

SUMMARY
The mitochondrial pathway of apoptosis is initiated by mitochondrial outer membrane permeabilization (MOMP). The BCL-2 family effectors BAX and BAK are thought to be absolute required for this process. Here we report that BCL-2 ovarian killer (BOK) is a bona fide yet unconventional effector of MOMP that can trigger apoptosis in the absence of both BAX and BAK. However, unlike the canonical effectors, BOK appears to be constitutively active and unresponsive to antagonistic effects of the antiapoptotic BCL-2 proteins. Rather, BOK is controlled at the level of protein stability by components of the endoplasmic reticulum–associated degradation pathway. BOK is ubiquitylated by the AMFR/gp78 E3 ubiquitin ligase complex and targeted for proteasomal degradation in a VCP/p97-dependent manner, which allows survival of the cell. When proteasome function, VCP, or gp78 activity is compromised, BOK is stabilized to induce MOMP and apoptosis independently of other BCL-2 proteins.
INTRODUCTION
The BCL-2 family proteins (hereafter BCL-2 proteins) are primary regulators of mitochondrial outer membrane permeabilization (MOMP), an essential event in mitochondrial apoptosis. MOMP results in the release of mitochondrial proteins such as cytochrome c, Smac, and Omi, that promote caspase activation and apoptosis (Green and Llambi, 2015). BAX and BAK are BCL-2 family effectors of MOMP. In their absence, cells are resistant to various apoptotic stimuli (Lindsten et al., 2000; Wei et al., 2001). Therefore, BAX and BAK are thought to be absolutely required for MOMP and the mitochondrial pathway of apoptosis. BAX and BAK activities are regulated though their interaction with BCL-2 family members. BH3-only proteins directly promote BAX and BAK activation, whereas antiapoptotic BCL-2 proteins antagonize proapoptotic proteins to prevent apoptosis (Chipuk et al., 2010).
BCL-2 ovarian killer (BOK) shares sequence homology with BAX and BAK but has not been directly proven to be a proapoptotic effector (Echeverry et al., 2013). Although BOK is frequently deleted in human cancers (Beroukhim et al., 2010), genetic deletion of Bok in mice has not revealed an overt phenotype (Ke et al., 2012). A recent study suggested that mouse embryonic fibroblasts (MEFs) lacking Bok are resistant to endoplasmic reticulum (ER) stress–induced apoptosis and that ER stress in vivo reduces apoptosis in Bok-deficient livers (Carpio et al., 2015). In contrast, another study (Ke et al., 2012) found that Bok-deficient thymocytes and MEFs show no defects in ER stress–induced apoptosis and in some cases show increased sensitivity to ER stress.
ER stress leads to accumulation of unfolded proteins in the ER, which triggers the unfolded protein response (UPR) via 3 signaling pathways controlled by IRE1α, ATF6, or PERK (Walter and Ron, 2011). Through its RNase activity, IRE1α activates the transcription factor XBP1 and regulates protein translation by degrading ER-localized mRNAs. The transcription factor ATF6 is activated through cleavage by Golgi-resident proteases S1P and S2P. The kinase PERK phosphorylates eIF2α, which in turn inhibits global protein translation while favoring the translation of UPR transcription factors such as ATF4 and CHOP, which coordinate UPR and ER-stress–induced apoptosis (Harding et al., 2000a; 2000b).
These 3 pathways collaborate to resolve the accumulation of unfolded proteins by increasing the ER protein-folding capacity, transient inhibition of de novo protein synthesis, and degradation of unfolded ER proteins through ER- associated degradation (ERAD) (Smith et al., 2011). During ERAD, unfolded proteins are retro-translocated from the ER lumen to the cytosol, where they are ubiquitylated and degraded by the proteasome. The failure to resolve ER stress can result in apoptosis via the mitochondrial pathway (Urra et al., 2013). However, the specific role of ERAD in regulating the apoptotic response to ER stress remains unknown.
Herein, we examined the regulation of BOK and its function in apoptosis. We found that BOK is an effector of MOMP that triggers membrane permeabilization and apoptosis in the absence of BAX and BAK. Strikingly, BOK did not require activation by BH3-only proteins and was not inhibited by antiapoptotic proteins BCL-2, BCL-xL, and MCL-1. BOK proapoptotic activity was controlled through ubiquitylation and ERAD by the E3 ubiquitin ligase AMFR/gp78, the AAA-ATPase VCP/p97, and the proteasome.
RESULTS
BOK Triggers Apoptosis in Response to Proteasome Inhibition
Because existing Bok-deficient models have yielded conflicting results (Carpio et al., 2015; Ke et al., 2012), we generated a null allele of Bok in which exons 2–5 were removed, thereby deleting all coding sequences (Figures S1A–C). The expression pattern in wild-type (WT) tissues was similar to that reported previously (Ke et al., 2012) (Figure S1D), and Bok−/− mice had no overt phenotype. MEFs derived from Bok−/− mice had no abnormalities in apoptosis induced by several agents, including the ER stressors tunicamycin (TN) and thapsigargin (Figure S1E). Importantly, we could not detect endogenous BOK expression in WT-MEFs (Figure S1F). Thus, we studied BOK function under enforced expression. To follow BOK expression over time, we designed a doxycycline (dox)-inducible expression system wherein BOK is fused to a Venus fluorescent protein (Figure S1G). Dox induction of the Venus-BOK fusion in MEFs did not result in detectable Venus fluorescence unless cells were treated with proteasome inhibitors (Figure 1A). In contrast, other stressors could not stabilize the Venus-BOK fluorescence (Figure 1A). Further, together with NOXA and MCL-1, BOK was one of the most unstable BCL-2 family proteins (Figure 1B). Thus, like NOXA and MCL-1 (Fernández et al., 2005; Nijhawan et al., 2003; Qin, 2005), BOK might be regulated by proteasomal degradation. We developed a system for dox-inducible expression of BOK in WT MEFs, monitored as Venus2ABOK (Figure S1G), in which Venus and BOK are produced in equimolar amounts as independent polypeptides. The Venus reporter and Bok mRNA were detected upon dox treatment (Figures 1C and S1H), but BOK protein was detected only when proteasome activity was inhibited (Figure 1C). Consistent with this finding, dox-induced BOK expression did not affect the rate or extent of apoptosis after treatment with several stressors but increased apoptosis in cells treated with the proteasome inhibitor MG132 (Figures 1D and S1I).
Figure 1. BOK triggers apoptosis in response to proteasome inhibition.

A. IncuCyte quantification of Venus-BOK in WT MEFs treated with dox, MG132 (10 μM), bortezomib (1 μM), carfilzomib (1 μM), actinomycin D (1 μM), etoposide (100 μM), UV (10 mJ/cm2), staurosporine (1 μM), thapsigargin (1 μM) or TN (1 μg/mL), and Q-VD-POh (40 μM). B. Immunoblot of FLAG-tagged BCL-2 family proteins transfected in 293T cells in the presence of 40 μM Q-VD-POh. C. Immunoblot of Venus and BOK. WT MEFs expressing Venus2ABOK were treated as in (A). D. IncuCyte quantification of SYTOX Green–stained WT MEFs expressing Venus2ABOK treated as in (A), in the absence of Q-VD-POh. E. Clonogenic survival of cancer cells treated with scrambled (SCR) or BOK-targeting siRNA and 10 μM MG132 for 30 h. F. Flow cytometry quantification of Annexin V–stained HCT116, T98G, and A172 cells transfected with siRNA and treated with 10 μM MG132 for 30 h (mean ± SD of 3 independent experiments). G. IncuCyte quantification of SYTOX Green–stained HCT116 cells treated with siRNA and MG132 (10 μM), ABT-737 (30 μM), staurosporine (2 μM) or a combination of TNFα (50 ng/mL) and cycloheximide (CHX, 2 μg/mL). IncuCyte data are represented as mean of triplicate samples ± SD and representative of 3 independent experiments (see also Figure S1).
Next, we screened several tumor cell lines in which endogenous BOK was silenced by using siRNA and examined clonogenic survival after transient treatment with MG132 (Figure 1E; downregulation efficiency shown in Table S1). Several lines showed increased survival in response to proteasome inhibition when endogenous BOK was silenced (Figure 1E, left panel). Of note, BOK was not detected in multiple myeloma cell lines U266 and H929 (Figure S1J), and siRNA silencing of BOK did not affect MG132-induced cell death in U266 cells (Figure S1K). MG132-induced apoptosis was significantly inhibited by BOK siRNA treatment in HCT116, T98G, and A172 cells (Figure 1F). Because the most striking response was observed in HCT116, it was used for further studies. Proteasome inhibition in HCT116 cells induced the stabilization of endogenous BOK (Figures S1L and S1M). Although the silencing of endogenous BOK in HCT116 cells inhibited MG132-induced cell death, it had no effect on other canonical apoptotic stimuli such as staurosporine, TNF plus cycloheximide, or the BH3-mimetic ABT-737 (Figure 1G). Therefore, BOK proapoptotic activity is likely regulated by proteasomal degradation and not by other proapoptotic stimuli.
BOK Triggers Apoptosis Independently of BAX and BAK
Most apoptotic stimuli cannot trigger MOMP or apoptosis in the absence of BAX and BAK (Lindsten et al., 2000; Wei et al., 2001). However, our finding that BOK is stabilized by proteasome inhibition but not by other stressors that require BAX and/or BAK to engage apoptosis prompted us to study whether BOK expression sensitizes bax−/−bak−/− (DKO) MEFs to apoptosis induced by proteasome inhibitors. Similar to WT MEFs, DKO MEFs did not express detectable levels of endogenous BOK unless BOK expression was enforced and the proteasome was inhibited (Figure S2A, lower panel). Treatment with different proteasome inhibitors induced cell death in DKO MEFs, but only under enforced BOK expression (Figure 2A). Other stressors (e.g., UV irradiation, actinomycin D, etoposide, staurosporine, TN, and thapsigargin) did not induce BOK stabilization or apoptosis in DKO MEFs under enforced BOK expression (Figures 2A and S2B). In contrast, enforced expression of BAX or BAK sensitized DKO MEFs to apoptosis induction in response to all stressors tested (Figure S2B). Of note, BOK-dependent cell death in DKO MEFs was caspase dependent (Figure S2A) and had characteristic features of apoptosis (membrane blebbing and Annexin V staining; Figure 2B, Movies S1 and S2).
Figure 2. BOK triggers apoptosis independently of BAX and BAK.

A. IncuCyte quantification of SYTOX Green-stained bax−/−bak−/− MEFs expressing Venus2ABOK and treated as in 1(A), in the absence of Q-VD-POh. B. Confocal microscopy images of bax−/−bak−/− MEFs expressing Venus or Venus2ABOK treated with dox, Annexin V Alexa Fluor® 647, and 10 μM MG132 for 3 h. C. IncuCyte quantification of SYTOX Green–stained WT or bax−/−bak−/− HCT116 cells treated with scrambled (SCR) or BOK-targeting siRNA and TNFα (50 ng/mL) plus cycloheximide (CHX, 2 μg/mL) or MG132 (10 μM). D. Flank tumor volume of CD-1 nude mice injected with bax−/−bak−/− MEFs expressing Venus (left flank) or Venus2ABOK (right flank) and treated with bortezomib at days 4 and 8 (right panel). E. Summary of the high-throughput screening of 4282 biologically active compounds for caspase activation in bax−/−bak−/− MEFs expressing Venus2ABOK. IncuCyte data are represented as mean of triplicate samples ± SD and representative of 3 independent experiments (see also Figure S2 and Movies S1 and S2).
HCT116 cells lacking BAX and BAK (DKO HCT-116), which are resistant to apoptosis induction by many stressors (Wang and Youle, 2011), did not engage in apoptosis in response to TNF plus cycloheximide (Figure 2C, left panel) but underwent BOK-dependent apoptosis upon proteasome inhibition by MG132 (Figures 2C, S2C, and S2D). Strikingly, the kinetics of cell death in WT and DKO HCT116 cells were similar after MG132 treatment, indicating that endogenous BOK is the main effector of apoptosis in this context (Figure 2C, right panel)
Proteasome inhibitors have shown promise in treating some malignancies in vivo (Nencioni et al., 2007). Thus, we determined whether the proapoptotic capacity of BOK could mediate antitumor effects of proteasome inhibitors independently of BAX and BAK. DKO MEFs with or without dox-inducible BOK expression that were implanted into flanks of CD-1 nude mice generated tumors at the same rate in the absence of proteasome inhibition (Figure 2D, left panel). DKO tumors (not expressing endogenous BOK even under proteasome inhibition; Figure S2A) were resistant to treatment with the proteasome inhibitor bortezomib, but induction of BOK expression sensitized the tumors to bortezomib treatment and inhibited tumor growth (Figure 2D, right panel). Thus, the antitumor activity of proteasome inhibition in this system is due to BOK stabilization and induction of apoptosis.
To further explore the conditions under which BOK can be stabilized to trigger apoptosis, we performed high-throughput screening of 4282 unique compounds with known biologic activity in DKO MEFs with enforced BOK expression to identify compounds activating caspase in a BOK-dependent manner (Figures 2E, S2E, S2F, and S2G). Most of the validated hits were known proteasome inhibitors (Figure 2E), but the screen also identified 3 novel natural compounds anthotecol, obtusaquinone, and pomiferin as new proteasome inhibitors (Figure S2G). Further, 2 other agents quinacrine and chlorothalonil activated caspase in BOK-expressing cells. Although the latter compounds had no proteasome inhibitory activity (Figure S2G), they stabilized Venus-BOK expression (Figure S2E) and induced BOK-dependent cell death in DKO MEFs (Figure S2F) and were therefore investigated further.
BOK Is Regulated by Components of the ERAD Pathway
The stabilization of BOK expression by proteasome inhibitors might occur through BOK ubiquitylation by an E3-ubiquitin ligase and targeting for proteasomal degradation. Indeed, as reported for MCL-1 (Nijhawan et al., 2003), MG132 treatment allowed the detection of BOK ubiquitylation (Figure 3A).
Figure 3. BOK stability is regulated by the ERAD pathway.

A. Ubiquitylation profile of Flag-tagged BOK, BAK, or MCL-1 immunoprecipitated from transiently transfected HCT116 cells treated with 40 μM Q-VD-POh ±10 μM MG132. B. Ubiquitylation sites in murine BOK identified by MS (peptide coverage underlined). C. Clonogenic survival of bax−/−bak−/− HCT116 cells transiently transfected with an empty pMX-IRES-GFP or expressing FLAG-BOKWT or FLAG-BOK6K-R and sorted for equivalent GFP intensity (lower panel). Upper panel: immunoblot of FLAG and GFP. D. Flow cytometry quantification of Annexin V–stained bax−/−bak−/− MEFs expressing Venus2ABOKWT or Venus2ABOK6K-R and treated with dox for 24 h (mean ± SD of 3 independent experiments) E. BOK interacting network as identified by tandem affinity purification and MS analysis. F. Immunoprecipitation of Flag-BOKFL and Flag-BOKΔC from lysates of bax−/−bak−/− MEFs treated with dox, MG132 (10 μM), and Q-VD-OPh (40 μM). G. Confocal microscopy images of HeLa cells cotransfected with Venus-BOK and gp78-Cerulean in presence of MG132 (10 μM) and Q-VD-OPh. Pearson correlation coefficient ± SD is indicated. H. Size exclusion chromatography of cell lysates from MEFs expressing Venus2ABOK and treated with dox, MG132 (10 μM), and Q-VD-OPh (40 μM). I. Ubiquitylation profile of Flag-tagged BOK immunoprecipitated from bax−/−bak−/− MEFs treated with scrambled (SCR) or gp78 targeting siRNA and dox, MG132 (10 μM), and Q-VD-OPh (40 μM) (see also Figure S3).
Tandem-affinity purification and mass spectrometry (MS) analysis revealed that 4 of 6 lysine residues in BOK are conjugated by ubiquitin (Ub) in MG132-stabilized BOK (Figure 3B). Arginine substitution of all 6 lysines in BOK yielded a more stably expressed protein (Figure 3C, upper panel) that had increased killing activity upon transient expression (Figure 3C, lower panel and 3D). Therefore, Ub-dependent regulation of BOK stability is an important determinant of its proapoptotic activity.
Next, we identified the E3-ligase responsible for BOK ubiquitylation by tandem affinity purification and proteomic screening. Because the C-terminal region of BOK associates with membranes (see below), which could result in potential artifactual interactions, we considered only interacting partners pulled down by both full-length BOK (BOKFL) and a BOK version lacking the C-terminus (BOKΔC). Both BOKFL and BOKΔC were degraded in an MG-132–inhibited manner (Figure S6D). Strep/FLAG-BOKFL and Strep/FLAG-BOKΔC were precipitated from MG-132–treated DKO MEFs and subjected to proteomic analysis. We identified several BOK-associated proteins that are components of the ERAD pathway (Christianson et al., 2012) (Figure S3A). The ERAD pathway is critical for protein homeostasis by targeting unfolded ER proteins for proteasomal degradation (Smith et al., 2011). In mammals, ubiquitylation of known ERAD substrates is primarily catalyzed by 2 ubiquitin ligase complexes inserted in the ER membrane and assembled around E3 ligases Hdr1 and gp78/AMFR (Fang et al., 2001; Kikkert, 2003). Ubiquitylated substrates are then extracted from the ER by the VCP/p97 complex and processed for proteasomal degradation (Meyer et al., 2012). Remarkably, our proteomic analysis revealed a BOK interaction network comprising 3 major ERAD compartments (Christianson et al., 2012; Fang et al., 2001): the 26S proteasome; VCP and its cofactors Ufd1 and Npl4; and components of the gp78 E3 ubiquitin ligase complex, gp78, Erlin-1, Erlin-2, UBAC-2, and UBXD8 (Figure 3E and S3A).
The association of BOK with its partners was further validated by immunoprecipitation and immunoblotting analysis (Figure 3F). The interaction between endogenous BOK and gp78 was observed after immunoprecipitation in untreated and MG132–treated HCT116 (Figure S3B). Mutation of the 6 lysines of BOK did not alter its interaction pattern (Figure S3C). Importantly, the C-terminal domain of BOK, which controls its subcellular localization (see below), was not sufficient for its interaction with gp78 (Figures S3D), suggesting that the cytosolic region of BOK is recognized by the ERAD pathway.
Live confocal microscopy analysis revealed that BOK and gp78 have a strong subcellular colocalization (Pearson’s correlation coefficient 0.79; Figure 3G). Gel filtration chromatography revealed that both gp78 and Erlin-2 co-eluted with BOK, further supporting that BOK associates with the gp78 E3 ubiquitin ligase complex (Figure 3H). Silencing of gp78 dramatically reduced BOK ubiquitylation (Figure 3I), which suggests that gp78 is a major catalyzer of BOK ubiquitylation.
During ERAD, gp78 catalyzes the ubiquitylation of unfolded ER proteins (Fang et al., 2001), which are then extracted from the ER and presented to the proteasome by action of the AAA-ATPase VCP/p97 (Meyer et al., 2012) (Fig. 4A). Importantly, the silencing of gp78 or VCP stabilized BOK (Figure S4A) and induced cell death in DKO MEFs in the absence of proteasome inhibitors (Figures 4B and S4B). Inhibition of VCP by eeyarestatin I (ESI) (Wang et al., 2008; 2010) or NMS-873 (Magnaghi et al., 2013) stabilized BOK expression, but at a slower rate and to a lesser extent than did MG132 (Figures 4C and 4D). Consistent with this, these inhibitors induced caspase-dependent apoptosis in both WT and DKO MEFs under enforced BOK expression (Figures 4E). NMS-873 also induced cell death in DKO HCT116 cells, which was largely prevented by silencing BOK (Figure 4F; ESI induced non-apoptotic cell death in HCT116 and was therefore not further examined; data not shown). Therefore, conditions in which gp78 or VCP function is impaired promote BOK stabilization and lead to cell death.
Figure 4. ERAD disruption triggers BOK-induced apoptosis.

A. Schematic representation of ERAD components regulating BOK degradation. B. Clonogenic survival of bax−/−bak−/− MEFs expressing Venus2ABOK transfected with scrambled (SCR), gp78-, or VCP-targeting siRNA and treated with dox for 24 h. C. IncuCyte quantification of Venus-BOK in bax−/−bak−/− MEFs treated with dox and MG132 (10 μM), ESI (1μM), and NMS-873 (10μM) in the presence of 40 μMQ-VD-OPh. D. Immunoblot of Venus and BOK. bax−/−bak−/− MEFs expressing Venus2ABOK were treated as in (C). E. Flow cytometry quantification of Annexin V–stained WT and bax−/−bak−/− MEFs expressing Venus2ABOK and treated with dox and MG132 (10 μM), ESI (1 μM) or NMS-873 (10 μM) for 8 h (mean ± SD of 3 independent experiments). F. IncuCyte quantification of SYTOX Green–stained bax−/−bak−/− HCT116 cells treated with scrambled (SCR) or BOK-targeting siRNA and 10 μM NMS-873. IncuCyte data represented as mean of triplicate samples ± SD and representative of 3 independent experiments (see also Figure S4).
Inhibition of PERK during ER Stress Induces ERAD Disruption and BOK-Induced Apoptosis
To test our observations that BOK is a sensor for ERAD dysfunction during ER stress, we determined the effect of compromised ERAD function on BOK expression and activity. During ER stress, accumulation of unfolded proteins in the ER activates the UPR through IRE1α, PERK, and ATF6 to upregulate ERAD components (Smith et al., 2011). Therefore, we reasoned that a failure of these pathways during ER stress might impair the adaptive ERAD response and overwhelm the system. We found that Venus-BOK expression was not stabilized by inducing ER stress with TN alone (Figure 5A), but its combination with a specific PERK inhibitor GSK2656157 (PERKi) (Atkins et al., 2013) stabilized BOK (Figure 5A). The stability of Venus-BOK was not affected by treatment with the IRE1α inhibitor 4μ8C or the S1P protease (essential for ATF6 activation) inhibitor PF429242 alone or in combination with TN. Consistent with this, DKO HCT116 cells underwent cell death only when treated with TN plus PERKi, and not TN in combination with inhibitors of components of the other UPR-associated signaling pathways (Figure 5B). Cell death induced by TN plus PERKi was caspase dependent and attenuated by silencing BOK (Figures 5B, S5A, and S5B). Therefore, PERK inhibition during TN-induced ER stress results in BOK stabilization and cell death induction in the absence of proteasome inhibition.
Figure 5. Inhibition of PERK during ER stress induces ERAD disruption and BOK-induced apoptosis.

A. IncuCyte quantification of Venus-BOK in bax−/−bak−/− MEFs treated with dox, Q-VD-OPh (40 μM), TN (1 μg/mL) and 4μ8C (IRE1αi, 50 μM), GSK2656157 (PERKi, 500 nM), or PF429242 (S1Pi, 10 μM). B. IncuCyte quantification of SYTOX Green– stained bax−/−bak−/− HCT116 cells transfected with scrambled (SCR) or BOK targeting siRNA and treated as in (A), in the absence of QV-VD-OPh. C, D. Immunoblot of lysate from bax−/−bak−/− MEFs expressing Venus2ABOK and treated as in (A). E. Immunoprecipitation of FLAG-BOK from lysate of bax−/−bak−/− MEFs treated with dox, Q-VD-OPh (40 μM), and MG132 (10 μM), ESI (1 μM) or TN (1 μg/mL) plus GSK2656157 (PERKi, 500 nM). NB: Immunoprecipitated samples were normalized for equivalent BOK loading. F. Immunoblot of bax−/−bak−/− MEF lysates treated with Q-VD-OPh, TN (1 μg/mL), GSK2656157 (PERKi, 500 nM), and salubrinal (100 μM) in complete DMEM or HBSS medium (-A.A). G. IncuCyte quantification of Venus-BOK in bax−/−bak−/− MEFs treated as in (F). H. IncuCyte quantification of SYTOX Green–stained bax−/−bak−/− HCT116 cells treated as in (F), in the absence of Q-VD-OPh. I. Global protein ubiquitylation of lysates from bax−/−bak−/− MEFs expressing FLAG-BOK and treated with dox, Q-VD-OPh (40 μM), and MG132 (10 μM), quinacrine (20 μM), or chlorothalonil (2 μM). J. Immunoprecipitation of FLAG-BOK from lysates of bax−/−bak−/− MEFs treated as in (I). NB: Immunoprecipitated samples were normalized for equivalent BOK loading. IncuCyte data are represented as mean of triplicate samples ± SD and representative of 3 independent experiments (see also Figure S5).
Bok mRNA was up-regulated in TN- or MG132-treated HCT116 cells (Figure S5C), which suggests that during ER stress, BOK-induced cell death is regulated at not only the protein stability level but also the transcriptional level. However, increases in Bok mRNA levels were similar after treatment with TN alone or TN plus PERKi, but only the combination induced cell death in DKO HCT116 cells. Therefore, protein stabilization is a critical step for BOK-induced apoptosis in the absence of BAX and BAK.
Strikingly, cells treated with the combination of TN and PERKi, but not alone or in combination with other inhibitors, showed rapid loss of Erlin-1 and -2 (Figure 5C). This finding suggests that PERK activity is critical to sustain the integrity of the gp78 complex during ER stress. We determined whether this was due to transcriptional regulation of Erlin-1 and -2 by PERK signaling pathways. Although mRNAs encoding UPR proteins BiP, Edem, and ERdj4 were induced by ER stress (Rodriguez et al., 2012), those for Erlin-1 and -2 were not affected by TN, alone or in combination with PERKi (Figure S5D). However, MG132 blocked the loss of Erlin-1 and -2 induced by the combination of TN and PERKi (Figure 5D), indicating that proteasomal activity mediates the degradation of these gp78 complex components under these conditions. To assess how the degradation of Erlin-1 and -2 affects gp78 function, we precipitated BOK from cells treated with MG132, ESI, or the combination of TN and PERKi (Figure 5E). BOK stabilization by MG132 or ESI led to co-precipitation of BOK with gp78, Erlin-1, and Erlin-2 (Figure 5E). In contrast, BOK stabilization by TN and PERKi led to degradation of Erlin-1 and -2 (Figure 5E, left panel), and gp78 did not associate with BOK (Figure 5E, right panel). This suggests that Erlin-1 and -2 are critical to recruit BOK to the gp78 complex. Accordingly, downregulation of Erlin-1 and -2 by siRNA silencing induced cell death in DKO MEFs upon BOK expression (Figure S5E).
Our findings indicate that PERK signaling is critical to maintain gp78-dependent ERAD function during ER stress. Accumulation of unfolded proteins in the ER leads to PERK activation and eIF2α phosphorylation, which in turn inhibits global protein translation while favoring the translation of a small subset of proteins (e.g., ATF4 and CHOP) critical for the UPR (Harding et al., 2003; Lange et al., 2008). We therefore tested whether collapse of the gp78 complex induced by PERK inhibition during ER stress could be prevented by downstream phosphorylation of eIF2α. We used the phosphatase inhibitor salubrinal, which inhibits eIF2α by preventing its dephosphorylation (Boyce, 2005) (Figure S5F). Alternatively, we subjected cells to amino acid deprivation, which promotes eIF2α phosphorylation, independently of PERK activation, via the kinase GCN2 (Dever et al., 1993) (Figure S5F). Neither salubrinal nor amino acid starvation alone inhibited TN-induced ER stress (detected by monitoring PERK activation under those conditions, Figure S3G), but both independently rescued Erlin-1 and -2 stability when used with TN plus PERKi (Figure 5F). Consistent with this pattern of Erlin-1 and -2 stabilization, salubrinal or amino acid starvation prevented the stabilization of BOK and apoptosis induced by TN plus PERKi (Figure 5G, 5H) but not by MG132 (Figure S3H). Together, these observations suggest that functionality of the gp78/Erlin E3 ubiquitin ligase complex is maintained during ER stress through a PERK/eIF2α signaling axis. Similar results were obtained when protein synthesis was inhibited by cycloheximide upon TN plus PERKi treatment (Figures 5G, 5H, and S5A). Therefore, inhibition of protein translation by the PERK/eIF2α signaling pathway during ER stress appears critical to maintain Erlin levels and prevent BOK expression.
We showed that regulation of BOK stability is critical aspect of the cellular response to ER stress, and we mostly used proteasome inhibition to dissect this pathway. However, the identification of BOK inducers chlorothalonil and quinacrine from our high-throughput screen, which lack proteasome inhibitory activity (Figures 2E, S2G, and 5I), suggested that they stabilize BOK upstream of the proteasome. Both compounds decreased the interaction of BOK and gp78, which suggests that they directly impair BOK recruitment to the gp78 E3 ubiquitin ligase complex (Figure 5J).
Taken together, our findings show that BOK can be independently stabilized by disrupting 3 distinct ERAD components: the proteasome, the VCP complex, and the gp78 E3 ubiquitin ligase complex. Thus, BOK might act as an apoptotic sensor that triggers cell death when the ERAD-proteasome degradation system is impaired.
BOK Is a Constitutively Active BCL-2 Family Effector That Is Not Regulated by BCL-2 Proteins
Upon activation by BH3-only proteins (e.g. BID or BIM) the BCL-2 pro-apoptotic effectors, BAX and BAK cause MOMP and the release of mitochondrial inter-membrane space proteins that function to engage caspase activation (Chipuk et al., 2010). Owing to its sequence homology with BAX and BAK, BOK has been speculated to be a proapoptotic BCL-2 family effector protein, although experimental evidence supporting this function has been lacking (Echeverry et al., 2013). We found that BOK stabilization by proteasome inhibition in DKO MEFs induced MOMP, causing release of the mitochondrial intermembrane space marker Omi-mCherry (Figures 6A and 6B; Movies S3 and S4). Similarly, proteasome and ERAD inhibitors (MG132 and NMS-873, respectively) but not canonical apoptosis inducers (ABT-737, staurosporine, and TNF plus cycloheximide) triggered MOMP in DKO HCT116 cells (Figure 6C).
Figure 6. BOK is a constitutively active BCL-2 family effector.

A. Confocal microscopy images of bax−/−bak−/− MEFs expressing Omi-mCherry and Venus2ABOK treated with dox, Q-VD-OPh (40 μM), and MG132 (10 μM) for 3 h. B. MOMP was assessed through flow cytometry quantification of loss of Omi-mCherry from digitonin-permeabilized bax−/−bak−/− MEFs treated as in (A). C. bax−/−bak−/− HCT116 expressing Omi-mCherry were processed as in (B) (left histogram: treatment with MG132 (10 μM), NMS-873 (10 μM); right histogram: treatment with ABT-737 (30 μM), staurosporine (STS, 2 μM), TNFα (50 ng/mL) plus cycloheximide (CHX, 2 μg/mL). D. Flow cytometry quantification of Annexin V–stained bax−/−bak−/− MEFs expressing BOK targeted constructs and treated with dox and 10 μM MG132 for 8 h (mean ± SD of 3 independent experiments). E. Permeabilization of LUV containing 0% or 5% DGS-NTA(Ni), upon incubation with BAXFL (100 nM), BAKΔC-8xHIS (1 μM) or BOKΔC-8xHIS (1 μM) and cleaved BID (n/cBID, 20 nM). F. NTA(Ni)-LUV permeabilization upon incubation with BH3 peptides (5 μM) and BAKΔC-8xHIS (1 μM) or BOKΔC-8xHIS (50 nM). LUV data are represented as mean of triplicate samples and representative of 3 independent experiments (see also Figures S6 and S7 and Movies S3 and S4).
As previously reported (Echeverry et al., 2013), we observed that BOK localizes to both the ER and mitochondria (Pearson’s correlation coefficient 0.71 and 0.53, respectively) (Figure S6A). We studied whether this localization is important for degradation of BOK and its function in the mitochondria. BOK subcellular localization was controlled by its C-terminal domain, as shown by a Venus-C-terminalBOK fusion faithfully mimicking localization of the full-length protein (Figure S6B). We generated BOK variants in which the C-terminus was either ablated, resulting in cytosolic localization (BOKCyto/ΔC); replaced with the C-terminus of Cb5 for strict ER localization (BOKER); or replaced with the C-terminus of BCL-xL for mitochondrial localization (BOKMito). Localization of Venus-BOK targeted constructs was assessed by imaging (Fig. S6C). In all cases, targeted BOK constructs were degraded (Figure S6D) and therefore inactive in causing apoptosis when expressed in DKO MEFs (Figure 6D). MG132 addition triggered apoptosis in WT and BOKMito cells, but BOKCyto and BOKER cells could not induce cell death in DKO MEFs (Figure 6D) despite being stabilized by MG132 (Figure S6D). These findings indicate that BOK must localize to the mitochondria to induce MOMP and apoptosis.
The pore-forming activity of BAX and BAK can be shown in vitro by following the permeabilization of large unilamellar vesicles (LUVs) (Kuwana et al., 2002; Lovell et al., 2008). BAX is a cytosolic protein, whereas BAK is a natural tail-anchored membrane protein. Therefore, in vitro LUV permeabilization requires tethering of BAK to the lipid bilayer, which is achieved using BAKΔC-8xHIS and Ni-coupled lipids [DGS-NTA(Ni)] in the LUVs (Oh et al., 2010). Since BOK is also anchored in membranes via its C-terminus (Figures S6B and S6C), we examined the effects of BOKΔC-8xHIS in Ni-NTA-LUVs. As expected, activation of full-length BAX or BAKΔC-8xHIS with cleaved BID (n/c-BID) (Kuwana et al., 2002) resulted in rapid permeabilization of LUVs. Importantly, this was not observed in the absence of BID or, in the case of BAKΔC-8xHIS, in the absence of DGS-NTA(Ni) (Figure 6E). In all cases, LUV permeabilization correlated with membrane insertion of the protein, as shown by carbonate extraction (Figure S6E).
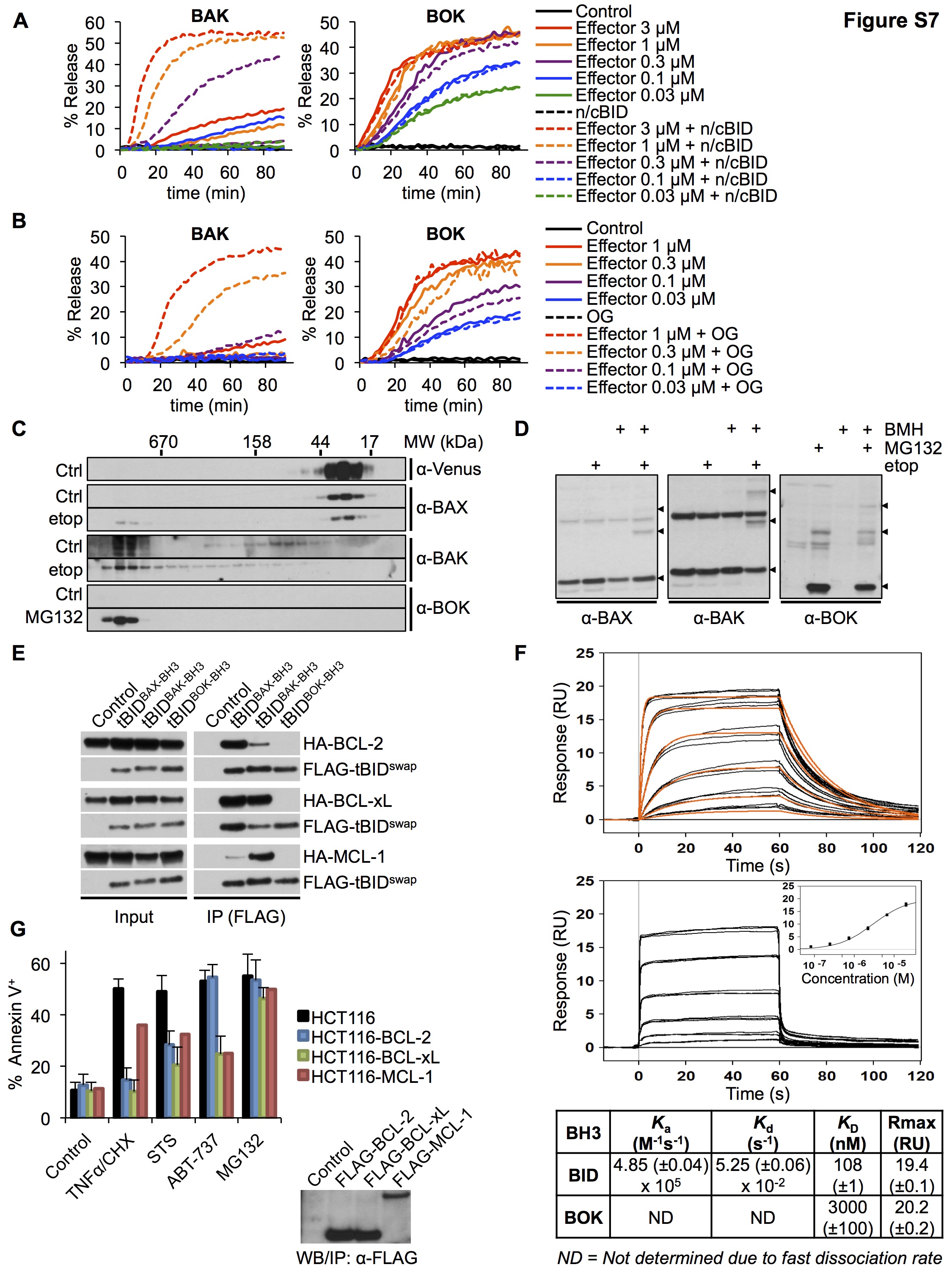
BOKΔC-8xHIS permeabilized LUVs (Figure 6E) and inserted into membranes (Fig. S6E) in the presence of DGS-NTA(Ni). Thus, BOK is a pore-forming protein and therefore a bona fide effector of the BCL-2 family. Surprisingly, BOK activity did not seem to require BID (Figures 6E). Titration of BOKΔC-8xHIS did not reveal any synergistic effect of BID on LUV permeabilization by BOK (Figure S7A), indicating that BID does not potentiate BOK pore-forming activity in vitro. It is possible, however, that another BH3-only protein activates BOK. Therefore, we tested 6 BH3 peptides for their ability to potentiate BOK activity. Only peptides of BID and BIM promoted LUV permeabilization by BAK, but no peptide affected LUV permeabilization by BOK (Figure 6F). This suggests that unlike BAX and BAK, BOK does not require direct activation to engage its pore-forming activity. To further test this possibility, we exploited the ability of nonionic detergents to activate BAX and BAK (Hsu and Youle, 1997). Treatment of BAK with n-octyl-D octylglucoside promoted its ability to permeabilize LUVs but did not affect LUV permeabilization by BOK (Figure S7B).
These observations suggest that BOK, when stabilized, is constitutively active. To address this possibility, we examined the oligomeric state of BAX, BAK, and BOK in cells. Size exclusion chromatography (Figure S7C) and cross-linking analysis (Figure S7D) revealed that in untreated cells BAX and BAK behave as monomers and transition to oligomers upon challenge with etoposide, as described previously (Antonsson et al., 2000; Eskes et al., 2000). In contrast, BOK was detectable only after MG132 treatment and revealed only a multimeric form. These results further support that idea that BOK, when stabilized, assumes an oligomeric state to function as a constitutively active BCL-2 effector protein that is regulated by rapid degradation in cells.
Antiapoptotic BCL-2 family proteins prevent MOMP and apoptosis by sequestering the active forms of BAX and BAK and of the BH3-only proteins that activate them (Llambi et al., 2011). Upon expression of BCL-2, BCL-xL, or MCL-1, BAX and BAK co-precipitated with the antiapoptotic proteins, with BCL-2 and MCL-1 showing preferences for BAX and BAK, respectively (Figure 7A), as previously described (Llambi et al., 2011; Simon N Willis, 2005; Zhai et al., 2008). However, as reported previously (Echeverry et al., 2013), we did not detect stabilized BOK in association with BCL-2 or BCL-xL and found that it only marginally boud to MCL-1 (Figure 7A). This interaction pattern was faithfully mimicked by chimeric tBID proteins, whose BH3 domain was exchanged with that of BAX, BAK, or BOK (Figure S7E). Since BOK was originally identified by an MCL-1 interaction in the yeast 2-hybrid system (Hsu et al., 1997), we measured the affinity of the BOK BH3 peptide for MCL-1 by surface plasmon resonance and found a KD of 3.44 μM compared with a KD of 134 nM for the BID BH3 binding (Figure S7F). Therefore, the absence of an interaction between BOK and antiapoptotic BCL-2 family members is probably due to the low affinity of its BH3 domain for these proteins.
Figure 7. BOK is not inhibited by antiapoptotic BCL-2 proteins.

A. Immunoprecipitation of FLAG-tagged BCL-2, BCL-xL, or MCL-1 from lysates of WT MEFs expressing BOK and treated with dox, MG132 (10 μM), and Q-VD-OPh (40 μM). B. NTA(Ni)-LUV upon incubation with n/cBID (20 nM) + BAKΔC-8xHIS (1 μM) or BOKΔC-8xHIS (1 μM) in combination with BCL-2 ΔC-8xHIS, BCL-xL ΔC-8xHIS, or MCL-1 ΔC-8xHIS. LUV data are represented as mean of triplicate samples ± SD and representative of 3 independent experiments. C. IncuCyte quantification of SYTOX Green–stained WT or bax−/−bak−/− MEFs expressing Venus2ABOK and overexpressing BCL-2, BCL-xL, or MCL-1 treated with the MG132 (10 μM) or staurosporine (1 μM). D. IncuCyte quantification of SYTOX Green–stained DKO HCT116 cells overexpressing BCL-2, BCL-xL, or MCL-1, treated with scrambled (SCR) or BOK-targeting siRNA and 10 μM MG132 (upper panel) or 10 μM NMS-873 (lower panel). E. Model of BOK-induced MOMP regulation by the ERAD pathway, independently of the BCL-2 protein family. IncuCyte data are represented as mean of triplicate samples ± SD representative of 3 independent experiments (see also Figure S7).
Accordingly, although each of the antiapoptotic proteins prevented LUV permeabilization with BAK plus BID, LUV permeabilization by BOK was unaffected by BCL-2 or BCL-xL and, at best, only slightly delayed by the presence of MCL-1 (Figure 7B). Overexpression of BCL-2, BCL-xl, or MCL-1 in WT MEFs effectively blocked BAX- or BAK-dependent cell death in response to staurosporine, but did not interfere with MG132-induced cell death in DKO MEFs that stably expressed BOK (Figure 7C). Similarly, HCT116 cells overexpressing the antiapoptotic proteins were resistant to various stressors (Figure S7G). However, cell death induced by MG132 or NMS-873 in HCT116 cells was completely resistant to the antiapoptotic effects of BCL-2, BCL-xL, or MCL-1 (Figures 7D), whereas silencing of endogenous BOK significantly inhibited apoptosis in all cell lines (Figure 7D). Therefore, unlike BAX and BAK, BOK is an effector of MOMP and apoptosis that is not regulated by either proapoptotic BH3-only proteins or the major antiapoptotic BCL-2 family proteins. Instead, BOK is likely regulated predominantly by the functions of gp78 and VCP to promote its rapid degradation (Figure 7E).
DISCUSSION
Although a role has been recently proposed for BOK in ER stress–induced cell death (Carpio et al., 2015; Echeverry et al., 2013), its function in the mitochondrial apoptotic pathway remains unclear. We show that like BAX and BAK, BOK is a bona fide effector of MOMP. Its pore-forming activity can trigger MOMP in the absence of BAX and BAK. Unlike BAX and BAK, however, BOK is constitutively active, independent of BH3-only proteins, and resistant to the antiapoptotic effect of BCL-2, BCL-xL, and MCL-1. Instead, BOK is controlled at the level of protein stability by the ERAD E3 ubiquitin ligase gp78 and its associated proteins Erlin-1, Erlin-2, and VCP, which target BOK for degradation by the proteasome to promote cell survival. When proteasome function, VCP, or gp78 activity is compromised, BOK is stabilized and induces MOMP to promote apoptosis.
Several mechanisms have been proposed to explain how the failure to resolve ER stress leads to terminal UPR, whereby the cell dies by apoptosis (Urra et al., 2013). One mechanism involves PERK-dependent induction of ATF4 and CHOP, which promotes expression of the BH3-only protein BIM, leading to apoptosis in a BAX- or BAK-dependent manner (Puthalakath et al., 2007). Paradoxically, and consistent with other reports (Back et al., 2009; Harding et al., 2000b), we found that PERK inhibition did not result in resistance to ER stress–induced apoptosis but rather to increased sensitivity to cell death induction (Fig. 5 and S5). Our study reveals a unique connection between PERK signaling and ERAD function. The inhibition of de novo protein synthesis by the PERK/eIF2α axis appears to be critical to sustain ERAD function under ER stress and increased unfolded protein burden. Failure of the PERK pathway in stressed cells leads to catastrophic failure of the gp78/Erlin complex (Figures 5 and S5).
Another example of terminal UPR is accumulation of death receptor-5 at the ER, which then engages MOMP through caspase-8 activation and cleavage of the BH3-only protein BID (Lu et al., 2014). Similarly to our study, this apoptotic pathway proceeds when a UPR signaling pathway fails (in this case IRE1α) during ER stress. This highlights how cell fate during ER stress is determined by apoptotic programs that are intricately “wired” into the UPR signaling pathways to trigger cell death.
Unlike conventional ERAD substrates that are degraded when misfolded, BOK seems to be a constitutive ERAD target that is continuously degraded in healthy cells. This notion is supported by the association of endogenous BOK with gp78 in HCT116 cells even in the absence of proteasome inhibition (Figure S3B). How the gp78 complex recruits BOK, however, is unclear. Erlin-1 and -2 appear to be critical for BOK degradation (Figures 5 and S5) and could serve as key adaptors for ERAD targeting. This is reminiscent of inositol-1,4,5-triphosphate receptors that are processed by ERAD in an Erlin-1 and -2 dependent manner (Pearce et al., 2009).
Some cells appeared to express BOK without succumbing to apoptosis, as seen in immunoblots of tissues from WT animals (confirmed in same tissues from Bok−/− animals) (Figure S1D). This suggests that BOK might be regulated by proteins other than BCL-2, BCL-xL, and MCL-1 or exist as a monomer until degraded by gp78 in such tissues. Alternatively, because BOK must localize to the mitochondria to trigger MOMP and apoptosis (Figure 6D), BOK sequestration at the ER might help keep its proapoptotic activity in check.
Proteasome inhibitors have been used in cancer therapy (Nencioni et al., 2007). Most studies on the mechanism of action of proteasome inhibitors in cancer cells have focused on the need for degradation in the NF-κB pathway (Nencioni et al., 2007). Our work reveals that BOK represents an alternative route for MOMP and apoptosis that is unaffected by the loss of proapoptotic or the overexpression of antiapoptotic BCL-2 proteins. This raises the promising possibility of stabilizing BOK in cancer cells, in which the canonical mitochondrial pathway is often impaired. However, unlike BAX and BAK that are widely expressed across cell types and tissues, BOK has a restricted expression pattern. For example, BOK does not appear to be transcribed significantly in multiple myeloma (the Cancer Cell Line Encyclopedia database; http://www.broadinstitute.org/ccle). Consistent with this, we did not find a key role for BOK in bortezomib-induced cell death in multiple myeloma cells (Figures S1J and S1K).
Nevertheless, our study suggests that cancer cells expressing high BOK mRNA levels may be particularly sensitive to treatment with proteasome inhibitors. Moreover, using specific ERAD inhibitors to trigger BOK-induced apoptosis in cancer cells can be a potential alternative therapy to proteasome inhibitors, which have pleiotropic side effects.
EXPERIMENTAL PROCEDURES
LUV Permeabilization Assay
LUVs were prepared as described previously (Asciolla et al., 2012; Lovell et al., 2008). Briefly, LUVs composed of 8% cardiolipin, 46% (or 41%) phosphatidylcholine, 25% phosphatidylethanolamine, 11% phosphatidylinositol, 10% phosphatidylserine, and when indicated, 5% DGS-NTA(Ni) (Aventi Polar Lipids, Inc.) were loaded with the fluorophore ANTS (12.5 mM) and the quencher DPX (45 mM) (Molecular Probes). LUVs were incubated with the indicated concentration of recombinant proteins or BH3 peptides in 10 mM HEPES pH 7.0, 200 mM KCl, and 5 mM MgCl2 at 37°C, and permeabilization was assessed by measuring ANTS fluorescence over time with a spectrophotometer. The percentage of release was calculated between the baseline provided by the LUVs-only control and the 100% release obtained by LUVs solubilized in 1% CHAPS.
Detergent-induced activation by n-octyl-β-D-glucoside (final concentration 0.015%) was performed as described previously (Asciolla et al., 2012). The following peptides were used: BIDBH3 (EDIIRNIARHLAQVGDSMDRSI), BIMBH3 (DMRPEIWIAQELRRIGDEFNAYYA), PUMABH3 (REIGAQLRRXADDLNAQYERRRQEE), BMFBH3 (HQAEVQIARKLQCIADQFHRLHVQQH), and NOXABH3 (QPSPARAPAELEVECATQLRAFGDALNFRQ).
Flow Cytometry
For Annexin V staining, cells were resuspended in Annexin V binding buffer (10 mM HEPES, 150 mM NaCl, 5 mM KCl, 1mM MgCl2, and 1.8 mM CaCl2) with Annexin V-APC (1/200; Caltag Laboratories). Annexin V–positive cells were quantitated by flow cytometry, using FACScan and FACsCalibur systems (BD Biosciences) and FlowJo Collectors’ Edition software (Tree Star). Flow cytometry data are represented as mean±SD of 3 independent experiments, unless otherwise stated.
To monitor Omi-mCherry release, MEFs or HCT116 cells were incubated for 10 min on ice in PBS with 50 μg/mL or 333 μg/mL digitonin (Sigma-Aldrich), respectively, and washed twice in cold PBS. mCherry fluorescence was quantified by flow cytometry by using LSRII (BD Biosciences) and analyzed with FlowJo Collectors’ Edition software.
IncuCyte Analysis
Venus-BOK stabilization and cell-death kinetics were monitored by the IncuCyte FLR or Zoom imaging system (Essen Bioscience). Dead cells were stained with 400 ng/mL propidium iodide (PI) or 25 nM SYTOX Green (Invitrogen). In cases where percentages are indicated, total cell number was quantified using SYTO16 Green. SYTOX Green-, PI-, SYTO16 Green-, or Venus-positive cells were quantified by the IncuCyte image analysis software (Essen Bioscience). Data were expressed as positive events per well or as percentages of the ratio of PI to SYTO16 Green counts (% cell death). IncuCyte data are representative of 3 independent experiments. Error bars represent the standard deviation from the mean for triplicate samples.
Flank Tumor Model
CD-1 nude mice were injected with 200 μL of a mixture containing 100 μL of Matrigel Matrix (BD Biosciences) and 100 μL of PBS containing 4×106 bax−/−bak−/− MEFs E1A-Ras-transformed and expressing dox-inducible Venus (left flank) or Venus2ABOK (right flank). Mice were given dox (1mg/mL doxycycline in a 0.5% sucrose solution) in their drinking water and treated or not treated with bortezomib (1 mg/kg body weight in PBS) through tail vein injection at 4 and 8 days after cell implantation. Tumor volume was measured weekly by ultrasound. All experiments were done in accordance with the Guide for the Care and Use of Laboratory Animals, and the St. Jude Institutional Animal Care and Use Committee approved all animal procedures.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Abbvie for providing ABT-737 and Richard Youle for providing the WT and DKO HCT116 lines. We thank Linda M. Hendershot, Helen M. Beere, Katherine Baran and Guiovanni Quarato for discussions and for careful reading of the manuscript. We thank Vani J. Shanker (Scientific Editing, St. Jude) for editing this manuscript; Richard Cross, Greig Lennon, and Parker Ingle (Microfluorimetry, St. Jude) for cell sorting analysis; Ashutosh Mishra and Vishwajeeth Pagala (Proteomics, St. Jude) for LC-MS/MS analysis; Cliff Guy (Confocal Imaging, St. Jude) for confocal microscopy analysis; Asli Goktug (Department of Chemical Biology and Therapeutics, St. Jude) for assistance with automation of HTS; Brett Waddell (Molecular Interaction Analysis, St. Jude) for SRP analysis; and Shantel Brown, Allison Weaver, Melissa D. Johnson, Ecklin Crenshaw, and Christopher Calabrese (Small Animal Imaging, St. Jude) for flank tumor imaging. This work was supported by NIH grants GM 096208 (DRG), GM 110034 (TC) and ALSAC.
Footnotes
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Conceptualization, F.L. and D.R.G.; Methodology, F.L. and D.R.G.; Investigation, F.L., Y.M.W., M.Y., and D.S.; Resources, B.V., S.G., M.J.P., J.H.Z., and S.A.B.; Writing–Original Draft, F.L. and D.R.G.; Writing–Review and Editing, F.L. and D.R.G.; Visualization, F.L., Y.M.W., and T.M.; Supervision, F.L., S.A.B., S.P., T.M., T.C., and D.R.G; Funding Acquisition, T.C. and D.R.G.
References
- Antonsson B, Montessuit S, Lauper S, Eskes R, Martinou JC. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem J. 2000;345(Pt 2):271–278. [PMC free article] [PubMed] [Google Scholar]
- Asciolla JJ, Renault TT, Chipuk JE. Examining BCL-2 family function with large unilamellar vesicles. J Vis Exp. 2012;68:e4291. doi: 10.3791/4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins C, Liu Q, Minthorn E, Zhang SY, Figueroa DJ, Moss K, Stanley TB, Sanders B, Goetz A, Gaul N, et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013;73:1993–2002. doi: 10.1158/0008-5472.CAN-12-3109. [DOI] [PubMed] [Google Scholar]
- Back SH, Scheuner D, Han J, Song B, Ribick M, Wang J, Gildersleeve RD, Pennathur S, Kaufman RJ. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab. 2009;10:13–26. doi: 10.1016/j.cmet.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce M. A Selective inhibitor of eIF2 dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- Carpio MA, Michaud M, Zhou W, Fisher JK, Walensky LD, Katz SG. BCL-2 family member BOK promotes apoptosis in response to endoplasmic reticulum stress. Proc Natl Acad Sci USA. 2015;112:7201–7206. doi: 10.1073/pnas.1421063112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, Richter CM, Tyler RE, Greenblatt EJ, Harper JW, Kopito RR. Defining human ERAD networks through an integrative mapping strategy. Nat Cell Biol. 2012;14:93–105. doi: 10.1038/ncb2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever TE, Chen JJ, Barber GN, Cigan AM, Feng L, Donahue TF, London IM, Katze MG, Hinnebusch AG. Mammalian eukaryotic initiation factor 2 alpha kinases functionally substitute for GCN2 protein kinase in the GCN4 translational control mechanism of yeast. Proc Natl Acad Sci USA. 1993;90:4616–4620. doi: 10.1073/pnas.90.10.4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echeverry N, Bachmann D, Ke F, Strasser A, Simon HU, Kaufmann T. Intracellular localization of the BCL-2 family member BOK and functional implications. Cell Death Differ. 2013;6:785–99. doi: 10.1038/cdd.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskes R, Desagher S, Antonsson B, Martinou JC. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol. 2000;20:929–935. doi: 10.1128/mcb.20.3.929-935.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S, Ferrone M, Yang C, Jensen JP, Tiwari S, Weissman AM. The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc Natl Acad Sci USA. 2001;98:14422–14427. doi: 10.1073/pnas.251401598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández Y, Verhaegen M, Miller TP, Rush JL, Steiner P, Opipari AW, Lowe SW, Soengas MS. Differential regulation of noxa in normal melanocytes and melanoma cells by proteasome inhibition: therapeutic implications. Cancer Res. 2005;65:6294–6304. doi: 10.1158/0008-5472.CAN-05-0686. [DOI] [PubMed] [Google Scholar]
- Green DR, Llambi F. Cell Death Signaling. Cold Spring Harb Perspect Biol. 2015;7 doi: 10.1101/cshperspect.a006080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000a;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000b;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Hsu SY, Kaipia A, McGee E, Lomeli M, Hsueh AJ. Bok is a pro-apoptotic Bcl-2 protein with restricted expression in reproductive tissues and heterodimerizes with selective anti-apoptotic Bcl-2 family members. Proc Natl Acad Sci USA. 1997;94:12401–12406. doi: 10.1073/pnas.94.23.12401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YT, Youle RJ. Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem. 1997;272:13829–13834. doi: 10.1074/jbc.272.21.13829. [DOI] [PubMed] [Google Scholar]
- Ke F, Voss A, Kerr JB, O’Reilly LA, Tai L, Echeverry N, Bouillet P, Strasser A, Kaufmann T. BCL-2 family member BOK is widely expressed but its loss has only minimal impact in mice. Cell Death Differ. 2012;19:915–925. doi: 10.1038/cdd.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikkert M. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. Journal of Biological Chemistry. 2003;279:3525–3534. doi: 10.1074/jbc.M307453200. [DOI] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- Lange PS, Chavez JC, Pinto JT, Coppola G, Sun CW, Townes TM, Geschwind DH, Ratan RR. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J Exp Med. 2008;205:1227–1242. doi: 10.1084/jem.20071460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llambi F, Moldoveanu T, Tait SWG, Bouchier-Hayes L, Temirov J, McCormick LL, Dillon CP, Green DR. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell. 2011;44:517–531. doi: 10.1016/j.molcel.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, Andrews DW. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell. 2008;135:1074–1084. doi: 10.1016/j.cell.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Lu M, Lawrence DA, Marsters S, Acosta-Alvear D, Kimmig P, Mendez AS, Paton AW, Paton JC, Walter P, Ashkenazi A. Cell death. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science. 2014;345:98–101. doi: 10.1126/science.1254312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnaghi P, D’Alessio R, Valsasina B, Avanzi N, Rizzi S, Asa D, Gasparri F, Cozzi L, Cucchi U, Orrenius C, et al. Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat Chem Biol. 2013;9:548–556. doi: 10.1038/nchembio.1313. [DOI] [PubMed] [Google Scholar]
- Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol. 2012;14:117–123. doi: 10.1038/ncb2407. [DOI] [PubMed] [Google Scholar]
- Nencioni A, Grünebach F, Patrone F, Ballestrero A, Brossart P. Proteasome inhibitors: antitumor effects and beyond. Leukemia. 2007;21:30–36. doi: 10.1038/sj.leu.2404444. [DOI] [PubMed] [Google Scholar]
- Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, Wang X. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003;17:1475–1486. doi: 10.1101/gad.1093903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh KJ, Singh P, Lee K, Foss K, Lee S, Park M, Lee S, Aluvila S, Park M, Singh P, et al. Conformational changes in BAK, a pore-forming proapoptotic Bcl-2 family member, upon membrane insertion and direct evidence for the existence of BH3-BH3 contact interface in BAK homo-oligomers. J Biol Chem. 2010;285:28924–28937. doi: 10.1074/jbc.M110.135293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce MMP, Wormer DB, Wilkens S, Wojcikiewicz RJH. An endoplasmic reticulum (ER) membrane complex composed of SPFH1 and SPFH2 mediates the ER-associated degradation of inositol 1,4,5-trisphosphate receptors. J Biol Chem. 2009;284:10433–10445. doi: 10.1074/jbc.M809801200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- Qin JZ. Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res. 2005;65:6282–6293. doi: 10.1158/0008-5472.CAN-05-0676. [DOI] [PubMed] [Google Scholar]
- Rodriguez DA, Zamorano S, Lisbona F, Rojas-Rivera D, Urra H, Cubillos-Ruiz JR, Armisen R, Henriquez DR, Cheng EH, Letek M, et al. BH3-only proteins are part of a regulatory network that control the sustained signalling of the unfolded protein response sensor IRE1. Embo J. 2012:1–14. doi: 10.1038/emboj.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334:1086–1090. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urra H, Dufey E, Lisbona F, Rojas-Rivera D, Hetz C. When ER stress reaches a dead end. Biochim Biophys Acta. 2013;1833:3507–3517. doi: 10.1016/j.bbamcr.2013.07.024. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang C, Youle RJ. Predominant requirement of Bax for apoptosis in HCT116 cells is determined by Mcl-1’s inhibitory effect on Bak. Oncogene. 2011;31:3177–3189. doi: 10.1038/onc.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Li L, Ye Y. Inhibition of p97-dependent protein degradation by Eeyarestatin I. J Biol Chem. 2008;283:7445–7454. doi: 10.1074/jbc.M708347200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Shinkre BA, Lee JG, Weniger MA, Liu Y, Chen W, Wiestner A, Trenkle WC, Ye Y. The ERAD inhibitor Eeyarestatin I is a bifunctional compound with a membrane-binding domain and a p97/VCP inhibitory group. PLoS ONE. 2010;5:e15479. doi: 10.1371/journal.pone.0015479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai D, Jin C, Huang Z, Satterthwait AC, Reed JC. Differential Regulation of Bax and Bak by anti-apoptotic Bcl-2 Family proteins Bcl-B and Mcl-1. J Biol Chem. 2008;283:9580–9586. doi: 10.1074/jbc.M708426200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.