the truncated 5-membered pyrrolo ring of 4 is compensated via the 3-atom bridge, compared to the 2- atom bridge of folic acid. Thus, the overall dimensions of both folic acid and 4 are comparable, permitting all important interactions with FRα to remain unaltered. The heteroatom in the bridge does not play an important role in binding to FRα for folic acid or 4. Figure 2S (A, B) shows electron density maps for 4 in complex with FRα.
As 4 was shown to exhibit nanomolar to sub-nanomolar potencies at GARFTase in our in situ and in vitro assays, we also determined the structure of a ternary complex of human GARFTase, 4 and substrate GAR (Figure 13; Table 1S, Supporting Information) to further understand the molecular determinants of inhibitor binding. Based on computational modeling (Figure 6A), we hypothesized that 4 would bind in a mode similar to that of 3, with the only difference being in the positions of the glutamyl α- and γ-carboxylates. Indeed, the pyrrolo[2,3- d]pyrimidine portion of 4 makes the predicted polar contacts with backbone atoms from GARFTase residues Ile898, Leu899, Ala947, Glu948 and Val950, whereas the L-glutamate of 4 (via the α-carboxylate) is positioned to make a bidentate interaction with Arg897 (Figure 13A). The heteroatom position, N10 in 4, also makes the predicted intermolecular interaction with GARFTase, mediated by an ordered water molecule that is coordinated by residues Asn913, His915 and Asp951, and the terminal NH2 of GAR. Based on the crystal structure of the ternary complex of GARFTase with 4 and GAR, and our molecular modeling studies, we predict that other heteroatoms at the 10-position, as seen in 8 (O) and 9 (S) will form hydrogen bonds to the ordered water, as well. N10-substituted compounds, on the other hand (i.e., 5, 6 and 7), would likely displace the ordered water and perhaps GAR, as well, while making direct polar contacts with residues Asn913, His915 and Asp951.
Figure 13. Crystal structures of human GARFTase.

Panel A, crystal structure of 4 (orange); panel B, 318 (green) with GAR (pink) in the folate binding pocket of GARFTase. GARFTase is shown in ribbon except for interacting side chains and backbone atoms, which are represented as sticks. For clarity, an alternate side-chain conformation of Arg871 has been removed in panel B. Figure 2S (C,D) (Supporting Information) shows electron density maps for 4 with GARFTase.
We previously reported that the computationally docked structures of our potent targeted molecules had different conformational requirements for attachment to FRα and GARFTase (or AICARFTase).35, 51 Superimposition of the crystallographic bound structures of 4 in FRα and GARFTase also shows that diametrically opposite conformations of the side chain of 4 are required for FRα (up) and GARFTase (down) binding. Thus, flexibility in the side chain of 4 is required for attachment to FRα and GARFTase to afford targeted transport and selective tumor inhibition.
In vivo antitumor efficacy with 4 with IGROV1 cells
Based on the in vitro efficacies of 4 versus 3 toward KB human tumor cells, we extended this analysis to IGROV1 human ovarian tumor cells. IGROV1 cells express ~40% of FRα levels and exhibit ~30% of PCFT activity of KB cells.18 We initially compared in vitro efficacies of 4 to 3 toward IGROV1 cells as a prelude to an in vivo efficacy trial with IGROV1 xenografts (Figure 14, panel A). Compound 3 was selected for comparison, reflecting its highly potent activity toward a broad spectrum of tumors including IGROV1.31 Results are also shown for compound 1 with a benzylic CH2 at position 10. With 3, an IC50 of 0.95 [+/−0.35 (SE)] nM was measured that shifted to 213 (+/−8) nM in the presence of 200 nM folic acid18, likely in part due to a non-mediated uptake component and to PCFT. Compound 4 was somewhat less potent (likely reflecting decreased non-mediated and PCFT-mediated uptake), with an IC50 of 2.70 (+/−1.15) nM, and protection by folic acid was substantially increased (IC50=872 +/− 106 nM). Similar results were seen with compound 1 toward IGROV1 cells (IC50 of 4.1 (+/−1.7) nM), although the inhibition was further reversed (IC50 >1000 nM) in the presence of excess folic acid.
Figure 14. In vitro and in vivo efficacies of compounds 4 and 3 toward the IGROV1 ovarian tumor.

Upper panel: Representative growth inhibition experiments for IGROV1 tumor cells treated with a range of concentrations of 3 or 4 in the absence (solid symbols and unbroken lines) or the presence (open symbols and broken lines) of 200 nM folic acid are shown. Results for compound 1 are included for comparison. The methods are described in the Experimental Section. Lower panel: Female NCR SCID mice (11 weeks old; 20 g average body weight) were maintained on a folate-deficient diet ad libitum for 14 days, prior to bilateral subcutaneous tumor engraftment. The mice were non-selectively randomized into 5 mice/group, then compounds 3 (28 mg/kg/inj; Q3dx5) or 4 (96 or 150 mg/kg/inj; Q3dx4) [dissolved in 5% ethanol (v/v), 1% Tween-80 (v/v), 0.5% NaHCO3 and deionized H2O; pH 6.5] were administered beginning on day 2. Mice were weighed daily and tumor measurements were recorded twice per week. Results are shown as mean values +/− standard errors. Abbreviation: cpd, compound; Rx, treatment.
For the in vivo drug efficacy trial, female NCR severe-combined immunodeficient (SCID) mice implanted bilaterally with subcutaneous IGROV1 ovarian tumors. Mice were provided a folate-deficient diet ad libitum to reduce serum folate concentrations to levels similar to those reported in humans.52 A parallel control cohort included mice fed standard (folate-replete) chow. For the trial, both the control and low-folate groups were implanted subcutaneously with IGROV1 tumor fragments. Compound 3 (Q3dx5; 28 mg/kg/inj) or compound 4 (Q3dx4; 96 and 150 mg/kg/inj) was administered intravenously beginning on day 2 post-implantation. The highest doses of 3 and 4 administered approximated the maximally tolerated doses for these compounds. The tumors were measured (twice weekly) and the overall health and body weights of the mice were recorded daily.
For the mice maintained on the folate-deficient diet, 3 (140 mg/kg total dose) was efficacious, with a 35% T/C on day 20, T-C of 9 days, and 1.4 log10 kill, as previously described.31 Compound 4 was tested at two dose levels. The highest non-toxic total dose of 600 mg/kg produced significant antitumor activity (12% T/C; T-C=15 days; 2.3 log10 kill), whereas the lower dose (384 mg/kg total dose) showed an efficacy similar to compound 3 (29% T/C; T-C=9 days, 1.4 log10 kill). These results demonstrate not only a dose-response but an efficacious depth-of-activity (favorable therapeutic index) for compound 4. For all treatments, the regimens were well tolerated, and the only dose-limiting symptom (weight loss) was completely reversible. Body weight loss (nadir of 15% for compound 3 and 17.0–17.3% for compound 4) occurred while the mice were undergoing treatment. However, following treatment, full weight recovery occurred within 48 hours. For the matched control group maintained on a standard diet, antitumor activity was ablated; i.e., compound 3 (140 mg/kg total dose) and compound 4 (600 mg/kg total dose) produced T/C values of 68% and 94% respectively. No weight loss or other adverse symptoms were observed.
The results from the in vivo efficacy trial with FRα- and PCFT-expressing IGROV1 human ovarian tumors substantiate our in vitro drug efficacy results and demonstrate that at equitoxic dose levels, 4 is more efficacious than 3, albeit with a higher dose requirement. Compound 4 showed a good depth-of-activity and there were no acute or long term toxicities other than completely reversible weight loss.
Conclusions
This study describes an in-depth analysis of the structure-activity determinants of 6-substituted pyrrolo[2,3-d]pyrimidine benzoyl antifolates as tumor-targeted therapeutics. The focus is on structural determinants involving heteroatom replacements of the carbon vicinal to the side chain phenyl ring in compound 1 by S, O, or N, and for the N-replacement, the impact of N-formyl, N-acetyl, or N-trifluoroacetyl moieties on cell proliferation and anti-tumor efficacy. The emphasis is on determinants of transport specificity by FRα and FRβ, and by PCFT, over RFC, and on inhibition of de novo purine nucleotide biosynthesis at GARFTase, the first folate-dependent step in this pathway.
Our results document several salient findings including (i) dramatically increased in vitro anti-proliferative activity resulting from heteroatom bridge substitutions toward CHO cell lines engineered to express human FRα or FRβ and FRα-expressing human tumor cells, over the corresponding C analog 1. (ii) For the N-heteroatom unsubstituted and substituted forms (compounds 4–7), in vitro drug efficacies were significantly greater toward FRβ-expressing CHO cells than toward CHO cells expressing a comparable amount of FRα. This likely reflects differences in rates of FR-mediated internalization and/or dissociation of bound ligands for FRβ versus FRα, as FR-targeted activities for these analogs were only modestly reflected in relative binding affinities to FRα and FRβ. FRα binding of 4 was confirmed by x-ray crystallography which validated the molecular modeling predictions for this series. Compared to compound 1, PCFT targeting was reduced by the heteroatom insertions, as reflected in decreased growth inhibition and decreased competition for [3H]MTX uptake with human PCFT-expressing R2/PCFT4 CHO cells.
We established de novo purine nucleotide biosynthesis as the targeted pathway and GARFTase as the enzyme target for the heteroatom-substituted antifolates, as measured by nucleoside/AICA protection from growth inhibition, and by in situ metabolic assays with [14C]glycine. Inhibition of cellular GARFTase in KB cells occurred at concentrations similar to those that inhibit cell proliferation. Results of the metabolic assays were corroborated by in vitro assays with purified GARFTase that showed greater enzyme inhibition by analogs with N (4, 5, 6, and 7) and O (8) insertions than that with a C-10 (1). For compound 4, a crystal structure was determined with GARFTase, providing further validation of this enzyme target and demonstrating clear contacts with the N10 heteroatom analogous to that in 10-CHOTHF. The disparity in drug concentrations for in vitro enzyme inhibition versus GARFTase inhibition in the cell-based metabolic assay likely reflects the importance of membrane transport and polyglutamate synthesis, as determinants of drug efficacy in intact cells. The potent in vitro inhibition of GARFTase by compounds 3, 4, 6 and 8 in their monoglutamate forms suggests that these antifolates would be effective against tumor cells resistant to PMX and other clinically used antifolates due to loss of polyglutamate synthesis, reflecting down regulation and/or deficiency of folylpolyglutamate synthetase.53
We extended our studies with engineered CHO and KB human tumor cells to IGROV1 human tumor cells, a model of serous ovarian cancer with significantly lower levels of FRα and PCFT.18 In a head-to-head comparison with 3, a thieonyl 6-substituted pyrrolo[2,3-d]pyrimidine and among the most potent FR- and PCFT-targeted agents we have identified,31 4 was more efficacious at an equitoxic dose, albeit with a higher dose requirement.
In conclusion, we established that heteroatom substitutions, including S, O and N, in the bridge region of 6-substituted 3-atom bridge pyrrolo[2,3-d]pyrimidine analogs related to 1 and 3 exert pronounced effects on the activity of this series, associated with increased targeting via FRs. Compared to clinically used antifolates such as PMX, analogs 4–9 are selectively transported into tumor cells, resulting in potent inhibition of GARFTase and cell proliferation, and would be expected to possess significantly lower toxicity toward normal tissues. In an in vivo study with IGROV1 human tumor cells, compound 4 was significantly more efficacious than 3, the most efficacious analog we have reported previously. Reflecting these characteristics, these novel tumor-targeted antifolates warrant further evaluation as anticancer agents.
EXPERIMENTAL SECTION
A rotary evaporator was used to carry out evaporation in vacuo. Final compounds and intermediates were dried in a CHEM-DRY drying apparatus over P2O5 at 80 °C. A MEL-TEMP II melting point with a FLUKE 51 K/J electronic thermometer apparatus was used and uncorrected to record melting points. A Bruker WH-400 (400 MHz) spectrometer or a Bruker WH-500 (500 MHz) spectrometer was used to record proton nuclear magnetic resonance spectra (1H NMR). Tetramethylsilane was used as an internal standard to express the chemical shift in ppm (parts per million): s, singlet; d, doublet; t, triplet; q, quartet; quin, quintet m, multiplet; and bs, broad singlet. Chemical names follow IUPAC nomenclature. Whatman Sil G/UV254 silica gel plates with a fluorescent indicator were used for performing thin-layer chromatography (TLC), and the spots were visualized under 254 nm and 365 nm illumination. All analytical samples were homogeneous on TLC in three different solvent systems. Solvents used for TLC were measured in volume. Columns of silica gel (230–400 mesh) (Fisher, Somerville, NJ) were used for chromatography. In spite of 24–48 h of drying in vacuo, fractional moles of water found in the analytical samples of antifolates could not be prevented and were confirmed by their presence in the 1H NMR spectra. Chemicals and solvents were purchased from Aldrich Chemical Co. or Fisher Scientific Co. and were used as received. Elemental analysis (C, H, N, F, S) was performed by Atlantic Microlab, Inc. (Norcross, GA). Element compositions were within 0.4% of the calculated values and confirmed >95% purity for all the compounds submitted for biological evaluation (Supporting Information is in Table 1S)
(4-{[2-(2-Amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)ethyl]amino}benzoyl)-L-glutamic acid (4)
To a 100 mL rbf compound 15 (100 mg, 0.32 mmol), N-methylmorpholine (0.64 mmol), 2-chloro-4,6-dimethoxy-1,3,5-triazine (0.64 mmol) and anhydrous DMF (7 mL) were added. The resulting mixture was stirred at room temperature under anhydrous condition for 1.5 hours. N-mehtylmorpholine (0.64 mmol) and L-glutamate di-tert-butyl hydrochloride (0.47 mmol) were added in reaction mixture. The resulting mixture was then stirred at room temperature under anhydrous condition for 12 hours. After evaporation of solvent under reduced pressure, MeOH (20 mL) was added followed by silica gel (1 gm). The resulting plug was loaded on to a silica gel column (3.5 cm × 2 cm) and eluted with CHCl3 followed by 3% MeOH in CHCl3. Fractions with Rf = 0.45 (MeOH/CHCl3 5:1) were pooled and evaporated to afford 16 (106 mg, 0.19 mmol, yield 60%) as solid. Compound 16 (106 mg, 0.19 mmol) was dissolved in dichloromethane (10 mL) and trifluoroacetic acid (2 mL) was added. The mixture was stirred at room temperature for 2 hours. TLC showed the disappearance of the starting material (Rf = 0.45) and one major spot at the origin (MeOH/CHCl3 5:1). The solvent was remove under reduced pressure and the residue was dissolved in 1 N NaOH. The suspension was filtered and filtrate was acidified to pH 4 with 1 N HCl. The resulting suspension was frozen in dry iceacetone bath, thawed to 4–5 °C in the refrigerator, and filtered. The residue was washed with a small amount of cold water and dried in vacuum using P2O5 to afford the target compound 4 (63 mg, yield 74%) as yellow powder; m.p. 163 °C; 1H NMR (DMSO-d6) δ 1.93–2.07 (m, 2H, β-CH2), 2.32–2.33 (m, 2H, γ-CH2), 2.74–2.77 (t, 1H, CH2CH2NH, J = 6.0 Hz), 3.29–3.32 (t, 2H, CH2CH2NH, J = 6.0 Hz), 4.32–4.38 (m, 1H, α-CH), 5.99 (d, 1H, C5-CH, J = 2.0 Hz), 6.03 (d, 2H, 2-NH2, exch.), 6.27 (s, 1H, CH2CH2NHAr, exch.), 6.60–6.62 (d, 2H, Ar-CH, J = 8 Hz), 7.66–7.68 (d, 2H, Ar-CH, J = 8 Hz), 8.13–8.15 (d, 1H, Ar-CONH, J = 8 Hz, exch.), 10.18 (bs, H, 3-NH, exch.), 10.93 (s, H, 7-NH, exch.). Anal. Calcd for (C20H22N6O6·1.75 H2O) C, H, N.
(4-{N-[2-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6- yl)ethyl]formamido}benzoyl)-L-glutamic acid (5)
To a solution of 4 (110 mg, 0.25 mmol) in 97% formic acid (5 mL) was added acetic anhydride (0.07 mL, 15.75 mmol), and the reaction mixture was stirred at 25 °C for 3 hours. The solvent was removed under reduced pressure and the residue dissolved in 1 N NaOH at 0 °C. The filtrate was acidified to pH 4 with 0.5 N HCI and stored at 0 °C for 2 hours. The yellow solid was collected by filtration and dried over P2O5 to give 40 mg (35%) of 5; m.p. 163 °C; 1H NMR (DMSO-d6) δ 1.95–2.13 (m, 2H, β-CH2), 2.34–2.37 (t, 2H, γ-CH2, J = 7.2 Hz), 2.68–2.72 (t, 2H, CH2CH2NCOH, J = 7.2 Hz), 4.06–4.09 (t, 2H, CH2CH2NCOH, J = 7.2 Hz), 4.38–4.44 (m, 1H, α-CH), 5.91 (s, 1 H, C5-CH), 6.08 (bs, 2H, 2-NH2, exch.), 7.41–7.43 (d, 2H, Ar-CH, J = 8.4 Hz), 7.93–7.95 (d, 2H, Ar-CH, J = 8.4 Hz), 8.57 (s, CH2CH2NCOH, 1H), 8.64–8.66 (d, 1H, Ar-CONH, J = 7.6 Hz), 10.23 (bs, H, 3-NH, exch.), 10.93 (s, H, 7-NH, exch.). Anal. Calcd for (C21H22N6O7·2H2O·0.4CF3COOH) C, H, N.
(4-{N-[2-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6- yl)ethyl]acetamido}benzoyl)-L-glutamic acid (6)
4 (110 mg, 0.25 mmol) was added in 5 mL acetic anhydride, and the reaction mixture was stirred under anhydrous condition at 25 °C for 3 hours. The excess of acetic anhydride was removed under reduced pressure. The residue was suspended in cold water and basified using 1N NaOH. The suspension was then filtered and acidified to pH 4 with 0.5 N HCI and stored at 0 °C for 2 hours. The white solid was collected by filtration and dried over P2O5 to give 57 mg (47%) of 6; m.p. 163 °C; 1H NMR (DMSO-d6) δ 1.93– 2.14 (m, 5H, β-CH2 & CH2CH2NCOCH3), 2.35–2.39 (m, 2H, γ-CH2), 2.64–2.67 (t, 2H, CH2CH2NCOCH3, J = 8 Hz), 3.89–3.92 (t, 2H, CH2CH2NCOCH3, J = 8 Hz), 4.38–4.44 (m, 1H, α- CH), 5.91 (s, 1 H, ArH), 5.99 (s, 2H, 2-NH2, exch.), 7.34–7.36 (d, 2H, Ar-CH, J = 8 Hz), 7.93– 7.95 (d, 2H, Ar-CH, J = 8 Hz), 8.68–8.70 (d, 1H, Ar-CONH, J = 8 Hz, exch.), 10.16 (bs, H, 3-NH, exch.), 10.83 (s, H, 7-NH, exch.). Anal. Calcd for (C22H24N6O7·2H2O) C, H, N.
(4-{N-[2-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)ethyl]-2,2,2- trifluoroacetamido}benzoyl)-L-glutamic acid (7)
5 mL trifluoroacetic anhydride was add to a solution of 4 (110 mg, 0.25 mmol) in 10 mL dichloromethane, and the reaction mixture was stirred at 25 °C for 3 hours. The solvent was removed under reduced pressure and the residue was suspended in water. 1N NaOH was added dropwise to the suspension and pH was adjusted to 4. The suspension was stored at 0 °C for 2 hours. The yellow solid was collected by filtration and dried over P2O5 to give 83 mg (76%) of 7; m.p. 163 °C; 1H NMR (DMSO-d6) δ 1.96– 2.12 (m, 2H, β-CH2), 2.36–2.39 (t, 2H, γ-CH2, J = 7.2 Hz), 2.73–2.77 (t, 2H, CH2CH2NCOCF3, J = 7.6 Hz), 3.95–3.99 (t, 2H, CH2CH2NCOCF3, J = 7.6 Hz), 4.38–4.44 (m, 1H, α-CH), 6.00 (s, 1 H, C5-CH), 6.10 (bs, 2H, 2-NH2, exch.), 7.40–7.42 (d, 2 H, Ar-CH, J = 8 Hz), 7.94–7.96 (d, 2H, Ar- CH, J = 8 Hz), 8.77–8.79 (d, 1H, Ar-CONH, J = 7.4 Hz, exch.), 10.25 (bs, H, 3-NH, exch.), 10.91 (s, H, 7-NH, exch.), 12.33 (bs, 2H, 2COOH, exch.). Anal. Calcd for (C22H21N6O7F3·1H2O·0.06CF3COOH) C, H, N, F.
{4-[2-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)ethoxy]benzoyl}-L-glutamic acid (8)
To a 250 mL rbf, was added a mixture of 22 (100 mg, 0.32 mmol), Nmethylmorpholine (0.64 mmol), 2-chloro-4,6-dimethoxy-1,3,5-triazine (0.64 mmol) and anhydrous DMF (7 mL). After 1.5 hour N-mehtylmorpholine (0.64 mmol) and L-glutamic acid dimethylester hydrochloride (0.5 mmol). The reaction mixture was then stirred at room temperature for 12 hours. After evaporation of solvent under reduced pressure, MeOH (20 mL) was added followed by silica gel (1 gm). The resulting plug was loaded on to a silica gel column (3.5 × 2 cm2) and eluted with CHCl3 followed by 3 % MeOH in CHCl3. Fractions with Rf = 0.42 (CHCl3/CH3OH, 5:1) were pooled and evaporated to afford 23 (63 mg, yield 42%). Compound 20 (0.13 mmol) was dissolved in MeOH (10 mL) added 1N NaOH (10 mL) and the mixture was stirred under anhydrous condition at room temperature for 10 h. TLC showed the disappearance of the starting material (Rf = 0.42) and one major spot at the origin (MeOH/CHCl3 5:1). The reaction mixture was dissolved in water (10 mL), the resulting solution was cooled in an ice bath, and the pH was adjusted to 3–4 with dropwise addition of 1N HCl. The resulting suspension was frozen in dry ice-acetone bath, thawed to 4–5 °C in the refrigerator, and filtered. The residue was washed with a small amount of cold water and dried in vacuum using P2O5 to afford the target compound 8 (8) (55 mg, 0.12 mmol) as white powder; m.p. 162 °C; 1H NMR (DMSO-d6) δ 1.95–2.11 (m, 2H, β-CH2), 2.34–2.37 (t, 2H, γ-CH2, J = 6.0 Hz), 2.97–3.00 (t, 2H, CH2CH2O, J = 6.0 Hz), 4.24–4.27 (t, 2H, CH2CH2O, J = 6.0 Hz), 4.36–4.41 (m, H, α-CH), 6.00 (s, H, C5-CH), 6.01 (bs, 2H, 2-NH2, exch.), 7.03–7.05 (d, 2H, Ar-CH, J = 8.0 Hz), 7.86–7.88 (d, 2H, Ar-CH, J = 8.0 Hz), 8.45–8.47 (d, 1H, Ar-CONH, J = 8.0 Hz), 10.23 (s, H, 3-NH, exch.), 10.97 (s, H, 7-NH, exch.), 12.39 (bs, 2H, 2COOH, exch.). Anal. Calcd for (C20H21N5O7·2 H2O) C, H, N.
(4-{[2-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)ethyl]thio}benzoyl)-L-glutamic acid (9)
To a solution of the 3 (309 mg, 0.6 mmol) in MeOH (10 mL) was added 1N NaOH (10 mL) and the mixture was stirred at room temperature for 10 h. The reaction mixture was evaporated to dryness under reduced pressure. The residue was dissolved in distilled water (5 mL), and the pH was adjusted to 3–4 by dropwise addition of 1N HCl. The precipitate was filtered, washed with water and dried under vacuum with P2O5 to afford 260 mg (95%) of 9 as a light yellow powder. mp 184–185 °C; 1H NMR (DMSO-d6): δ 1.92–2.12 (m, 2H, β-CH2), 2.34– 2.37 (t, 2H, γ-CH2, J = 7.5 Hz), 2.82–2.85 (t, 2H, CH2CH2S, J = 7.5 Hz,), 3.29–3.32 (t, CH2CH2S J = 7.5 Hz, 2H, CH2), 4.37–4.42 (m, 1H, α-CH), 6.00 (s, 1H, C5-CH), 6.02 (s, 2H, 2-NH2, exch), 7.41–7.43 (d, J = 8.5 Hz, Ar-CH), 7.83–7.85 (d, J = 8.5 Hz, Ar-CH), 8.56–8.58 (d, J = 8.0 Hz, 1H, Ar-CONH, exch), 10.17 (s, 1H, 3-NH, exch), 10.91 (s, 1H, 7-NH, exch), 12.46 (bs, 2H, COOH, exch). Anal. Calcd for (C20H21N5O6S·1.0 H2O) C, H, N, S.
Ethyl 4-[(3-oxobutyl)amino]benzoate (12)
To a vigorously stirred solution of compound 11 (5 gm, 30 mmol) in anhydrous absolute ethanol (10 mL), methyl vinyl ketone 10 (2.5 mL) was added. The reaction mixture was heated under reflux for 4.5 hours and resultant mixture was cooled to 0 °C. The precipitated crystalline product was collected and washed with precooled ethanol and dried to afford 12 (4.2 gm, yield 59%) as white powder; m.p. 98 °C; Rf 0.57 (1:1 hexane:EtOAc); 1H NMR (CHCl3) δ 1.36–1.39 (t, 3H, OCH2CH3, J = 7.2 Hz), 2.19 (s, 3H, CH2COCH3), 2.79–2.82 (t, 2H, CH2CH2O, J = 6.0 Hz), 3.48–3.51 (t, 2H, CH2CH2O, J = 6.0 Hz), 4.31–4.36 (q, 2H, OCH2CH3, J = 7.2 Hz), 5.54 (bs, Ar-NH, exch.), 6.61–6.64 (d, 2H, Ar-CH, J = 8.4 Hz), 7.88–7.91 (d, 2H, Ar-CH, J = 8.4 Hz).
Ethyl 4-[(4-bromo-3-oxobutyl)amino]benzoate (13)
A suspension of 12 (3 gm, 12.77 mmol) in 8 mL of 33% hydrogen bromide-glacial acetic acid solution was treated with 1 mL of bromine in 2 mL of glacial acetic acid and stirred for 2.5 hours at 25 °C. Ether (100 mL) was added to the reaction mixture with swirling until separation of the syrupy hydrobromide was complete. The ether was decanted, and any ether remaining was evaporated from the syrup along with excess hydrogen bromide. After further drying by evaporation (bath 35 °C) of a solution in an equal volume of dichloromethane, the syrup weighed 3.3 gm. The syrup was then stirred with cold water and filtered to afford yellow solid. Yellow solid obtained was then washed with water (3 × 20 mL) and dissolved in dichloromethane. The solution was dried over sodium sulfate. Removal of solvent under reduced pressure at room temperature gave a beige colored semisolid. The crude product was thoroughly triturated with pre-cooled ether and the solid product was collected by filtration to afford 13 (1.75 gm, yield 33%); m.p. 142 °C; Rf 0.81 (1:1 hexane:EtOAc); 1H NMR (CHCl3) δ 1.37–1.41 (t, 3H, OCH2CH3, J = 7.2 Hz), 2.79–2.82 (t, 2H, CH2CH2O, J = 6.0 Hz), 3.48–3.51 (t, 2H, CH2CH2O, J = 6.0 Hz), 3.91 (s, 2H, CH2COCH2Br), 4.33–4.38 (q, 2H, OCH2CH3, J = 7.2 Hz), 6.82–6.84 (d, 2H, Ar-CH, J = 8.4 Hz ), 7.94–7.96 (d, 2H, Ar-CH, J = 8.4 Hz).
4-{[2-(2-Amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)ethyl]amino}benzoic acid (15)
A suspension of α-bromoketone 13 (11.89 gm, 38 mmol) in trifluoroacetic anhydride (100 mL) was stirred under anhydrous condition at room temperature to give a clear solution within 30 minutes. The stirring continued for an additional 2 hours and the solution was allowed to stand at room temperature overnight. After removal of excess trifluoroacetic anhydride under reduced pressure at 40–45 °C, the brownish oily residue was dissolved in dichloromethane (150 mL). The solution was washed with cold 2% HCI (100 mL), cold 5% NaHCO3 (100 mL), and finally with cold water (2 × 100 mL). The organic layer was then treated with a mixture of Florisil-charcoal- sodium sulfate (59 gm: 2.4 gm: 20 gm) and stirred for 0.5 hour at room temperature. The mixture was filtered and excess of solvent was evaporated under reduced pressure at 25– 30 °C to afford amine protected α-bromoketone (11.31 gm, yield 76%). Amine protected α- bromoketone (3.84 gm, 9.4 mmol) was then added to a suspension of 2,6-diaminopyrimidin-4- one (1.26 gm, 10 mmol) in anhydrous dimethylformamide (25 mL). The resulting mixture was stirred under anhydrous condition at room temperature for 3 days. After evaporation of solvent under reduced pressure, Methanol (20 mL) was added followed by silica gel (5 gm). The resulting plug was loaded on to a silica gel column (3.5 cm × 12 cm) and eluted with CHCl3 followed by 3% MeOH in CHCl3 and then 5% MeOH in CHCl3. Fractions with and Rf = 0.58 (TLC) (CHCl3/CH3OH, 5:1) were pooled and evaporated to afford 14 (1.03 gm, 2.36 mmol, yield; 25%). Compound 14 (306 mg, 0.7 mmol) was dissolved in MeOH (10 mL) and 1N NaOH (10 mL) was added. The mixture was stirred under anhydrous condition at room temperature for 10 hours. TLC showed disappearance of the starting material (Rf = 0.45) and one major spot at the origin (MeOH/CHCl3 5:1). The reaction mixture was dissolved in water (10 mL), the resulting solution was cooled in an ice bath, and the pH was adjusted to 3–4 with drop wise addition of 1N HCl. The resulting suspension was frozen in dry ice-acetone bath, thawed to 4–5 °C in the refrigerator, and filtered. The residue was washed with a small amount of cold water and dried in vacuum using P2O5 to afford compound 15 (157 mg, yield 88%) as yellow powder.
Compound 14; m.p. 157 °C; 1H NMR (DMSO-d6) δ 1.25–1.29 (t, 3H, OCH2CH3, J = 7.2 Hz), 2.75– 2.78 (t, 2H, CH2CH2NCOCF3, J = 6.0 Hz), 3.31–3.35 (t, 2H, CH2CH2NCOCF3, J = 6.0 Hz), 4.18– 4.23 (q, 2H, OCH2CH3, J = 7.2 Hz), 6.00 (s, 1H, C5-CH), 6.01 (bs, 2H, 2-NH2), 6.62–6.64 (d, 2H, Ar-CH, J = 8 Hz), 7.68–7.70 (d, 2H, Ar-CH, J = 8 Hz), 10.25 (s, H, 3-NH), 10.97 (s, H, 7-NH).
Compound 15; m.p. 177 °C; 1H NMR (DMSO-d6) δ 2.74–2.77 (t, 1H, CH2CH2NH, J = 6.0 Hz), 3.29–3.32 (t, 2H, CH2CH2NH, J = 6.0 Hz), 5.98 (s, 1H, C5-CH), 6.11 (bs, 2H, 2-NH2, exch.), 6.54 (t, 1H, CH2CH2NH, exch.), 6.60–6.62 (d, 2H, Ar-CH, J = 8 Hz), 7.65–7.68 (d, 2H, Ar-CH, J = 8 Hz), 10.25 (s, H, 3-NH, exch.), 10.94 (s, H, 7-NH, exch.). Anal. Calcd for (C15H15N5O3· 0.2CHCl3·2.5CH3COOH) C, H, N.
3-[4-(ethoxycarbonyl)phenoxy]propanoic acid (19)
To 100 mL of distilled water in an Erlenmeyer flask was added 4.15 gm (25 mmol) of 18 and the mixture was slowly heated to 80 °C under stirring with the dropwise addition of 25 mL of 1 N NaOH. The clear solution thus obtained was treated with 2 mL of β-propiolactone 17, and the mixture was allowed to stir at this temperature for 0.5 hours. The solution was then cooled to room temperature and acidified with 6N HCl to pH 3. The precipitated material was extracted with three 25 mL portions of ether, and the ether extract was washed twice with distilled water and extracted several times with 20 mL portions of saturated sodium bicarbonate until all the effervescence ceased on further addition of NaHCO3. The bicarbonate extracts were combined and acidified to pH 3 with 6 N HCl. The white precipitate thus obtained was washed with water and dried to afford compound 19 (3.18 gm, 52%); m.p. 146–147 °C; Rf 0.51 (Hexane/EtOAc 1:1); 1H NMR (CDCl3): δ 2.89–2.92 (t, 2 H, CH2CH2O, J = 6.0 Hz), 3.91 (s, 3H, OCH3), 4.31–4.34 (t, 2 H, CH2CH2O, J = 6.0 Hz), 6.94–6.95 (d, 2 H, Ar-CH, J = 7.5 Hz), 8.00–8.02 (d, 2 H, Ar-CH, J = 7.5 Hz).
4-[2-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)ethoxy]benzoic acid (22)
To 19 (2.24 gm,10 mmol) in a 250 mL flask was added oxalyl chloride (7.61 gm, 60 mmol) and anhydrous CH2Cl2 (20 mL). The resulting solution was refluxed for 1 hour and then cooled to room temperature. After the solvent was evaporated under reduced pressure, the residue was dissolved in 20 mL of Et2O. The resulting solution was added dropwise to an ice cooled diazomethane (generated in situ from 15 gm of diazald by using Aldrich Mini Diazald apparatus) in an ice bath over 10 min. The resulting mixture was allowed to stand for 30 min and then stirred for an additional 1 hour. To this solution was added 48% HBr (20 mL). The resulting mixture was refluxed for 1.5 hours. After the mixture was cooled to room temperature, the organic layer was separated, and the aqueous layer was extracted with Et2O (2 × 200 mL). The combined organic layer and Et2O extract was washed with two portions of 10% Na2CO3 solution and dried over Na2SO4. Evaporation of the solvent afforded 20 (2.82 gm, 9.4 mmol) in 94% yield. To a suspension of 2,6-diaminopyrimidin-4-one (1.26 gm, 10 mmol) in anhydrous DMF (25 mL) were added 20 (2.82 gm, 9.4 mmol). The resulting mixture was stirred under N2 at room temperature for 3 days. After evaporation of solvent under reduced pressure, MeOH (20 mL) was added followed by silica gel (5 gm). The resulting plug was loaded on to a silica gel column (3.5 cm × 12 cm) and eluted with CHCl3 followed by 3% MeOH in CHCl3 and then 5% MeOH in CHCl3 (CHCl3/CH3OH, 5:1). Fractions with Rf 0.58 (TLC) were pooled and evaporated to afford 21 (956 gm, yield 31%) as white powder. Compound 21 (0.7 mmol) was dissolved in MeOH (10 mL) added 1N NaOH (10 mL) and the mixture was stirred under anhydrous condition at room temperature for 10 hours. TLC showed the disappearance of the starting material (Rf = 0.58) and one major spot at the origin (MeOH/CHCl3 5:1). The reaction mixture was dissolved in water (10 mL), the resulting solution was cooled in an ice bath, and the pH was adjusted to 3–4 with dropwise addition of 1N HCl. The resulting suspension was frozen in dry ice-acetone bath, thawed to 4–5 °C in the refrigerator, and filtered. The residue was washed with a small amount of cold water and dried in vacuum using P2O5 to afford the target 22 (677 mg, yield 74%) as white powder; m.p.; 159 °C, 1H NMR (DMSO-d6) δ 2.97–2.99 (t, 2H, CH2CH2O, J = 6.0 Hz), 4.25–4.28 (t, 2H, CH2CH2O, J = 6.0 Hz), 6.01 (s, H, C5-CH), 6.26 (bs, 2H, 2-NH2, exch.), 7.04– 7.06 (d, 2H, Ar-CH, J = 8.0 Hz), 7.88–7.90 (d, 2H, Ar-CH, J = 8.0 Hz), 10.35 (s, H, 3-NH, exch.), 11.04 (s, H, 7-NH, exch.). Anal. Calcd for (C15H14N4O4·0.75 H2O) C, H, N.
Ethyl 2-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)acetate (27)
To a solution of 2,4-diamino-6-hydroxypyrimidine 25, (7.20 gm, 50 mmol) and s odium acetate (4.10 gm, 50 mmol) in water (150 mL) at reflux was added, dropwise, ethyl-4-chloroacetoacetate, 26 (7.41 mL, 55 mmol). Within an hour, a thick white precipitate appeared. The mixture was heated at reflux for 18 hours. The suspension was cooled to room temperature, filtered, washed with water (2 × 50 mL), acetone (2 × 50 mL) and dried to afford 7.31 gm (54%) of 27 as a buff-colored solid: m.p. >250 °C; 1H NMR (DMSO- d 6 ) δ 1.16–1.21 (t, 3H, OCH2CH3, J = 7.2 Hz), 3.57 (s, 2H, Ar -CH2), 4.04–4.11 (q, 2H, OCH2CH3, J = 7.2 Hz), 6.00 (s, 1H, C5-CH), 6.04(bs, 2H, 2-NH2, exch), 10.20 (bs, 1H, 3-NH, exch), 10.90 (bs, 1H, 7- NH, exch).
2-Amino-6-(2-hydroxyethyl)-3,7-dihydro-4H-pyrrolo[2,3-d]pyrimidin-4-one (28)
To a suspension of the ester 27 (1.0 gm, 4.24 mmol) in anhydrous tetrahydrofuran (25 mL) at 0 °C was added a 1 M solution of lithium triethylborohydride (Super-Hydride) in tetrahydrofuran (33 mL, 33.92 mmol). The solution was stirred for 30 minutes, after which water (30 mL) was added carefully, followed by acidification of the mixture to pH 5.0 with 5 N hydrochloric acid. Tetrahydrofuran was evaporated under vacuum, and the white precipitate obtained was refrigerated overnight, filtered and dried under vacuum over phosphorous pentoxide to afford 0.615 gm (75%) of 28 as a white solid: m.p. >275 °C; 1H NMR (DMSO- d 6 ) δ 2.60–2.64 (t, 2H, CH2CH2OH, J = 6.0 Hz), 3.55–3.61 (q, 2H, CH2CH2OH, J = 6.0 Hz), 4.61–4.64 (bs, 1H, CH2CH2OH), 5.88 (s, 1H, C5-CH), 5.96 (bs, 2H, 2-NH2, exch), 10.12 (bs, 1H, 3-NH, exch), 10.76 (bs, 1H, 7-NH, exch).
2-(2-Amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)ethyl methanesulfonate (29)
To a solution of the alcohol 28 (0.25 gm, 1.29 mmol) in N,N-dimethylformamide (20 mL) at 0 °C was added triethylamine (0.27 mL, 1.93 mmol) and methanesulfonyl chloride (0.16 gm, 1.42 mmol) and the solution stirred under nitrogen for 2 hours. The reaction mixture was filtered and the filtrate was evaporated to dryness. The residue was suspended in acetone, silica gel (0.50 gm) was added to the suspension and the acetone evaporated to form a plug, which was loaded on top of a silica gel column (20 cm × 2 cm) and eluted using a 5:1 mixture of chloroform:methanol. Fractions containing the product (Rf = 0.41, chloroform:methanol, 3:1) were pooled and the solvent evaporated to afford 0.28 gm (80%) of 29 as a white solid: m.p. >250 °C (dec.); 1H NMR (DMSO-d6) δ 2.89–2.93 (t, 2H, CH2CH2OSO2CH3, J = 6.0 Hz), 3.13 (s, 3H, CH2CH2OSO2CH3), 4.36–4.41 (t, 2H, CH2CH2OSO2CH3, J = 6.0 Hz), 6.01 (bs, 3H, 2-NH2 & CH), 10.17 (bs, 1H, 3-NH, exch), 10.91 (bs, 1H, 7-NH, exch).
Methyl 4-{[2-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6- yl)ethyl]thio}benzoate (30)
To a solution of 4-mercaptobenzoic acid methyl ester (907 mg, 5.4 mmol) in anhydrous DMF (20 mL) was added potassium carbonate (1.12 gm, 8.1 mmol), and the mixture was stirred at room temperature under nitrogen. After 1 h, methanesulfonic ester, 25, (730 mg, 2.7 mmol) was added all at once, and the resulting mixture was stirred for a further 6 h at room temperature. The solvent was evaporated under reduced pressure and the residue was dissolved in methanol. Silica gel (1 gm) was added, and the solvent was evaporated under reduced pressure. The resulting plug was loaded on the top of a silica gel column (2 cm × 15 cm) and eluted using a gradient of with 5–10% MeOH in CHCl3. Fractions that showed the desired product (TLC) were pooled and evaporated to dryness to afford 344 mg (37%) of 30 as a white powder. m.p. 179–180 °C; TLC Rf 0.58 (CHCl3/MeOH 5:1); 1H NMR (DMSO-d6): δ 2.84– 2.87 (t, 2H, CH2CH2S, J = 7.5 Hz), 3.31–3.34 (t, 2H, CH2CH2S, J = 7.5 Hz), 6.02 (s, 1H, C5-CH), 6.10 (s, 2H, 2-NH2, exch), 7.43–7.45 (d, Ar-CH, J = 8.5 Hz), 7.86–7.88 (d, Ar-CH, J = 8.5 Hz), 10.24 (s, 1H, 3-NH, exch), 10.96 (s, 1H, 7-NH, exch).
4-{[2-(2-Amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)ethyl]thio}benzoic acid (31)
To a solution of the 30 (344 mg, 1 mmol) in MeOH (10 mL) was added 1N NaOH (10 mL) and the mixture was stirred at 60 °C for 10 h. The reaction mixture was evaporated to dryness under reduced pressure. The residue was dissolved in distilled water (5 mL), and the pH was adjusted to 3–4 by dropwise addition of 1N HCl. The precipitate was filtered, washed with water and dried under vacuum with P2O5 to afford 330 mg (99%) of 31 as a light yellow powder. m.p. 189–190 °C; 1H NMR (DMSO-d6): δ 2.84–2.87 (t, 2H, CH2CH2S, J = 7.5 Hz), 3.31–3.34 (t, 2H, CH2CH2S, J = 7.5 Hz), 6.04 (s, 1H, C5-CH), 6.25 (s, 2H, 2-NH2, exch), 7.41–7.42 (d, Ar-CH, J = 8.5 Hz,), 7.85–7.87 (d, Ar-CH, J = 8.5 Hz,), 10.39 (s, 1H, 3-NH, exch), 11.04 (s, 1H, 7-NH, exch), 12.79 (s, 1H, COOH, exch).
Diethyl (4-{[2-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6- yl)ethyl]thio}benzoyl)-L-glutamate (32)
To a solution of 31, (330 mg, 1 mmol) in anhydrous DMF (20 mL) was added N-methylmopholine (0.2 mL, 1.8 mmol) and 2-chloro-4,6-dimethoxy- 1,3,5-triazine (317 mg, 1.8 mmol). The resulting mixture was stirred at room temperature for 2 h. To this mixture was added N-methylmopholine (0.2 mL, 1.8 mmol) and L-glutamic acid diethyl ester hydrochloride (360 mg, 1.5 mmol). The reaction mixture was stirred for an additional 4 h at room temperature and then evaporated to dryness under reduced pressure. The residue was mixed with 1 g of silica gel to make a plug and chromatographed on a silica gel column (2 × 15 cm) with 10% MeOH in CHCl3 as the eluent. Fractions that showed the desired spot (TLC) were pooled and evaporated to dryness to afford 309 mg (60%) of 32 as a light yellow powder. m.p. 84–85 °C; TLC Rf 0.59 (CHCl3/MeOH 5:1); 1H NMR (DMSO-d6): δ 1.15–1.21 (m, 6H, OCH2CH3), 1.97–2.15 (m, 2H, β-CH2), 2.43–2.46 (t, 2H, γ-CH2, J = 7.5 Hz), 2.82–2.85 (t, 2H, CH2CH2S, J = 7.5 Hz), 3.29–3.33 (t, 2H, CH2CH2S, J = 7.5 Hz), 4.03–4.13 (m, 4H, 2OCH2CH3), 4.41–4.45 (m, 1H, C5-CH), 6.00 (s, 1H, C5-CH), 6.15 (s, 2H, 2-NH2, exch), 7.41–7.43 (d, J = 8.5 Hz, Ar-CH), 7.83–7.85 (d, J = 8.5 Hz, Ar-CH), 8.71–8.72 (d, J = 7.5 Hz, 1H, CONH, exch), 10.29 (s, 1H, 3- NH, exch), 10.96 (s, 1H, 7-NH, exch).
Molecular Modeling and Computational Studies
The X-ray crystal structures of human FRα bound to folic acid (PDB: 4LRH, 2.80 Å)45 and human GARFTase bound to 3 (PDB: 4ZYW, 2.05 Å)18 were obtained from the protein database. Antifolate GARFTase docking studies were performed using LeadIT 2.1.6.54 Default settings were used to calculate the protonation state of the proteins and the ligands. Free rotation of water molecules in the ligand binding site (defined by amino acids within 6.5 Å from the crystal structure ligand) was permitted. Ligands for docking were sketched using MOE 2013.08,55 and energy was minimized using the MMF94X force field (limit of 0.05 kcal/mol). Molecules were docked using the triangle matching placement method and scored using default settings. The docked poses were visualized using CCP4MG.56
In order to validate the docking process (using LeadIT 2.1.6), the crystallized ligands (folic acid for FRα and compound 3 for GARFTase) were sketched using MOE, energy minimized and docked. Deviation of the best docked poses from the crystal structure conformation was calculated using an RMSD SVL code obtained from the ChemComp website (www.chemcomp.com). The best scored pose of folic acid in FRα had an RMSD of 0.81 Å. Compound 3 in the human GARFTase had an RMSD of 1.04 Å. Thus, LeadIT 2.1.6 was validated for our docking purposes in FRα and human GARFTase.
Compound 4 was docked into a structure of FRα with folic acid removed from the coordinate file (PDB:4LRH), using the standard flexible-ligand sampling algorithm57 implemented in DOCK6.72.58 Ligand placement was prioritized by the combined grid score (vdw+es) and the atom footprint similarity was compared to folic acid. Prior to docking, partial charges from the all-atom AMBER ff14SB359 were assigned to the receptor and semi-empirical AM1-BCC charges were derived for each antifolate using UCSF chimera.60
Reagents for biological studies
[3′, 5′, 7, 9-3H]Folic acid (25 Ci/mmol), [3′, 5′, 7-3H]MTX (20 Ci/mmol), and [14C(U)]-glycine (87mCi/mmol) were purchased from Moravek Biochemicals (Brea, CA). Unlabeled folic acid was purchased from the Sigma-Aldrich (St. Louis, MO). LCV [(6R,S)5-formyl tetrahydrofolate] was provided by the Drug Development Branch, National Cancer Institute (Bethesda, MD). The sources of the classical antifolate drugs were as follows: MTX, Drug Development Branch, National Cancer Institute; LMTX (5,10-dideaza-5,6,7,8- tetrahydrofolate) and PMX [N-{4-[2-(2-amino-3,4-dihydro-4-oxo-7H-pyrrolo[2,3-d]pyrimidin-5- yl)ethyl]benzoyl}-L-glutamic acid] (Alimta), Eli Lilly and Co. (Indianapolis, IN); RTX [N-(5-[N-(3,4- dihydro-2-methyl-4-oxyquinazolin-6-ylmethyl)-N-methyl-amino]-2-thienoyl)-L-glutamic acid], AstraZeneca Pharmaceuticals (Maccesfield, Cheshire, England); and PT523 [N(alpha)-(4-amino-4-deoxypteroyl)-N(delta)-hemiphthaloyl-L-ornithine],30 A. Rosowsky (Boston, MA). Other chemicals were obtained from commercial sources in the highest available purities.
Cell lines and assays of antitumor drug activities
The engineered CHO sublines including RFC-, PCFT- and FRα-null MTXRIIOuaR2-4 (R2), and RFC- (pC43-10), PCFT- (R2/PCFT4), or FRα-(RT16) and FRβ-(D4) expressing CHO sublines were previously described.29, 46–48 The CHO cells were cultured in α-minimal essential medium (MEM) supplemented with 10% bovine calf serum (Invitrogen, Carlsbad, CA), penicillin (1000 U/mL) streptomycin (1000 μg/mL) and 2 mM L-glutamine at 37° C with 5% CO2. All the R2 transfected cells (PC43-10, RT16, R2/hPCFT4) were cultured in complete α-MEM media plus 1 mg/mL G418. Prior to the cytotoxicity assays (see below), RT16 and D4 cells were cultured for 3 days in complete folatefree RPMI 1640 (without added folate), plus dialyzed fetal bovine serum (FBS) (Sigma-Aldrich) and penicillin/streptomycin. KB human nasopharengeal carcinoma cells were purchased from the American Type Culture Collection (Manassas, VA). IGROV1 ovarian carcinoma cells were a gift of Dr. Manohar Ratnam (Karmanos Cancer Institute). IGROV1 and KB cells were routinely cultured in folate-free RPMI 1640 medium, supplemented with 10% FBS (Sigma-Aldrich), penicillin-streptomycin solution, and 2 mM L-glutamine.
For growth inhibition studies, cells (CHO, KB, IGROV1) were plated in 96 well dishes (~2500– 5000 cells/well; total volume of 200 μl medium) with a range of antifolate concentrations (0–1000 nM).31, 61 The experiments with RT16, D4, KB, and IGROV1 cells used folate-free RPMI medium with 10% dialyzed FBS, antibiotics and L-glutamine; the medium was supplemented with 2 nM LCV. To confirm FR-mediated drug uptake, 200 nM folic acid was added to parallel incubations. For experiments with R2, PC43-10, and R2/PCFT4 cells, cells were routinely cultured in folate-free RPMI 1640 (pH 7.2)/10% dialyzed FBS with antibiotics and L-glutamine, supplemented with 25 nM LCV. Cells were incubated up to 96 h and viable cells were assayed with Cell-Titer Blue™ reagent (Promega, Madison, WI), with fluorescence measured with a fluorescence plate reader. Fluorescence data were analyzed for calculations of IC50s, corresponding to the drug concentrations that resulted in 50% loss of cell proliferation.
To confirm the targeted pathway/enzyme target, in vitro growth inhibition of KB tumor cells was measured in the presence of thymidine (10 μM) or adenosine (60 μM).29–31, 47 For de novo purine biosynthesis, additional protection experiments used AICA (320 μM) to distinguish inhibitory effects at GARFTase from those at AICARFTase.29–31, 47
FR binding assay
To measure relative binding affinities for antifolate drugs,29–31, 47 RT16 (expresses FRα) and D4 (FRβ) CHO cells were cultured in 60 mm dishes until they were ~80% confluent. Cells (2–4 × 106 cells) were sequentially rinsed at 4°C with Dulbecco’s phosphate-buffered saline (DPBS) (3x), followed by acidic buffer (10 mM sodium acetate, 150 mM NaCl, pH 3.5) (2x) (removes FR-bound folates), and finally HEPES-buffered saline (20 mM HEPES, 140 mM NaCl, 5 mM KCl, 2 mM MgCl2, 5 mM glucose, pH7.4) (HBS). Cells were incubated with [3H]folic acid (50 nM, specific activity 0.5 Ci/mmol) in HBS in the presence and absence of unlabeled folic acid, MTX (negative control), or the 6-pyrrolo[2,3-d]pyrimidine antifolates (range of concentrations up to 1000 nM) for 15 min at 0°C. Dishes were rinsed with HBS (0–4°C, 3x). Cells were solubilized with 0.5 N NaOH, and aliquots were measured for radioactivity and protein contents. Protein concentrations were quantified with Folin-phenol reagent.62 FR-bound [3H]folic acid was calculated as pmol/mg protein and binding affinities were calculated as the inverse molar ratios of unlabeled ligands required to inhibit [3H]folic acid binding by 50%. The relative affinity of folic acid was assigned a value of 1.
Transport assays
R2 and R2/hPCFT4 CHO sublines were grown in suspension as spinner cultures at densities of 2–5 × 105 cells/mL. Cells were collected by centrifugation, washed with DPBS, and the cell pellets (~2 × 107 cells) were suspended in transport buffer (2 mL) for cellular uptake assays. PCFT-dependent uptake of 0.5 μM [3H]MTX was assayed in cell suspensions over 2 min at 37° C in HBS at pH 6.8, or in 4-morpholinopropane sulfonic (MES)-buffered saline (20 mM MES, 140 mM NaCl, 5 mM KCl, 2 mM MgCl2, and 5 mM glucose) at pH 5.5 in the presence of 1 or 10 μM inhibitor. At the end of the incubations, transport was quenched with ice-cold DPBS, cells were washed three times with ice-cold DPBS, and cellular proteins were solubilized with 0.5 N NaOH. Levels of drug uptake were expressed as pmol/mg protein, calculated from direct measurements of radioactivity and protein contents of the cell homogenates. Proteins were quantified using Folin-phenol reagent.62 Transport results were normalized to levels in untreated controls. For determining Ki values for the 6-pyrrolo[2,3- d]pyrimidine antifolates compared to PMX, transport was measured over 2 min with 0.5 μM [3H]MTX and 0–5 μM of the unlabeled antifolate. Data were analyzed by Dixon plots.
In situ GARFTase assays
Inhibition of intracellular GARFTase in the presence of antifolate inhibitors was measured by following incorporation of [14C(U)] glycine into [14C]formyl GAR in cells treated with azaserine.30, 32, 33, 35, 63 KB cells in 60 mm dishes were washed with folate-free RPMI 1640 (L-glutamine-free) with 10% dialyzed FBS, penicillin-streptomycin and 2 nM LCV. Cells were incubated at 37° C for 1 h in folate and L-glutamine-free RPMI 1640 media/10% dialyzed FBS (supplemented with 2 nM LCV), with antifolate inhibitors over a range of concentrations. Azaserine (4 μM final) was added and the cells were incubated for 30 min, after which L-glutamine (2 mM) and [14C(U)]glycine (final specific activity 0.1 mCi/L) were added. Cells were incubated an additional 16 h at 37° C, washed with ice-cold DPBS, trypsinized and collected. The cells were treated with 5% trichloroacetic acid (TCA) at 0 37° C, the samples centrifuged (4°C, 14,000 rpm) and the precipitated proteins solubilized (0.5 N NaOH). Proteins were quantified with the Folin-phenol protein method.62 The TCA supernatants were extracted with ether at 4° C, and the aqueous layer was fractionated by anion-exchange chromatography on ~1 mL columns of [AG1x8 (chloride form)]. Columns were washed with 10 mL of 0.5 N formic acid, followed by 10 mL of 4 N formic acid; the bound [14C]formyl GAR was eluted with 1 N HCl as eight 1 mL fractions. The 1 N HCl eluate was quantified for radioactivity and the percentages of radioactivity in the [14C]formyl GAR and non-specific [14C]fractions were calculated. Results were expressed as cpm [14C]formyl GAR/mg cell protein. IC50s corresponding to antifolate concentrations that inhibit accumulation of [14C]formyl GAR compared to untreated controls were calculated by plotting percent inhibition versus antifolate concentration.
In vitro GARFTase assays and Ki determinations
For in vitro enzymatic assays of GARFTase with pyrrolo[2,3-d]pyrimidine inhibitors, HisGARFTase (formyltransferase domain), containing an N-terminal cleavable hexahistidine tag, was purified as described previously.18 GARFTase catalytic activity was measured by following the formation of 5,8-dideazafolate spectrophotometically from 10-formyl-5,8-dideazafolate in the presence of varying concentrations of antifolate.64 Assays included 30 μM α,β-GAR, 5.4 μM 10-formyl-5,8- dideazafolate, and a range of antifolates in 0.1 M Hepes, pH 7.5 (150 μL total volume). Reactions were pre-incubated at 37° C in a UV transparent 96-well plate (Costar) and initiated by adding 150 μL of 20 nM GARFTase or buffer (reference wells). Measurements were recorded at 295 nm every fifteen seconds over twenty minutes using a BioTek Synergy H1M plate reader. Tripicate assays were performed. For determining the initial rate for each drug concentration, absorbance of the reference well was subtracted. Initial rate changes in absorbance at 295 nm were determined and correlation coefficients were compared over appropriate time spans to identify regions over which linear absorbance increases occurred for all replicates. Initial slopes were graphed against the antifolate concentrations and a hyperbola fit [y = (−a*x/(Ki + x)) + b, where “a” is the amplitude and “b” is the y-intercept] was used to calculate the Ki value for each compound (KaleidaGraph version 4.1).
In vivo efficacy study with IGROV1 human ovarian tumor xenografts
Protocols for drug treatments, toxicity evaluations, and data analysis using transplantable human cell line xenografts have been previously described.30, 31, 65–68 IGROV1 ovarian tumor cells were implanted subcutaneously (5 × 106 cells/flank) to establish a solid tumor xenograft model in female NCR SCID mice (NCI Animal Production Program). For drug efficacy studies, 11 week old mice (20 g average body weight) were provided food and water ad libitum. Mice were fed either a folate-deficient diet (Harlan-Teklad,TD.00434) or a standard (folate-replete) diet (Lab Diet, 5021; autoclavable mouse breeder diet). For the trial, mice were placed on their respective diets 14 days before tumor implant so that the serum folate levels for mice maintained on the folate-deficient diet would approximate those in humans.52 A Lactobacillus casei bioassay69 was used to measure serum folate levels. Mice were pooled for each dietary group and implanted bilaterally subcutaneously with 30–60 mg tumor fragments using a 12 gauge trocar, then unselectively distributed to the various drug treatment and control groups. Drugs were administered by tail vein injection beginning 2 days post-tumor implantation, when the tumors had established blood supply but were below the limit of palpation (63 mg). Tumors were measured with a caliper two-to-three times weekly and mice were weighed and assessed daily. Animals were sacrificed when the cumulative tumor size reached 1500 mg (while still asymptomatic). Tumor weights were estimated from two-dimensional measurements [i.e., tumor mass (in mg) = (a × b2)/2, where a and b are the tumor length and width in mm, respectively]; for calculating the endpoints, both tumor masses on each flank were added together. Quantitative end-points to assess anti-tumor activity include: (i) tumor growth delay [T-C, where T is the median time in days required for the treatment group tumors to reach a predetermined size (e.g., 1000 mg), and C is the median time in days for the control group tumors to reach the same size; tumor-free survivors are excluded from these calculations]; and (ii) tumor cell kill [log10 cell kill total (gross) = (T – C)/(3.32)(Td), where T - C is the tumor growth delay, as described above, and Td is the tumor volume doubling time in days, estimated from the best fit straight line from a log-linear growth plot of control group tumors in exponential growth (100 to 800 mg range)]. A T/C value (in percent) was also determined for each treatment group when treatment (T) and control (C) groups for the control groups reached 700–1200 mg in size (exponential growth phase). The median and mean values for each group was determined (including zeros). T/C is a non-quantitative determination of antitumor activity and the inverse of tumor growth inhibition. Rather, T/C is a measure of antitumor effectiveness based on the specific day of caliper measurement.
Crystallization and X-ray data collection and structure determination of human GARFTase
GARFTaseHis protein (formyltransferase domain; containing a non-cleavable C-terminal hexahistidine) purification and crystallization were performed as described in our previous work.18 Briefly, purified GARFTaseHis at a concentration of 10 mg/mL in 25 mM Tris (pH 8), 200 mM NaCl and 10 mM β-mercaptoethanol was incubated with a 3-fold molar excess of α, β-GAR and compound 4 (dissolved in DMSO) for 2 h at 4° C. Sitting drop vapor diffusion plates were set up with 1 μL protein, 1 μL crystallant and 0.2 μL of either 32 mM N-nonyl-β-D-thiomaltoside or 9 mM N-decyl-β-D-thiomaltoside (Hampton Research), using 0.1 M Tris (pH 7.5), 0.333 mM NaCl, 18% polyethylene glycol (PEG) 3350, and 2% PEG 400 as the crystallant solution. Plates were incubated at 4° C and crystals formed within a few days. Crystals were cryoprotected via addition of PEG 400 to 14% stepwise via incremental transfers, increasing PEG at 2% per step. Cyroprotectant solutions included α,β-GAR and compound 4 at the same concentration as the initial crystallization mixture. Data collection was performed at the Advanced Photon Source beamline 24-ID-E, part of the Northeastern Collaborative Access Team (NE-CAT). All data sets were processed to a P3221 space group using the RAPD data processing protocol implemented at NE-CAT. Molecular replacement was performed using one chain of 4ZZ1 with ligands and waters removed as a search model (PHENIX).70, 71 Subsequent model building and refinement were performed using Coot and PHENIX.72, 73 The final GARFTase/compound 4/β-GAR model was refined to give Rwork and Rfree of 17.8 % and 21.5 %, respectively.
Expression and purification of human FRα
Human FRα (23–234) in fusion with a human IgG Fc tag (FRα-FcH6)45 was expressed as stable clone in HEK293 cells maintained in 500 mL of Dubecco’s minimal essential medium (DMEM) supplemented with 5% FBS, 20 mM HEPES, 5 μM kifunensine, and 200 μM folic acid in 1 L roller bottles at 37° C, as previously described.45 Two liters of conditioned medium were collected, concentrated to 400 mL, and dialyzed against TBS buffer (20 mM Tris, pH8.0, 150 mM NaCl, and 5% glycerol) at 4° C overnight before loading on a 50 mL Ni-chelating Sepharose column (GE Healthcare). The column was washed with 300 mL buffer A (25 mM Tris, pH 8.0, 150 mM NaCl, 25 mM imidazole, and 5% glycerol) and eluted with buffer B (25 mM Tris, pH 8.0, 150 mM NaCl, and 500 mM imidazole). The peak fractions were pooled and buffer-exchanged against 20 mM citrate (pH 5.5), 150 mM NaCl, and 5% glycerol for deglycosylation using endoglycosidase Hf (New England Biolabs) at room temperature overnight. The endogenous folic acid ligand was dissociated from FRα-FcH6, as described.50 Briefly, the FRα-FcH6 buffer was exchanged against 25 mM acetate (pH 3.5) with 20% FBS and incubated with 0.5 mL active charcoal (Sigma-Aldrich) at room temperature for 15 min. Charcoal was then removed by centrifugation and the pH adjusted to pH 7.5 using 1 M Tris, pH 11. A 6-fold molar excess of compound 4 (relative to protein) was added. The mixture was incubated at 4° C overnight. The 4/FRα-FcH6 complex was then purified over a 5 mL Nichelating Sepharose column. The peak fractions were pooled, concentrated, buffer-exchanged against TBS buffer with 4 at a final 2x concentration of FRα-FcH6 and subjected to thrombin digestion at a 1:1000 mass ratio at 4° C overnight. The digestion product was loaded on a 5 mL Ni-NTA column to remove the FcH6 tag. The flow-through was collected and was finally separated by Sephadex S-200 gel filtration in buffer C (25 mM Tris, pH 8.0, 200 mM ammonium acetate, 1 mM EDTA). The protein eluted from the gel filtration column at a volume corresponding to the size of a monomer with a purity >95%, as judged by SDS/polyacrylamide gel electrophoresis.
Soluble FRα radioligand-binding assay
The affinities of folic acid and compound 4 were estimated by homologous (folic acid) and heterologous (antifolates) competition assays. First, the endogenous ligand was stripped, as described,45 and then exchanged against [3H]folic acid in 100 μL binding buffer (25 mM Tris, pH 8.0, 150 mM NaCl, 0.1% Triton X-100). The FRα/[3H]folic acid complex was then incubated with increasing concentrations of homologous or heterologous competitor, as indicated. Unbound ligands were removed by two 100 μL washes in binding buffer and remaining FRα/[3H]folic acid complex determined by scintillation counting.
FRα crystallization, data collection and structure determination
The purified FRα/compound 4 complex was supplemented with a two-fold molar excess of 4 and concentrated to ~6 mg/mL before setting up crystallization trials using a Phenix crystallization robot (Art Robbin Instruments) at 20 °C. Commercial screens (Hampton Research) were used for initial high-throughput sitting drop screening in 96-well intelliplates (Rigaku), with a mixture of 0.2 μl protein and 0.2 μl crystallization solution sitting in 50 μl reservoir solution. Once crystals appeared in the initial screen, optimization trays in 24-well VDX plates (Hampton Research) were set up manually using the hanging drop method at 20° C. Crystals were grown at 20° C containing 1 μl of the purified protein and 0.5 μl of well solution [10% (w/v) PEG 10K, 0.1 M BIS-TRIS (pH 6.5), 0.2 M potassium sodium tartrate tetrahydrate]. Crystals appeared in two-to-three weeks and grew to a dimension of ~100 μm length with a cubic shape.
Crystals were transferred to well solution with 20% (v/v) ethylene glycol as a cryoprotectant before flash freezing in liquid nitrogen. Data collection was performed at sector 21-ID-D (LS-CAT) of the APS synchrotron using single crystals and the diffraction data were processed using XDS,74 combined using Pointless, and merged using Scala of the CCP4 suite.75 According to Matthew’s coefficient calculation, the crystals have a large unit cell with eight molecules per asymmetric unit. Eight molecules in one asymmetric unit were located by molecular replacement using Phaser76 with the FRα/folic acid crystal structure with ligand removed (PDB code: 4LRH) as a search model. The eight molecules of the FRα models were refined against the diffraction data with 8-fold non-crystallographic symmetry restraints using the Refmac program of CCP4.77 The densities for compound 4 became clear after several rounds of model adjustments and refinements and six molecules of compound 4 were built into the final model. The final model was refined to give Rwork and Rfree of 22.2 % and 26.5 %, respectively (Table 2S, Supporting Information).
Supplementary Material
Acknowledgments
This work was supported in part by grants from the National Institutes of Health, CA53535 (LHM and ZH), CA125153 (AG), CA152316 (LHM and AG), CA166711 (AG, LHM and CED), GM094472 (CED), DK071662 (HEX), GM102545 (KM) and GM104212 (KM), National Science Foundation grant NSF91217311 (HEX), the Eunice and Milt Ring Endowed Chair for Cancer Research (LHM), the Duquesne University Adrian Van Kaam Chair in Scholarly Excellence (AG), the Van Andel Research Institute (HEX and KM), and Ministry of Science and Technology (China) grants 2012ZX09301001, 2012CB910403, 2013CB910600, XDB08020303, and 2013ZX09507001. Mr. Wilson was supported by T32 CA009531 (LHM). Ms. Deis was supported by T32 GM007757. The Animal Model and Therapeutics Evaluation Core (AMTEC; LP, KW, JK) was supported, in part, by NIH Center grant P30 CA022453 to the Karmanos Cancer Institute and the Wayne State University. We thank T.S. Widlanski for providing the α, β-GAR substrate for crystallography and enzyme inhibition experiments. All GARFTase crystallization experiments were carried out in the Indiana University METACyt Crystallization Automation Facility. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We thank staff members of the Life Science Collaborative Access Team of the Advanced Photon Source (APS) for assistance in collection of FRα diffraction data 0 at the beam lines of sector 21, which is in part funded by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). Use of APS was supported by the Office of Science of the US Department of Energy, under Contract No. DE-AC02-06CH11357.
ABBREVIATIONS USED
- AICA
5-aminoimidazole-4-carboxamide
- AICARFTase
5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase
- CHO
Chinese hamster ovary
- FBS
fetal bovine serum
- DMEM
Dulbecco’s minimal essential medium
- DPBS
Dulbecco’s phosphate-buffered saline
- FR
folate receptor
- 10-CHOTHF
N10-formyl tetrahydrofolate
- GAR
glycinamide ribonucleotide
- GARFTase
glycinamide ribonucleotide formyltransferase
- HBSS
Hank’s balanced salts solution
- HBS
HEPES-buffered saline
- LMTX
lometrexol
- LCV
leucovorin
- MTX
methotrexate
- MEM
minimal essential media
- PMX
pemetrexed
- PEG
polyethylene glycol
- PDX
pralatrexate
- PCFT
proton-coupled folate transporter
- PEG
polyethylene glycol
- RTX
raltitrexed
- RFC
reduced folate carrier
- SCID
severe-combined immunodeficient
- TAMs
tumor-associated macrophages
- TCA
trichloroacetic acid
- TLC
thin layer chromatography
Footnotes
Notes
The authors declare no competing financial interest.
PDB ID codes: FRα/Cpd 4 PDB ID: 51ZQ; GARFTase/Cpd 4/β-GAR PDB ID: 5J9F.
Authors will release the atomic coordinates and experimental data upon article publication.
Competing Interests: The authors declare that they have no competing interests. Atomic coordinates and structure factors for the reported crystal structures of FRα and GARFTase in complex with compound 4 has been deposited in the Protein Data Bank (PDB ID codes 51ZQ and 5J9F, respectively; see Table S2, Supporting Information). Authors will release the atomic coordinates and experimental data upon article publication.
Supporting Information Available: Elemental Analysis, X-ray diffraction data and refinement statistics and Figure 1S; graphs of antifolate concentration against initial slope of GARFTase activity; 1H NMR spectra of final compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Stokstad ELR. Historical Perspective on Key Advances in the Biochemistry and Physiology of Folates. Wiley-Liss; New York: 1990. pp. 1–21. [Google Scholar]
- 2.Matherly LH, Hou Z, Deng Y. Human reduced folate carrier: translation of basic biology to cancer etiology and therapy. Cancer Metastasis Rev. 2007;26:111–128. doi: 10.1007/s10555-007-9046-2. [DOI] [PubMed] [Google Scholar]
- 3.Elnakat H, Ratnam M. Distribution, functionality and gene regulation of folate receptor isoforms: Implications in targeted therapy. Adv Drug Deliv Rev. 2004;56:1067–1084. doi: 10.1016/j.addr.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Zhao R, Goldman ID. The molecular identity and characterization of a proton-coupled folate transporter--PCFT; biological ramifications and impact on the activity of pemetrexed. Cancer Metastasis Rev. 2007;26:129–139. doi: 10.1007/s10555-007-9047-1. [DOI] [PubMed] [Google Scholar]
- 5.Desmoulin SK, Hou Z, Gangjee A, Matherly LH. The human proton-coupled folate transporter: Biology and therapeutic applications to cancer. Cancer Biol Ther. 2012;13:1355–1373. doi: 10.4161/cbt.22020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qiu A, Jansen M, Sakaris A, Min SH, Chattopadhyay S, Tsai E, Sandoval C, Zhao R, Akabas MH, Goldman ID. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell. 2006;127:917–928. doi: 10.1016/j.cell.2006.09.041. [DOI] [PubMed] [Google Scholar]
- 7.Qiu A, Min SH, Jansen M, Malhotra U, Tsai E, Cabelof DC, Matherly LH, Zhao R, Akabas MH, Goldman ID. Rodent intestinal folate transporters (SLC46A1): secondary structure, functional properties, and response to dietary folate restriction. Am J Physiol Cell Physiol. 2007;293:C1669–1678. doi: 10.1152/ajpcell.00202.2007. [DOI] [PubMed] [Google Scholar]
- 8.Inoue K, Nakai Y, Ueda S, Kamigaso S, Ohta KY, Hatakeyama M, Hayashi Y, Otagiri M, Yuasa H. Functional characterization of PCFT/HCP1 as the molecular entity of the carrier-mediated intestinal folate transport system in the rat model. Am J Physiol Gastrointest Liver Physiol. 2008;294:G660–668. doi: 10.1152/ajpgi.00309.2007. [DOI] [PubMed] [Google Scholar]
- 9.Chladek J, Grim J, Martinkova J, Simkova M, Vaneckova J. Low-dose methotrexate pharmacokinetics and pharmacodynamics in the therapy of severe psoriasis. Basic Clin Pharmacol Toxicol. 2005;96:247–248. doi: 10.1111/j.1742-7843.2005.pto960318.x. [DOI] [PubMed] [Google Scholar]
- 10.Gonen N, Assaraf YG. Antifolates in cancer therapy: structure, activity and mechanisms of drug resistance. Drug Resist Updat. 2012;15:183–210. doi: 10.1016/j.drup.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 11.Monahan BP, Allegra CJ. Antifolates. Lippincott Williams and Wilkins; Philadelphia, PA: 2011. pp. 109–138. [Google Scholar]
- 12.Visentin M, Zhao R, Goldman ID. The antifolates. Hematol Oncol Clin North Am. 2012;26:629–648. doi: 10.1016/j.hoc.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wessels JA, Huizinga TW, Guchelaar HJ. Recent insights in the pharmacological actions of methotrexate in the treatment of rheumatoid arthritis. Rheumatology (Oxford) 2008;47:249–255. doi: 10.1093/rheumatology/kem279. [DOI] [PubMed] [Google Scholar]
- 14.Chattopadhyay S, Moran RG, Goldman ID. Pemetrexed: biochemical and cellular pharmacology, mechanisms, and clinical applications. Mol Cancer Ther. 2007;6:404–417. doi: 10.1158/1535-7163.MCT-06-0343. [DOI] [PubMed] [Google Scholar]
- 15.Racanelli AC, Rothbart SB, Heyer CL, Moran RG. Therapeutics by cytotoxic metabolite accumulation: pemetrexed causes ZMP accumulation, AMPK activation, and mammalian target of rapamycin inhibition. Cancer Res. 2009;69:5467–5474. doi: 10.1158/0008-5472.CAN-08-4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shih C, Thornton DE. Preclinical pharmacology studies and the clinical development of a novel multitargeted antifolate, MTA (LY231514) In: Jackman AL, editor. Anticancer Drug Development Guide: Antifolate Drugs in Cancer Therapy. Humana Press, Inc; Totowa, NJ: 1999. pp. 183–201. [Google Scholar]
- 17.Kugel Desmoulin S, Wang L, Hales E, Polin L, White K, Kushner J, Stout M, Hou Z, Cherian C, Gangjee A, Matherly LH. Therapeutic targeting of a novel 6-substituted pyrrolo [2,3-d]pyrimidine thienoyl antifolate to human solid tumors based on selective uptake by the proton-coupled folate transporter. Mol Pharmacol. 2011;80:1096–1107. doi: 10.1124/mol.111.073833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L, Wallace A, Raghavan S, Deis SM, Wilson MR, Yang S, Polin L, White K, Kushner J, Orr S, George C, O’Connor C, Hou Z, Mitchell-Ryan S, Dann CE, 3rd, Matherly LH, Gangjee A. 6-Substituted pyrrolo[2,3-d]pyrimidine thienoyl regioisomers as targeted antifolates for folate receptor alpha and the proton-coupled folate transporter in human tumors. J Med Chem. 2015;58:6938–6959. doi: 10.1021/acs.jmedchem.5b00801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson MR, Hou Z, Yang S, Polin L, Kushner J, White K, Huang J, Ratnam M, Gangjee A, Matherly LH. Targeting nonsquamous nonsmall cell lung cancer via the proton-coupled folate transporter with 6-substituted pyrrolo[2,3-d]pyrimidine thienoyl antifolates. Mol Pharmacol. 2016;89:425–434. doi: 10.1124/mol.115.102798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salazar MD, Ratnam M. The folate receptor: what does it promise in tissue-targeted therapeutics? Cancer Metastasis Rev. 2007;26:141–152. doi: 10.1007/s10555-007-9048-0. [DOI] [PubMed] [Google Scholar]
- 21.Xia W, Low PS. Folate-targeted therapies for cancer. J Med Chem. 2010;53:6811–6824. doi: 10.1021/jm100509v. [DOI] [PubMed] [Google Scholar]
- 22.Puig-Kroger A, Sierra-Filardi E, Dominguez-Soto A, Samaniego R, Corcuera MT, Gomez-Aguado F, Ratnam M, Sanchez-Mateos P, Corbi AL. Folate receptor beta is expressed by tumor-associated macrophages and constitutes a marker for M2 antiinflammatory/ regulatory macrophages. Cancer Res. 2009;69:9395–9403. doi: 10.1158/0008-5472.CAN-09-2050. [DOI] [PubMed] [Google Scholar]
- 23.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 24.Vergote I, Leamon CP. Vintafolide: A novel targeted therapy for the treatment of folate receptor expressing tumors. Ther Adv Med Oncol. 2015;7:206–218. doi: 10.1177/1758834015584763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vergote IB, Marth C, Coleman RL. Role of the folate receptor in ovarian cancer treatment: evidence, mechanism, and clinical implications. Cancer Metastasis Rev. 2015;34:41–52. doi: 10.1007/s10555-014-9539-8. [DOI] [PubMed] [Google Scholar]
- 26.Naumann RW, Coleman RL, Burger RA, Sausville EA, Kutarska E, Ghamande SA, Gabrail NY, Depasquale SE, Nowara E, Gilbert L, Gersh RH, Teneriello MG, Harb WA, Konstantinopoulos PA, Penson RT, Symanowski JT, Lovejoy CD, Leamon CP, Morgenstern DE, Messmann RA. PRECEDENT: a randomized phase II trial comparing vintafolide (EC145) and pegylated liposomal doxorubicin (PLD) in combination versus PLD alone in patients with platinum-resistant ovarian cancer. J Clin Oncol. 2013;31:4400–4406. doi: 10.1200/JCO.2013.49.7685. [DOI] [PubMed] [Google Scholar]
- 27.Reddy JA, Bloomfield A, Nelson M, Dorton R, Vetzel M, Leamon CP. Preclinical development of EC1456: a potent folate targeted tubulysin small molecule drug conjugate. Proceedings AACR. 2014;55:199. [Google Scholar]
- 28.Gibbs DD, Theti DS, Wood N, Green M, Raynaud F, Valenti M, Forster MD, Mitchell F, Bavetsias V, Henderson E, Jackman AL. BGC 945, a novel tumor-selective thymidylate synthase inhibitor targeted to alpha-folate receptor-overexpressing tumors. Cancer Res. 2005;65:11721–11728. doi: 10.1158/0008-5472.CAN-05-2034. [DOI] [PubMed] [Google Scholar]
- 29.Deng Y, Wang Y, Cherian C, Hou Z, Buck SA, Matherly LH, Gangjee A. Synthesis and discovery of high affinity folate receptor-specific glycinamide ribonucleotide formyltransferase inhibitors with antitumor activity. J Med Chem. 2008;51:5052–5063. doi: 10.1021/jm8003366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L, Cherian C, Desmoulin SK, Polin L, Deng Y, Wu J, Hou Z, White K, Kushner J, Matherly LH, Gangjee A. Synthesis and antitumor activity of a novel series of 6- substituted pyrrolo[2,3-d]pyrimidine thienoyl antifolate inhibitors of purine biosynthesis with selectivity for high affinity folate receptors and the proton-coupled folate transporter over the reduced folate carrier for cellular entry. J Med Chem. 2010;53:1306–1318. doi: 10.1021/jm9015729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L, Kugel Desmoulin S, Cherian C, Polin L, White K, Kushner J, Fulterer A, Chang MH, Mitchell-Ryan S, Stout M, Romero MF, Hou Z, Matherly LH, Gangjee A. Synthesis, biological, and antitumor activity of a highly potent 6-substituted pyrrolo[2,3- d]pyrimidine thienoyl antifolate inhibitor with proton-coupled folate transporter and folate receptor selectivity over the reduced folate carrier that inhibits beta-glycinamide ribonucleotide formyltransferase. J Med Chem. 2011;54:7150–7164. doi: 10.1021/jm200739e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang L, Cherian C, Kugel Desmoulin S, Mitchell-Ryan S, Hou Z, Matherly LH, Gangjee A. Synthesis and biological activity of 6-substituted pyrrolo[2,3-d]pyrimidine thienoyl regioisomers as inhibitors of de novo purine biosynthesis with selectivity for cellular uptake by high affinity folate receptors and the proton-coupled folate transporter over the reduced folate carrier. J Med Chem. 2012;55:1758–1770. doi: 10.1021/jm201688n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitchell-Ryan S, Wang Y, Raghavan S, Ravindra MP, Hales E, Orr S, Cherian C, Hou Z, Matherly LH, Gangjee A. Discovery of 5-substituted pyrrolo[2,3-d]pyrimidine antifolates as dual-acting inhibitors of glycinamide ribonucleotide formyltransferase and 5- aminoimidazole-4-carboxamide ribonucleotide formyltransferase in de novo purine nucleotide biosynthesis: Implications of inhibiting 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase to ampk activation and antitumor activity. J Med Chem. 2013;56:10016–10032. doi: 10.1021/jm401328u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Cherian C, Orr S, Mitchell-Ryan S, Hou Z, Raghavan S, Matherly LH, Gangjee A. Tumor-targeting with novel non-benzoyl 6-substituted straight chain pyrrolo[2,3- d]pyrimidine antifolates via cellular uptake by folate receptor alpha and inhibition of de novo purine nucleotide biosynthesis. J Med Chem. 2013;56:8684–8695. doi: 10.1021/jm401139z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Golani LK, George C, Zhao S, Raghavan S, Orr S, Wallace A, Wilson MR, Hou Z, Matherly LH, Gangjee A. Structure-activity profiles of novel 6-substituted pyrrolo[2,3- d]pyrimidine thienoyl antifolates with modified amino acids for cellular uptake by folatereceptors alpha and beta and the proton-coupled folate transporter. J Med Chem. 2014;57:8152–8166. doi: 10.1021/jm501113m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mendelsohn LG, Worzalla JF, Walling JM. Preclinical and clinical evaluation of the glycinamide ribonucleotide formyltransferase inhibitors lometrexol and LY309887. In: Jackman AL, editor. Anticancer Drug Development Guide: Antifolate Drugs in Cancer Therapy. Humana Press, Inc; Totowa, NJ: 1999. pp. 261–280. [Google Scholar]
- 37.Boritzki TJ, Barlett CA, Zhang C, Howland EF. AG2034: A novel inhibitor of glycinamide ribonucleotide formyltransferase. Invest New Drugs. 1996;14:295–303. doi: 10.1007/BF00194533. [DOI] [PubMed] [Google Scholar]
- 38.Bissett D, McLeod HL, Sheedy B, Collier M, Pithavala Y, Paradiso L, Pitsiladis M, Cassidy J. Phase I dose-escalation and pharmacokinetic study of a novel folate analogue AG2034. Br J Cancer. 2001;84:308–312. doi: 10.1054/bjoc.2000.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Budman DR, Johnson R, Barile B, Bowsher RR, Vinciguerra V, Allen SL, Kolitz J, Ernest CS, 2nd, Kreis W, Zervos P, Walling J. Phase I and pharmacokinetic study of LY309887: A specific inhibitor of purine biosynthesis. Cancer Chemother Pharmacol. 2001;47:525–531. doi: 10.1007/s002800000272. [DOI] [PubMed] [Google Scholar]
- 40.Ray MS, Muggia FM, Leichman CG, Grunberg SM, Nelson RL, Dyke RW, Moran RG. Phase I study of (6R)-5,10-dideazatetrahydrofolate: A folate antimetabolite inhibitory to de novo purine synthesis. J Natl Cancer Inst. 1993;85:1154–1159. doi: 10.1093/jnci/85.14.1154. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y, Desharnais J, Marsilje TH, Li C, Hedrick MP, Gooljarsingh LT, Tavassoli A, Benkovic SJ, Olson AJ, Boger DL, Wilson IA. Rational design, synthesis, evaluation, and crystal structure of a potent inhibitor of human GAR Tfase: 10- (Trifluoroacetyl)-5,10-dideazaacyclic-5,6,7,8-tetrahydrofolic Acid†,‡. Biochemistry. 2003;42:6043–6056. doi: 10.1021/bi034219c. [DOI] [PubMed] [Google Scholar]
- 42.Meanwell NA. Synopsis of some recent tactical application of bioisosteres in drug design. J Med Chem. 2011;54:2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 43.Molecular Operating Environment (MOE) C. C. G. I; 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7: 2014. [Google Scholar]
- 44.Dahms TES, Sainz G, Giroux EL, Caperelli CA, Smith JL. The apo and ternary complex structures of a chemotherapeutic target: human glycinamide ribonucleotide transformylase†,‡. Biochemistry. 2005;44:9841–9850. doi: 10.1021/bi050307g. [DOI] [PubMed] [Google Scholar]
- 45.Chen C, Ke J, Zhou XE, Yi W, Brunzelle JS, Li J, Yong EL, Xu HE, Melcher K. Structural basis for molecular recognition of folic acid by folate receptors. Nature. 2013;500:486–489. doi: 10.1038/nature12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong SC, Proefke SA, Bhushan A, Matherly LH. Isolation of human cDNAs that restore methotrexate sensitivity and reduced folate carrier activity in methotrexate transportdefective Chinese hamster ovary cells. J Biol Chem. 1995;270:17468–17475. doi: 10.1074/jbc.270.29.17468. [DOI] [PubMed] [Google Scholar]
- 47.Deng Y, Zhou X, Kugel Desmoulin S, Wu J, Cherian C, Hou Z, Matherly LH, Gangjee A. Synthesis and biological activity of a novel series of 6-substituted thieno[2,3- d]pyrimidine antifolate inhibitors of purine biosynthesis with selectivity for high affinity folate receptors over the reduced folate carrier and proton-coupled folate transporter for cellular entry. J Med Chem. 2009;52:2940–2951. doi: 10.1021/jm8011323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flintoff WF, Davidson SV, Siminovitch L. Isolation and partial characterization of three methotrexate-resistant phenotypes from Chinese hamster ovary cells. Somatic Cell Genet. 1976;2:245–261. doi: 10.1007/BF01538963. [DOI] [PubMed] [Google Scholar]
- 49.Kugel Desmoulin S, Wang Y, Wu J, Stout M, Hou Z, Fulterer A, Chang MH, Romero MF, Cherian C, Gangjee A, Matherly LH. Targeting the proton-coupled folate transporter for selective delivery of 6-substituted pyrrolo[2,3-d]pyrimidine antifolate inhibitors of de novo purine biosynthesis in the chemotherapy of solid tumors. Mol Pharmacol. 2010;78:577–587. doi: 10.1124/mol.110.065896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wibowo AS, Singh M, Reeder KM, Carter JJ, Kovach AR, Meng W, Ratnam M, Zhang F, Dann CE., 3rd Structures of human folate receptors reveal biological trafficking states and diversity in folate and antifolate recognition. Proc Natl Acad Sci U S A. 2013;110:15180–15188. doi: 10.1073/pnas.1308827110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y, Mitchell-Ryan S, Raghavan S, George C, Orr S, Hou Z, Matherly LH, Gangjee A. Novel 5-substituted pyrrolo[2,3-d]pyrimidines as dual inhibitors of glycinamide ribonucleotide formyltransferase and 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase and as potential antitumor agents. J Med Chem. 2015;58:1479–1493. doi: 10.1021/jm501787c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alati T, Worzalla JF, Shih C, Bewley JR, Lewis S, Moran RG, Grindey GB. Augmentation of the therapeutic activity of lometrexol -(6-R)5,10-dideazatetrahydrofolate- by oral folic acid. Cancer Res. 1996;56:2331–2335. [PubMed] [Google Scholar]
- 53.Assaraf YG. Molecular basis of antifolate resistance. Cancer Metastasis Rev. 2007;26:153–181. doi: 10.1007/s10555-007-9049-z. [DOI] [PubMed] [Google Scholar]
- 54.2.1.6: L., BioSolveIT. GmbH. Augustin, S; Germany: www.biosolveit.de. [Google Scholar]
- 55.Molecular Operating Environment (MOE) Chemical Computing Group Inc; 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7: 2013. [Google Scholar]
- 56.McNicholas S, Potterton E, Wilson KS, Noble ME. Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr D Biol Crystallogr. 2011;67:386–394. doi: 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mukherjee S, Balius TE, Rizzo RC. Docking validation resources: protein family and ligand flexibility experiments. J Chem Inf Model. 2010;50:1986–2000. doi: 10.1021/ci1001982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Allen WJ, Balius TE, Mukherjee S, Brozell SR, Moustakas DT, Lang PT, Case DA, Kuntz ID, Rizzo RC. DOCK 6: Impact of new features and current docking performance. J Comput Chem. 2015;36:1132–1156. doi: 10.1002/jcc.23905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 61.Kugel Desmoulin S, Wang L, Polin L, White K, Kushner J, Stout M, Hou Z, Cherian C, Gangjee A, Matherly LH. Functional loss of the reduced folate carrier enhances the antitumor activities of novel antifolates with selective uptake by the proton-coupled folate transporter. Mol Pharmacol. 2012;82:591–600. doi: 10.1124/mol.112.079004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 63.Friedkin M, Roberts D. Conversion of uracil deoxyriboside to thymidine of deoxyribonucleic acid. J Biol Chem. 1956;220:653–660. [PubMed] [Google Scholar]
- 64.Caperelli CA, Giroux EL. The human glycinamide ribonucleotide transformylase domain: purification, characterization, and kinetic mechanism. Arch Biochem Biophys. 1997;341:98–103. doi: 10.1006/abbi.1997.9947. [DOI] [PubMed] [Google Scholar]
- 65.Corbett TH, Valeriote FA, Demchik L, Lowichik N, Polin L, Panchapor C, Pugh S, White K, Kushner J, Rake J, Wentland M, Golakoti T, Hetzel C, Ogino J, Patterson G, Moore R. Discovery of cryptophycin-1 and BCN-183577: Examples of strategies and problems in the detection of antitumor activity in mice. Invest New Drugs. 1997;15:207–218. doi: 10.1023/a:1005875015011. [DOI] [PubMed] [Google Scholar]
- 66.Padmanabhan N, Fichtner L, Dickmanns A, Ficner R, Schulz JB, Braus GH. The yeast HtrA orthologue Ynm3 is a protease with chaperone activity that aids survival under heat stress. Mol Biol Cell. 2009;20:68–77. doi: 10.1091/mbc.E08-02-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Polin LCT, Roberts BJ, Lawson AJ, Leopold WR, III, White K, Kusner J, Paluch J, Hazeldine S, Moore R, Rake J, Horwitz JP. Transplantable syngeneic tumors: solid tumors of mice. In: Teicher B, editor. Tumor Models in Cancer Research. Humana Press, Inc; Totowa, NJ: 2011. pp. 43–78. [Google Scholar]
- 68.Polin L, Valeriote F, White K, Panchapor C, Pugh S, Knight J, LoRusso P, Hussain M, Liversidge E, Peltier N, Golakoti T, Patterson G, Moore R, Corbett TH. Treatment of human prostate tumors PC-3 and TSU-PR1 with standard and investigational agents in SCID mice. Invest New Drugs. 1997;15:99–108. doi: 10.1023/a:1005856605726. [DOI] [PubMed] [Google Scholar]
- 69.Varela-Moreiras G, Selhub J. Long-term folate deficiency alters folate content and distribution differentially in rat tissues. J Nutr. 1992;122:986–991. doi: 10.1093/jn/122.4.986. [DOI] [PubMed] [Google Scholar]
- 70.Adams PD, Gopal K, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Pai RK, Read RJ, Romo TD, Sacchettini JC, Sauter NK, Storoni LC, Terwilliger TC. Recent developments in the PHENIX software for automated crystallographic structure determination. J Synchrotron Radiat. 2004;11:53–55. doi: 10.1107/s0909049503024130. [DOI] [PubMed] [Google Scholar]
- 71.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 72.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 73.Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, Adams PD. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr D Biol Crystallogr. 2008;64:61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bailey S. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 76.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.