

Abstract
Summary: Rule-based modeling is invaluable when the number of possible species and reactions in a model become too large to allow convenient manual specification. The popular rule-based software tools BioNetGen and NFSim provide powerful modeling and simulation capabilities at the cost of learning a complex scripting language which is used to specify these models. Here, we introduce a modeling tool that combines new graphical rule-based model specification with existing simulation engines in a seamless way within the familiar Virtual Cell (VCell) modeling environment. A mathematical model can be built integrating explicit reaction networks with reaction rules. In addition to offering a large choice of ODE and stochastic solvers, a model can be simulated using a network free approach through the NFSim simulation engine.
Availability and implementation: Available as VCell (versions 6.0 and later) at the Virtual Cell web site (http://vcell.org/). The application installs and runs on all major platforms and does not require registration for use on the user’s computer. Tutorials are available at the Virtual Cell website and Help is provided within the software. Source code is available at Sourceforge.
Contact: vcell_support@uchc.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
The traditional approach for modeling reaction networks requires explicit specification of all molecular species and reactions. Therefore, a reaction network developed manually by the modeler can include only a limited number of species and reactions and cannot capture the combinatorial complexity of many cell signaling systems (Mayer et al., 2009).
The rule-based modeling approach overcomes this limitation by providing a compact model description that is capable of accounting for all the potential molecular complexes and interactions among them that can be generated during a response to a signal, without explicit enumeration. In this approach, molecular interactions and their effects are represented in the form of reaction rules that serve as generators of molecular species and reactions (Hlavacek et al., 2006). A model is defined as an initial set of species with non-zero amounts (seed species), a set of reaction rules operating on seed species, and a set of observables that define computed outcomes of simulations. Every rule in the set is applied to those species among the seed species that can be selected as reactants, generating proper well-defined reactions and an extended set of species (seed species and species that are products of these reactions). The set of rules is again applied to this extended set of species to generate even more reactions and more species. During network generation (Hlavacek et al., 2006) this process is iteratively repeated until no new species and reactions are generated, or until a specified termination condition is reached, producing a reaction network that can be then simulated using deterministic or stochastic methods. During network-free simulations (Sneddon et al., 2011) individual particles rather than populations of identical species are stochastically tracked over time without the need to explicitly enumerate all possible species and reactions.
The rule-based modeling approach was implemented in numerous modeling and simulation tools, such as BioNetGen software (Blinov et al., 2004) and Simmune (Meier-Schellersheim et al., 2006) for simulations following network generation, KaSim (Danos et al., 2007) and NFSim (Sneddon et al., 2011) for ‘network-free’ simulations.
Rule-based models are often described using scripting languages such as BioNetGen Language (BNGL, Faeder et al., 2009) used by BioNetGen and NFSim, Kappa language (used by KaSim), ML Rules (Maus et al., 2011), Python scripts (Lopez, et al, 2013) etc. Modeling with scripting languages requires considerable mathematical and/or computational expertise and has a steep learning curve. Writing such scripts, although easier than writing a program in a computer language and usually aided by a set of tutorials, may be difficult for beginner modelers without programming experience.
Writing a script is also an error-prone process, where a simple typo can lead to a valid model that implements an unexpected mechanism different from the intended one. While a reaction network model can be verified by checking every species and reaction, such verification may be impossible for a model specified by rules because the generated network may include thousands of species and reactions and it can be too large for manual tracking and validation. Thus, preventing errors is a critical issue in rule-based modeling.
Here, we present a novel tool that makes rule-based modeling easier by (i) providing a graphical user interface for specification and simulation of rule-based models, eliminating the need to use a scripting language; (ii) preventing errors in model specification by constructing model elements through verified components and extensive built-in automatic updating and verification algorithms of the model after any edits.
Additionally, we allow user to link specified reaction networks and rules together, where a species can be a part of a reaction network and participate in a rule at the same time. Ordinary species and reactions are not required to be encoded in a script as trivial rules; rather, they can be specified and visualized graphically. Another feature is an explicit unit system, where a modeler is guided to distinguish between molecules and concentrations, thus avoiding situations when an inexperienced modeler may use concentrations in place of particle numbers for stochastic simulations. The tool is implemented within the Virtual Cell (VCell) modeling and simulation framework (Loew and Schaff, 2001; Moraru et al., 2008).
2 Rule-based modeling with VCell
2.1 Rule-based model specification
The new VCell features allow specification of entirely rule-based models as well as inclusion of rule-based features into standard models. A modeler starts by specifying Molecules that have binding Sites for binding to other molecules. Each Site may have potential modification States. Specifying and renaming of Molecules, Sites and States is done graphically through right click on shapes. Generally, each molecule may define a pool of species—e.g. a molecule with nine phosphosites which can be in either phosphorylated or unphosphorylated state may define up to 29 = 512 distinct species (if phosphorylated and unphosphorylated states of all sites are allowed in any combination).
Once Molecules are specified, they can form parts of Species and Observables, and participate in Reaction Rules. Species in the rule-based modeling paradigm can be composed of one or more molecules, with bonds connecting binding sites within a molecule or between different molecues. While a molecule may have multiple states, each state in a species must be uniquely defined—otherwise the species will present a pool of different chemical entities and will not be valid. For example, the second reactant (marked as Shc) in Figure 1 is a species, while the first reactant (EGFR) is not.
Fig. 1.
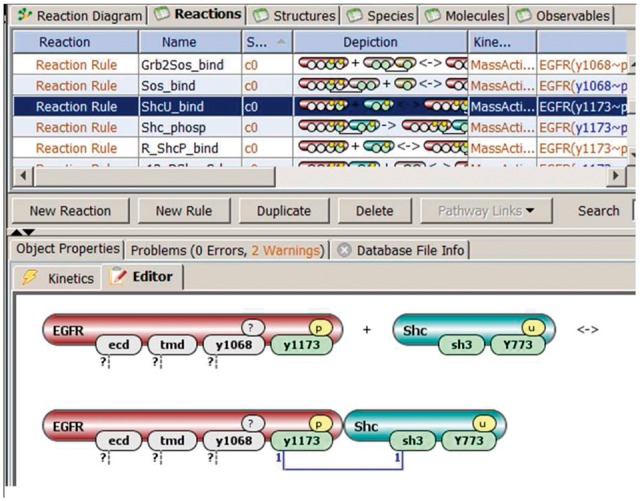
Specification of Reaction Rules. Binding of two molecules. One has four binding and modification sites, but only one of them y1173 affects interaction: it must be unbound and phosphorylated. Another has two binding sites, both must be unbound, but the site Y773 must be unphosphorylated. Sites in grey (also have a question mark) do not affect interaction and may be bound to other molecules, or be in different states. Dashed lines with a question mark specify that such sites may be either bound to their respective binding partners or remain unbound, with no effect on the reaction Thus this reaction rule specifies an individual reaction for each state of Y1068 site of EGFR molecule, and possibly many more if EGFR can be bound to other molecules. The result of interaction is formation of a bond (marked with 1) between two sites
Observables define features of molecules that are tracked during simulations, such as phosphorylation of specific Sites or combination of specific Molecules. Observables are defined through one or more of species patterns that select species with certain features. The first reactant (EGFR) in Figure 1 is a species pattern that selects multiple species, all of them have a site y1173 phosphorylated and unbound. Sites and their states that do not affect interaction are shown in grey.
Reaction Rules (Fig. 1) specify interactions among Molecules by defining features that are required for participation in the reaction, and features that are changing during a reaction. Association of two molecules is specified as the formation of a bond between two binding sites. Reactants and products in a reaction rule can be either species or species patterns.
2.2 New features for model specification and verification
The new interface is intuitive for both beginner and advanced users because it includes both graph-based and table-based functionality. Edits are being done graphically in a Graphical Editor. BioNetGen Language (BNGL) code is shown in Tables and can be exported for further edits and use in stand-alone tools supporting the format. All model elements are visualized as easily understood graphical glyphs in a table which enables a quick model overview, as well as in a fully detailed view of the selected row in a pane below.
Species, Reaction Rules and Observables are constructed through a selection of previously specified Molecules. The software automatically guards against accidental errors. Potential problems are tracked in real time: for example, when a molecule is used in a Species, Sites with multiple States are shown in red so a modeler must select a specific State to make a species into a pool of identical entities.
To avoid errors while matching reactants to products, they are located one underneath another. All changes in reactants are propagated down to products, while changes in products are allowable only when consistent with states of reactants. For example, if a site state is unspecified in a reactant molecule, it can not be assigned a specific state in the corresponding product molecule.
Our tool makes modifying rule-based models easy, as any change to Molecules, Sites or States is propagated to all other elements of the model. This is in sharp contrast with scripting languages where any single change must be consistently propagated by manual edits throughout the whole document.
2.3 Model simulation
Once a model is specified, it can be simulated in multiple ways within the VCell platform. A user can create deterministic or stochastic applications for simulation of a reaction network transparently generated with the BioNetGen network-generating engine. The size and properties of the network are controlled by user-defined settings that include the maximal number of iterations and a maximal size of a generated species (maximal number of molecules comprising it). To prevent ‘run-away’ rules that can generate a network too large for efficient simulations, these parameters are set by default to low values. A user is warned if the network is too large or not fully generated, and then is guided by VCell on how to verify the size of the network and adjust parameters, before he/she is able to start simulations.
Reaction rules can not be represented as a reaction network unless a complete network generation is performed (Blinov and Moraru, 2012). Thus, they are not shown in the VCell Reaction Diagram. However, all generated species and reactions can be visualized using the same notations as in Figure 1, and a resulting reaction network can be seen as a separate model. Vice versa, a reaction network specified in VCell can be converted into a rule-based model by defining molecular structure for participating species and adding rules.
Alternatively, a user can create a network-free application that will use the NFSim engine to perform agent-based stochastic simulations which compute time courses for Observables and initial Species. It can simulate any combination of reactions and reaction rules, even for entirely non-rule-based models, although its use may not be optimal in the latter case. On the other hand, network-free application is the only viable option to simulate models where the specified rules would generate infinite or extremely large reaction networks.
3 Implementation
3.1 Software implementation
The new rule-based features have been retrofitted to the existing VCell-layered design which decouples the biological model from the generated mathematical description required for VCell applications (spatial/non-spatial and deterministic/stochastic). We extended the VCell physiology layer with several new elements (Molecules, Reaction Rules, Observables), while other elements required significant redesign (Species, Reactions). The mathematics layer was changed to merge rules with reactions and a new mathematical representation was created to support the NFSim simulation engine.
BioNetGen and NFSim are integrated through separate native format layers (BNGL/NFSim XML) which isolate syntax changes from internal VCell data structures and algorithms. This allows us to easily maintain compatibility with, and support new features in future releases of these tools. The current latest versions of both BioNetGen (2.2.6) and NFSim (1.11) executables are distributed within the VCell 6.0 installation package (alongside the existing large number of other ODE, PDE and SSA solvers included with VCell).
3.2 Model exchange
VCell stores models internally in Virtual Cell Markup Language format (VCML), which is similar to the Systems Biology Markup Language (SBML), and includes information on all molecules and species, reactions and rules, initial conditions, simulation parameters and optional geometry specifications. A VCell model can be exported and imported as SBML, but the rule-based part of it can currently only be exported if flattened (if the full network is generated). The VCell team participates in the development of the SBML Level 3 Multistate, Multicomponent and Multicompartment Species extension (Hucka et al., 2015), which will provide full support for rule-based model components once finalized. Additionally, VCell can export and import rule-based models in BNGL format, and our compliance with this format will be maintained in the future, as it is required internally when invoking the BioNetGen engine. Alternatively, a set of rules together with reactions can be exported as NFSim XML—the internal format used by the NFSim solver that has regulat reactions and species added to a rule-based description.
Due to this extensive import–export functionality, the new tool is compatible and synergistic with stand-alone BioNetGen and NFsim simulators; thus, rule-based models can be specified in VCell and sent out for simulations in standalone versions of these tools outside the VCell framework, and vice versa.
Supplementary Material
Funding
This work was funded by the NIH [P41-GM103313, R01-GM095485].
Conflict of Interest: none declared.
References
- Blinov M.L., Moraru I.I. (2012). Leveraging modeling approaches: reaction networks and rules In: Advances in Systems Biology, pp. 517–530. Springer, New York. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinov M.L. et al. (2004) BioNetGen: software for rule-based modeling of signal transduction based on the interactions of molecular domains. Bioinformatics, 2020, 3289–3291. [DOI] [PubMed] [Google Scholar]
- Danos V. et al. (2007) Rule-based modelling of cellular signalling In: CONCUR. Vol. 4703 of Lecture Notes in Computer Sciences, pp. 17–41. Springer, Berlin. [Google Scholar]
- Faeder J.R. et al. (2009) Rule-based modeling of biochemical systems with BioNetGen. Systems biology, 500, 113–167. [DOI] [PubMed] [Google Scholar]
- Hlavacek W.S. et al. (2006) Rules for modeling signal-transduction systems. Science’s STKE, 2006, re6. [DOI] [PubMed] [Google Scholar]
- Hucka M. et al. (2015) The Systems Biology Markup Language (SBML): language specification for level 3 version 1 core. J. Integr. Bioinf., 12, 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez C.F. et al. (2013) Programming biological models in Python using PySB. Mol. Syst. Biol., 9, 646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loew L.M., Schaff J.C. (2001) The Virtual Cell: a software environment for computational cell biology. Trends Biotechnol., 19, 401–406. [DOI] [PubMed] [Google Scholar]
- Maus C. et al. (2011) Rule-based multi-level modeling of cell biological systems. BMC Syst. Biol., 5, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer B.J. et al. (2009) Molecular machines or pleiomorphic ensembles: signaling complexes revisited. J. Biol., 8, 81.1–81.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier-Schellersheim M. et al. (2006) Key role of local regulation in chemosensing revealed by a new molecular interaction-based modeling method. PLoS Comput. Biol., 2, e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraru I.I. et al. (2008) Virtual cell modelling and simulation software environment. IET Syst. Biol., 22, 352–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneddon M.W. et al. (2011) Efficient modeling, simulation and coarse-graining of biological complexity with NFsim. Nat. Methods, 88, 177–183. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.