

Abstract
Background
The life of schizophrenia patients is severely affected deficits in working memory. In various brain regions, the reciprocal interactions between excitatory glutamatergic neurons and inhibitory GABAergic neurons are crucial. Other neurotransmitters, in particular dopamine, serotonin, acetylcholine, and norepinephrine, modulate the local balance between glutamate and GABA and therefore regulate the function of brain regions. Persistent alterations in the balances between the neurotransmitters can result in working memory deficits.
Methods
Here we present a heuristic computational model that accounts for interactions among neurotransmitters across various brain regions. The model is based on the concept of a neurochemical interaction matrix at the biochemical level and combines this matrix with a mobile model representing physiological dynamic balances among neurotransmitter systems associated with working memory.
Results
The comparison of clinical and simulation results demonstrates that the model output is qualitatively very consistent with the available data. In addition, the model captured how perturbations migrated through different neurotransmitters and brain regions. Results showed that chronic administration of ketamine can cause a variety of imbalances, and application of an antagonist of the D2 receptor in PFC can also induce imbalances but in a very different manner.
Conclusions
The heuristic computational model permits a variety of assessments of genetic, biochemical, and pharmacological perturbations and serves as an intuitive tool for explaining clinical and biological observations.
General Significance
The heuristic model is more intuitive than biophysically detailed models. It can serve as an important tool for interdisciplinary communication and even for psychiatric education of patients and relatives.
Keywords: interaction matrix, mesoscopic model, mobile, neurotransmitter, schizophrenia, systems biology, working memory deficit
Introduction
Schizophrenia is a chronic mental disorder affecting 0.5 – 2.5% of the population worldwide [1]. The illness is characterized by symptoms such as hallucinations, delusions, emotional distress and withdrawal [2]. In addition, patients are greatly affected by an impairment of cognitive function, such as deficits in working memory, language and executive function, and episodic memory. It has been argued that a deficit in the ability to represent goal information in working memory is one of the common mechanisms underlying cognitive impairments in schizophrenia [3].
The brain areas that seem to be crucially involved in this function are the dorsolateral prefrontal cortex (DLPFC) and the basal ganglia. These regions receive neurotransmitter-mediated inputs from other areas, including the parietal cortex, thalamus, and striatum as it was reviewed by Carlsson [4]. In these regions, local neuronal microcircuits are connected via excitatory synapses driven by glutamate (Glu) and inhibitory synapses driven by gamma-aminobutyric acid (GABA). The local balance of Glu and GABA is crucial for the stability of electrical activity of these micro-circuits. Furthermore, strong, stable gamma oscillations are closely related to normal cognitive function, such as sensory processing [5]. Other neurotransmitters, in particular dopamine (DA), norepinephrine (NE), serotonin (5-HT) and acetylcholine (ACh), regulate the function of the involved regions [6, 7]. Alterations in the balance among these neurotransmitters can result in cognitive deficits in schizophrenia and other disorders. The maintenance of these balances is a complex process because the neurotransmitter systems related to the involved brain regions do not function independently but interact with each other in a complicated manner. This intra-synaptic interplay of transmitter release, reuptake, decay, diffusion, and receptor occupation in the context of schizophrenia is insufficiently understood and therefore has not been efficiently explored with macroscopic computational models [8].
From a pharmacological perspective, one could say that the balance among neurotransmitters is critical, delicate, and dynamic, and that its disruption may result in various mental disorders. The concept of a chemical imbalance as a cause of disease is not new, as even Hippocrates used the “balance of juices” as a criterion to describe health status. Following Fritze’s suggestion to use the concept of multiple balances between neurotransmitters in studies of brain health [9], Tretter formulated a neurochemical interaction matrix that represents clinical observations of the actions of psychoactive drugs and pharmaceutical interventions [10]. On this basis he developed the concept of a “neurochemical mobile” that represents a hierarchy of counteracting influences among neurotransmitters at the physiological and behavioral level.
Specifically, different pairs of neurotransmitters form a set of hierarchical balances. The resulting mobile, in its simplest form, includes the neurotransmitters DA, 5-HT, and NE on one side, and ACh, Glu, and GABA on the other (Fig. 1). Anatomically, the substrates in this mobile can be localized in each neuronal local network; for instance, in the midbrain with centers for NE (locus coeruleus), 5-HT (nucleus raphe), ACh (pedunculopontine tegmental nucleus and nucleus basalis), DA (ventral tegmental area), and with local GABA neurons that all interact with Glu transmission from cortex, thalamus, and others (see Fig. 2). We recently represented this simple mobile as a set of ordinary differential equations and used it to investigate the multiple dynamics of alcohol dependence [11, 12].
Figure 1. The neurochemical mobile is a hierarchical scale system of balanced rods.

The neurochemical mobile represents a hierarchical, functional organization of neurotransmitters, along with their relative functional contributions and dynamic imbalances in human brain. It visualizes synergisms or antagonisms between neurotransmitter systems at the physiological, behavioral, and pathological levels. The lengths of the arms of each rod are in reality different and reflect corresponding relative signal intensities. The neurotransmitters, under normal, healthy condition, are perfectly balanced at all levels of the mobile. Thus, strong deviations from balances point to (temporarily) abnormal states or disease. Abbreviations: dopamine (DA), serotonin (5-HT), norepinephrine (NE), acetylcholine (ACh), glutamate (Glu), gamma-aminobutyric acid (GABA).
Figure 2. Macro-anatomy of the neurochemical interaction matrix.
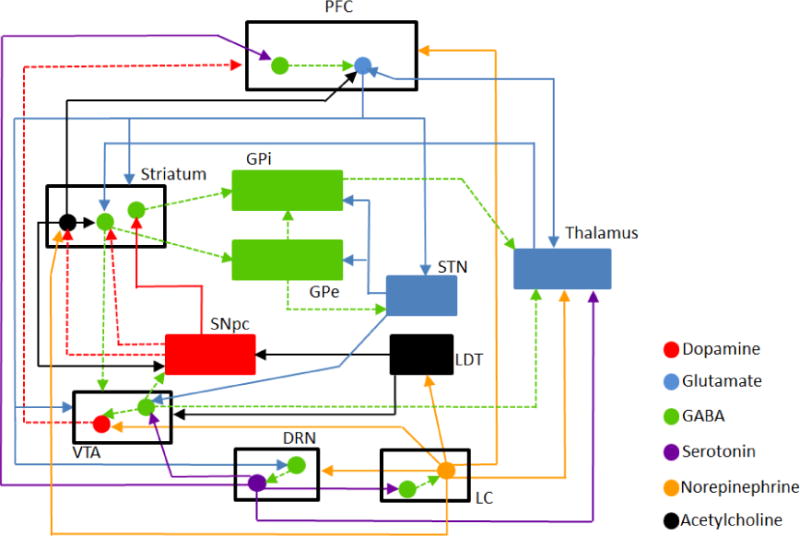
The interactions among the six neurotransmitter systems in multiple regions across the human brain form a complicated network, which is shown here with different arrows. An arrow pointing to a circle codes for a projection onto the specific type of neurons. By contrast, if an arrow points to a box, the projections affect either all types of neurons inside this box, if there are multiple circles, or the entire region, if the box represents a single type of neurons within this brain region. Modeled brain regions include: prefrontal cortex (PFC), striatum, global pallidus internal (GPi), global pallidus external (GPe), subthalamic nucleus (STN), thalamus, substantia nigra pars compacta (SNpc), laterodorsal tegmental nucleus (LDT), ventral tegmental area (VTA), dorsal raphe nucleus (DRN), and locus coeruleus (LC). Solid circles in different colors represent different types of neurons as shown in the legend. Note that the two green circles in striatum represent GABAergic neurons having DA receptors of D1 or D2 type. Arrows with solid lines represent activation, while arrows with dashed lines represent inhibition. Arrows in different colors represent different neurotransmitter projections and follow the same coloring scheme as for the circles.
A neurochemical mobile, with its balances and imbalances among various neurotransmitters, can easily integrate competing hypotheses about global neurotransmission in schizophrenia, such as:
The hyperfunction hypothesis of (sub-cortical) dopamine [13];
The hypofunction hypothesis of (prefrontal) glutamate [14];
The hypofunction hypothesis of (cortical) GABA [15];
The hyperfunction hypothesis of serotonin [16].
In some respects, the hypotheses have specific heuristic value for elective anatomical explanations of positive or negative symptoms of schizophrenia [17], or for different modes of behavior, such as subcortically controlled automatic behavior and cortically controlled goal-related, decision-based behavior [18]. However, the coexistence of these hypotheses suggests that schizophrenia is associated with a defect in the neuro-circuitry among multiple brain regions. This inference is supported by various experimental and behavioral studies. For example, Goldman-Rakic et al. showed with electrophysiological experiments in monkeys that excitatory glutamatergic neurons in PFC maintain information [19] and that the activity of these glutamatergic neurons is regulated by lateral GABAergic interneurons [20]. In addition, DA can regulate PFC microcircuits and enable working memory as a modulatory neurotransmitter [21, 22]. Thus, considering the singular hypotheses simultaneously leads to two conclusions. First, the disease involves imbalances between DA, 5-HT, Glu and GABAergic transmissions, and these imbalances are dynamic. Second, perturbations of neurotransmitters in regard to working memory deficits affect different anatomical regions, at least in animal models of schizophrenia (see Fig. 2).
As a case in point, overstimulation of DA receptors in the prefrontal cortex (PFC) [19] and DA depletion in striatum [23] both worsen working memory performance. Ketamine, an NMDA antagonist, increases DA release in PFC and can impair working memory [24]. The frontal GABA level changes during working memory tasks [25], and GABA is found to be reduced post mortem in schizophrenia patients [26]. Plasma levels of Glu and GABA are also reduced in schizophrenia patients, while the GABA/Glu ratio is elevated [27]. After six weeks of risperidone treatment, Glu and GABA do not return to normal, but the ratio is comparable to that in controls, indicating the importance of the balance between these neurotransmitters. 5-HT, which is released from the dorsal raphe to VTA and cortex, is believed to counteract the effect of DA during working memory [28]. Nicotine, through its effect on the ACh receptor, can interact with the DA system and actually improve working memory [29]. As Pogarell suggested, “neuropsychiatric disorders comprise multiple tuned circuits, including excitatory or inhibitory neuronal pathways and various feedback loops” [17]. He concluded that multimodal strategies of integrative systems biology might be the most promising tools for characterizing the dynamic balances.
Although cognitive function is severely affected, current drugs for schizophrenia are mainly antipsychotic and do not primarily target or improve cognitive deficits [26]. This lack is interesting, because schizophrenia was first defined as “dementia praecox,” which recognized the cognitive features of the disorder as primary. However, with the advent of antipsychotic drugs, the definition shifted more toward psychosis than cognition. The insufficiency of efficacious treatment options may be due, at least in part, to the fact that the interactions and balances among all neurotransmitters involved in working memory are complex and not truly understood at a mechanistic, molecular level. In addition, the interactions are affected by specific receptor subtypes. For example, DA released to the striatum can bind to specific receptors of D1 or D2 subtypes on the surface of medium spiny neurons. These D1 and D2 receptor subtypes are segregated in most neurons, with about one third expressing D1, D2, or both. DA binding stimulates and inhibits the corresponding direct and indirect pathways, respectively. D1 furthermore interacts intracellularly with NMDA. Importantly, all neurotransmitter systems are interconnected, so that a perturbation affecting one system propagates through the interacting network and affects the levels of all other neurotransmitters. The ultimate outcome can be an imbalance in the entire system and result in impaired working memory. Therefore, any impairment of working memory may have different root causes and should be attributed to the network as a whole.
This view of a complex neural network underlying working memory function and its disorders has been explored with a number of computational models; some representatives of such models were reviewed in [30] and are briefly mentioned in the following. One class of models focuses on details of the neuronal circuitry and, for instance, studies the role of the ratio of D1 to D2 receptors [31] or attractor properties of imaging signals [32]. Other models attempt to integrate biophysical observations [33, 34]; they may aim at simulating the actions of antipsychotic drugs, using dynamic models of macro-anatomical connections that are relevant to schizophrenic symptoms [35]. Yet other models have focused on the role of dopamine metabolism and analyzed potential sites for interventions to schizophrenia [36–41]. Three entire special issues of Pharmacopsychiatry, Vol. 39 (2006), Vol. 40 (2007) and Vol. 45 (2012), were dedicated to such models.
In this study, we propose a moderately complicated, heuristic and intuitive model that focuses on the dynamics of interactions among neurotransmitters without resorting to details of neurobiological mechanisms. Of course, the human brain contains a huge number of interconnected neuronal loops both within and between regions, but to consider all of these interconnections in a model would not be feasible. Instead, what this heuristic model purports to do (as do many other models of complex brain circuits) is focus on the connections that are most relevant to function and disrupted in selected disease states, with particular emphasis on schizophrenia. There is no question that other phenomena, such as different receptor subtypes or local, oscillatory micro-circuits [42, 43], modulate or fine-tune these responses, but they may not be essential for understanding the primary drivers of the proposed interactions and their consequences. Thus, to tame the enormous complexity, we assembled the structure of the interaction matrix for our model from numerous diagrams in the literature, acknowledging that the model captures the main modes of neurotransmission but is certainly not complete (see Discussion and Conclusions). The resulting composite diagram in Fig. 2 provides a visual bridge between microscopic and macroscopic modeling, and the corresponding mesoscopic model exhibits moderate structural complexity but has exploratory function and seems to be accurate, robust, and intuitive enough to become an effective tool for interdisciplinary communication.
The model is composed of two levels: one consists of a biochemical model capturing the matrix of interactions between neurotransmitters, while the other represents the mobile for physiological and behavioral balances between neurotransmitters. The dynamically changing state of the interaction matrix feeds into the mobile model, which visualizes local or systemic imbalances among the neurotransmitters. Each component of the neurochemical mobile represents the “relative functional weight” of a neurotransmitter, a term which summarily captures its physiological and behavioral importance. The concept of a mobile makes it clear that it is typically not a single deviation in a neurotransmitter that leads to a deficit, but that different brain sections with multiple neurotransmitter systems throughout the brain are involved in different diseases.
Materials and Methods
Clinical and biological data
Clinical and biological information related to working memory deficit in schizophrenia was collected from the literature [44–70]. Specifically, we searched for chronic effects of pharmacological perturbations and lesions presented in peer-reviewed journals using the keywords: chronic perturbation, working memory, and schizophrenia, combined with any of the pertinent brain regions, namely prefrontal cortex, striatum, substantia nigra pars compacta (SNpc), or a particular neurotransmitter and its antagonists, agonists, or reuptake inhibitors. The results of this literature survey are compiled in Table 1, which describes pharmacological agents, their biochemical actions on neurotransmitter systems, targeted brain regions, and chronic effects in observed brain regions.
Table 1.
Clinical data of chronic pharmacological perturbations and comparisons with simulation results
| Pharmacological agent | Pharmacological action# | Targeted regions | Chronic effects$ | Sim% | Ref |
|---|---|---|---|---|---|
| Ethanol (5 weeks) | Increased GABA binding (positive allosteric modulator) | Brain-wide | No change in DA or ACh release to PFC; decrease release of Glu and NE to PFC | √ | [44] |
| PCP (7 days, daily) | Glu (NMDA receptor antagonist) | Brain-wide | Enhanced activation of DA neurons in VTA | √ | [45] |
| Ketamine (7 or 10 days, daily) | Glu (NMDA receptor antagonist) | Brain-wide | increased DA release to striatum and mPFC; increased 5-HT release to mPFC; increased Glu release to mPFC | √ | [46–48] |
| Nicotine (7 or 13 weeks) | ACh (nAChR agonist) | Brain-wide | Increased DA release to striatum; increased Glu release to PFC; decreased ACh release to striatum | √ | [49–51] |
| 5,7-DHT | 5-HT (lesion) | Intracerebro-ventricular | Increased firing of DA neurons in VTA | √ | [52] |
| Tph2 (heterozygote) | 5-HT (reduced 5-HT synthesis) | Brain-wide | Decreased GABA in PFC | √ | [56] |
| Fluoxetine (2 or 3 weeks, daily) | 5-HT (reuptake inhibitor) | Brain-wide | No change in DA release to PFC; increased 5-HT release to PFC; decreased firing of NE neurons in LC; decreased firing of pyramidal PFC cells | √ | [53–55] |
| HT2c (null mutant) | 5-HT (5-HT2c receptor elimination) | Brain-wide | Increased DA release from SNpc to striatum | √ | [57] |
| Olanzapine (21 days) | 5-HT (5-HT2 antagonist) & DA (D2 antagonist) | Brain-wide | Increased firing of NE neurons in LC | √ | [54] |
| Clozapine (21 days) | 5-HT (5-HT2 antagonist) & DA (D2 antagonist) | Brain-wide | Increased DA release to mPFC; no change in DA release to striatum | √ | [58–60] |
| Venlafaxine (3 weeks, daily) | 5-HT & NE (reuptake inhibitor) | Brain-wide | Increased NE, DA, and 5-HT release to PFC | √ | [62] |
| Desipramine (2 weeks, daily) | NE (reuptake inhibitor) | Brain-wide | No change in DA release to striatum; increased DA and NE release to PFC | √ | [49, 62] |
| Nomifensine (22 days, daily) | NE & DA (reuptake inhibitor) | Brain-wide | Increased DA release to striatum; no change in DA release to PFC; Increased ACh release to PFC and striatum; no change in Glu release to PFC and striatum; no change in GABA release to PFC | √ | [63] |
| Mirtazpine (21 days) | NE (alpha2 antagonist) | Brain-wide | Increased firing of 5-HT and NE neurons | √ | [65, 66] |
| 6-OHDA | DA (lesion) | SNpc | Increased firing rate of pyramidal neurons in PFC; increased firing rate of neurons in STN | √ | [64, 67–70] |
| 6-OHDA | DA (lesion) | VTA | Increased firing of NE neurons | √ | [52] |
| 6-OHDA | DA (lesion) | Intracerebro-ventricular | Decreased spontaneous firing of 5-HT neurons | √ | [52] |
: Action sites show which neurotransmitter systems are targets of the pharmacological agent, and mechanisms (in parentheses) indicate the exact mechanisms of actions.
: Abbreviations: Phencyclidine (PCP), 5,7-Dihydroxytryptamine (5,7-DHT), medial prefrontal cortex (mPFC). For other abbreviations, please refer to the legend of Figure 1.
: Comparisons between clinical data and model simulations marked with a check mark indicate qualitative consistency.
Neurochemical interaction matrix
The interactions among neurotransmitters are based on information from the literature and databases that are mentioned in the following text. They can be visualized as a directed connectivity graph, which is shown in Figure 2, where the connecting arrows represent neuronal projections. DA mainly has two types of projections that are relevant for schizophrenia: one from SNpc to striatum and the other from ventral tegmental area (VTA) to PFC [19, 25, 71–82]. The projection from SNpc to striatum allows for binding of DA to D1 and D2 receptor subtypes, which regulate direct and indirect pathways, respectively. To account for these opposing effects, two nodes are used in the graph for the different medium spiny GABAergic neurons. Glu is mainly released from PFC to multiple brain regions, which include striatum, VTA, thalamus, subthalamic nucleus (STN), and dorsal raphe nucleus (DRN) [24, 71–73, 75–78, 81, 83, 84]. GABAergic neurons release GABA, which is associated with direct and indirect pathways connecting striatum, global pallidus, STN, and thalamus [71, 72, 76–78, 81, 83]. 5-HT is released from DRN and regulates PFC, VTA, thalamus, and locus coeruleus (LC) [74, 75, 85–89]. PFC, VTA, and laterodorsal tegmental nucleus (LDT), thalamus, and DRN receive NE from LC [90–94]. In addition, ACh projections are from basal forebrain to PFC and from LDT to SNpc [95–97].
The diagram in Figure 2 was converted into a neurochemical interaction matrix, which was implemented as a set of nonlinear ordinary differential equations (ODEs), where each node was modeled as a dependent variable [12]. As a minimally biased format for mathematical functions governing the processes in the equations, we followed the tenets of Biochemical Systems Theory [98–101] and represented the processes as power-law functions. The parameters in the interaction matrix were assigned with typical default values of 0.5 for positive effects and −0.3 for negative effects (cf. Ch. 5 of [98]). Extensive sensitivity analysis revealed that these settings were not particularly sensitive and that the model overall exhibits remarkable robustness.
A typical simulation consisted of changing a parameter (e.g., the binding affinity of a neurotransmitter to its receptor) and studying how this change propagates through the interaction matrix. The changes in the state of the neurotransmitter profile are dynamic and usually reach a new steady state. During the dynamic up and down in one or all neurotransmitters, the concentration profile is used as an input to the mobile. MATLAB code for the model simulations can be found in the Supplements and on our lab website (www.bst.bme.gatech.edu/research11.php); it contains the ODEs, initial values for all variables, and all parameter values.
Neurotransmitter mobile
Dynamic changes in the interaction matrix are reported and fed as input directly into the neurotransmitter mobile, by means of a function call in MATLAB. The mobile consists of a hierarchical system of balance scales, with the two ends of a lever representing neurotransmitters or mobile sub-systems, which may have different concentrations. As described elsewhere [11], the length of each lever arm relative to its counterpart can be computed since the mobile is assumed to be perfectly balanced under normal, healthy conditions, which allows two lever arms of a balance scale to have different lengths. When one of the neurotransmitters is perturbed by an environmental challenge or a pharmacological agent, the mobile may show an imbalance. To represent this imbalance properly, without the rotation angle reaching ±90°, a resistant force is imposed on the end of each lever arm. This force is proportional to the vertical displacement of the end of a lever arm from its corresponding balanced position. In this way, the mobile properly visualizes dynamic imbalances. For the characterization of a state of imbalance, we define counterclockwise and downward as positive directions for rotation and force, respectively. The mobile offers an intuitive visualization of the dynamically changing state of the neurochemical interaction system. The mathematical details of the computation of the tilting of levers were derived directly from physical and mechanical principles and presented in the Supplements of [11].
As a demonstration, consider PFC, which is critical to working memory. Because it contains both GABAergic and glutamatergic neurons, we use two mobiles for these neurons. GABAergic neurons receive inputs of DA from VTA, 5-HT from DRN, and NE from LC. Therefore, the related mobile reflects these three nodes. By contrast, glutamatergic neurons are regulated by five of the six neurotransmitter systems, so that the appropriate mobile is structurally very similar to the overall mobile in Figure 1. The overall mobile itself is used to visualize the balances among neurotransmitters throughout the entire brain and has six nodes with each of them representing a different neurotransmitter system that sums over all relevant brain regions.
Typical simulations
The model simulations fall into two categories. In the first, we compare model results qualitatively with clinical observations. As a first example, consider an injection of PCP, which leads to activation of DA neurons in VTA. PCP is an NMDA functional antagonist; therefore, the impact of Glu at its targets is reduced. This reduction is modeled by lowering the parameter value (kinetic order) for Glu action after binding. For a second example, consider 5,7-DHT, which causes lesions in 5-HT neurons. It is modeled by lowering the parameter value (rate constant) associated with 5-HT synthesis. Third, Fluoxetine is a 5-HT reuptake inhibitor. Its action is modeled by lowering the kinetic order for the removal of 5-HT from synapses.
The second type of simulation addresses analyses of pharmacological perturbations and their effects on the mobile balances. As an example, administration of the NMDA receptor antagonist ketamine results in increased releases of DA, 5-HT, and Glu in the mPFC. This situation is simulated by lowering the binding affinity of Glu in the interaction matrix model. This change diffuses through the interaction model and is fed into the appropriate mobile models.
Results
We used the combined interaction matrix and mobile model to simulate numerous perturbations that correspond to documented disease states or pharmacological interventions. The results of these simulations were qualitatively compared against clinical and biological data collected from literature (Table 1). Most of these data had been collected from rodents that were chronically injected with some pharmacological agents.
The comparison of clinical and simulation results demonstrates that the model outputs are qualitatively very consistent with the available data. For example, simulations of a chronic injection of the NMDA receptor antagonist ketamine showed qualitatively the same responses of the in vivo system as those observed in rats and mice. This finding is remarkable, as the model parameters are fixed and not adjusted in each simulation to capture specific data (see Methods).
The chronic pharmacological perturbations documented in the literature not only cover all neurotransmitter systems, but most of these perturbations have a brain-wide extension so that all brain regions associated with the same neurotransmitter are affected. Unfortunately, the available data do not probe every single projection, but they do cover a host of interactions associated with working memory in schizophrenia. The qualitative consistency of model results with biological data provides some validation of the model and suggests that the interaction matrix has a reasonable structure that simulates clinical and biological observations quite well even with default parameter values for the neurochemical interaction processes.
To visualize the results from the interaction model, we fed the results of the interaction model into the mobile model which allows exploration of the biochemical mechanisms associated with working memory deficits in schizophrenia. According to the data, several perturbations can impair working memory performance. Here, we show two examples, namely brain-wide, chronic ketamine administration (case 1) and application of an antagonist of the dopamine D2 receptor in PFC (case 2).
Ketamine is an NMDA receptor antagonist, and when it is chronically administered, the two PFC mobiles, as well as the overall mobile, show clear imbalances (Fig. 3). Here, the input is the chronic action of ketamine and the output is the balance configuration of the mobile, which consists of the entire hierarchy of scales. An imbalance of a rod, such as the rod between 5-HT (left end) and DA (right end) in Figure 3A, exhibits clockwise tilting in response to ketamine, which means that this region receives imbalanced signals where 5-HT exceeds DA. Such a change forces the rod to assume a tilted position that differs from the original under normal, healthy condition. The stronger the imbalance, the larger is the tilting angle.
Figure 3. In response to chronical ketamine administration, the overall and sub-mobiles exhibit significant imbalances.

All three mobiles show imbalance in response to chronic ketamine administration, which is an NMDA receptor antagonist. The same coloring scheme as in Figure 1 is used here (red: DA; purple: 5-HT; orange: NE; black: ACh; blue: Glu; green: GABA). A: Mobile for GABAergic neurons in PFC with three nodes (5-HT, DA, and NE); B: Mobile for glutamatergic neurons in PFC with five nodes (DA, NE, ACh, Glu, and GABA); C: Overall mobile for the whole brain with six nodes for all neurotransmitter systems across whole brain. As the results show, DA/5-HT outweighs NE (A), DA/NE outweighs ACh/Glu/GABA (B), and DA/5-HT/NE (C) similarly outweighs ACh/Glu/GABA.
One notes that there are notable differences among these imbalances, with the overall mobile being perturbed most. A surprising finding here is that the right side of the mobile, due to the complicated dynamics of the interaction matrix, exhibits an essentially undisturbed balance between ACh and Glu/GABA (Fig. 3C), although ketamine is an NMDA receptor antagonist. A similar result is observed for the mobile representing glutamatergic neurons (Fig. 3B). It becomes apparent that DA/5-HT (Fig. 3A), DA/NE (Fig. 3B), or DA/5-HT/NE (Fig. 3C) outweighs the right sides of the mobiles. This conclusion suggests that the elevated modulator DA is a major driver, if not even the dominant contributor to the imbalances shown in all three mobiles.
By contrast, application of an antagonist of the D2 receptor in PFC does not cause a noticeable imbalance on GABAergic neurons (Fig. 4A), while both the overall mobile and the glutamatergic neurons in PFC show imbalances (Fig. 4B–C). Different to the case of ketamine, the right side, which includes Glu/GABA/NE, now outweighs the side of DA/ACh. Interestingly, the dynamics of the interaction matrix leads to a state where the local release of DA and NE (Fig. 4B) to glutamatergic neurons in PFC, as well as overall levels of DA, 5-HT, and NE (Fig. 4C), are well balanced.
Figure 4. In response to application of antagonist of DA D2 receptor in PFC, the overall and sub-mobiles exhibit significant imbalances.

Two of the three mobiles show imbalances in response to application of an antagonist of DA D2 receptor in PFC. The same coloring scheme as in Figure 1 is used here (red: DA; purple: 5-HT; orange: NE; black: ACh; blue: Glu; green: GABA). A: Mobile for GABAergic neurons in PFC with three nodes (5-HT, DA, and NE) and does not involve Glu; B: Mobile for glutamatergic neurons in PFC with five nodes (DA, NE, ACh, Glu, and GABA); C: Overall mobile for the whole brain with six nodes for all neurotransmitter systems across whole brain. As the results show, the DA D2 antagonist does not cause imbalance on GABAergic neurons in PFC. Interestingly, projections of DA and ACh (B) to glutamatergic neurons in PFC, or overall levels of DA, 5-HT, and ACh (C), are balanced.
In addition to these two scenarios of impairing working memory performance, we explored other changes in neurotransmitter systems that may be associated with working memory deficits. Table 2 shows the balance states of the three mobiles following fifteen different perturbations, each of which targets a different node in the interaction matrix. Interestingly, these three mobiles show distinct responses. The overall mobile always shows imbalances in some of its scales. In contrast, the mobile for GABAergic neurons in PFC always keeps its balance except for two situations. Meanwhile, the mobile for glutamatergic neurons in PFC exhibits imbalances following some perturbations but not others. Among the influential perturbations, two actions target Glu neurons in PFC and thalamus, two target DA neurons in VTA and SNpc, and two affect ACh neurons in LDT and 5-HT neurons in DRN. These results in Table 2, which are not always intuitive, demonstrate that the entire, brain-wide interaction system needs to be considered.
Table 2.
Perturbations on neurotransmitters and their effects on balance of mobiles
| Perturbation | Mobile for GABAergic neurons# | Mobile for Glutamatergic neurons$ | Overall Mobile% |
|---|---|---|---|
| ACh antagonist on Glu neurons in PFC | ⟺ | DA/NE down GABA down |
5-HT/DA/NE down 5-HT down GABA down |
| DA D2 antagonist on GABAergic neurons in striatum | ⟺ | ⟺ | ACh/Glu/GABA down |
| Activation of DA D1 on GABAergic neurons in striatum | ⟺ | ⟺ | GABA down ACh/Glu/GABA down |
| NMDA receptor agonist on GABAergic neurons in GPi | ⟺ | ⟺ | GABA down ACh/Glu/GABA down |
| NMDA receptor antagonist on GABAergic neurons in GPe | ⟺ | ⟺ | Glu down |
| Reuptake inhibitor of GABA release to Glu neurons in STN | ⟺ | ⟺ | ACh/Glu/GABA down GABA down |
| 5-HT2 agonist on Glu neurons in Thalamus | ⟺ | ACh/Glu/GABA down Glu down |
ACh/Glu/GABA down Glu down |
| NE α1 antagonist on DA neurons in VTA | NE down 5-HT down |
ACh/Glu/GABA down NE down |
ACh/Glu/GABA down 5-HT down |
| 5-HT2 antagonist on GABA neurons in VTA | ⟺ | ⟺ | 5-HT/DA/NE down DA down Glu down |
| Reuptake inhibitor of ACh release to DA in SNpc | ⟺ | DA/NE down GABA down |
5-HT/DA/NE down 5-HT down GABA down |
| NE α1 antagonist on ACh neurons in LDT | ⟺ | ACh/Glu/GABA down | ACh/Glu/GABA down 5-HT down Glu down |
| Reuptake inhibitor of GABA release to 5-HT neurons in DRN | DA down | DA/NE down Glu down |
ACh/Glu/GABA down DA down GABA down |
| Reuptake activation of GABA release to NE neurons in LC | ⟺ | ⟺ | 5-HT/DA/NE down Glu down |
| NMDA receptor antagonist on GABAergic neurons in DRN | ⟺ | ⟺ | 5-HT/DA/NE down 5-HT down Glu down |
| 5-HT2 agonist on GABA neurons in LC | ⟺ | ⟺ | ACh/Glu/GABA down GABA down |
: the symbol “⟺“ indicates balance of a mobile.
: Text descriptions, such as DA/NE down, indicate an imbalanced state of a mobile; specifically, DA/NE down means the scale with DA and NE tilts down on the side of these neurotransmitters.
: For abbreviations, please refer to the legend of Figure 1.
One might add that many perturbations listed in Table 2 may temporarily tilt some of the rods but that they do not cause permanent imbalances in healthy subjects. By contrast, this normalization in healthy subjects (i.e., the return to balance) fails to occur in schizophrenia. This failure is highly consistent with what most researchers in psychiatry view as (allostatic) disease states – the failure of homeostatic compensation.
Studies have shown that increased ACh levels, nicotine nasal spray, and smoking can improve working memory performance in schizophrenia patients [102–104]. We explored the effect of changing the ACh level on mobile imbalances caused by chronic ketamine administration. Because ACh directly affects DA neurons, we specifically targeted the projection of ACh to Glu neurons in PFC and focused on the mobile for glutamatergic neurons in PFC. Figure 5 shows that an ACh agonist can restore the balance between DA and NE on the left side of the overall scale and ACh, Glu and GABA on the right side. However, the imbalances in DA and NE, and Glu and GABA are not compensated by the ACh agonist. One notes that the balance between ACh and Glu/GABA is not affected by ketamine or the ACh agonist. Therefore, it could be an indirect effect of the ACh agonist on DA that partly ameliorates the imbalance stemming from chronic ketamine administration.
Figure 5. ACh agonist reduces imbalances caused by chronic ketamine administration.

The mobile shows that an ACh agonist reduces imbalances caused by chronic ketamine administration. The mobile shown is for glutamatergic neurons in PFC. The same coloring scheme as in Figure 1 is used here (red: DA; purple: 5-HT; orange: NE; black: ACh; blue: Glu; green: GABA). As shown, the ACh agonist can restore the balance between DA/NE on the left side of the overall scale and ACh/Glu/GABA on the right side. The increased imbalance between Glu and GABA may be due to the action of ACh or to influences from other brain regions not included in the model.
Discussion and Conclusions
Schizophrenia has a wide spectrum of clinical symptoms, of which the positive symptoms are most obvious and best studied. By contrast, its debilitating cognitive symptoms, e.g., working memory deficits, have received much less attention, even though they are pervasive and their onset often occurs earlier than other symptoms. Current pharmacotherapies are not particularly efficacious with respect to restoring the functionality of impaired working memory. The reason may be that the neurotransmitter levels associated with schizophrenia change dynamically and in a complex manner. This conclusion, which is due to the rich set of multiple interacting neurotransmitter systems, renders it evident that tools of investigation should be capable of truly capturing the systemic nature of the disease. In this article, we implemented such tools in the form of a moderately complicated mesoscopic model that appears to capture the interactions quite well and visualizes them in the form of mobiles, which intuitively portray physiological imbalances among neurotransmitters.
In line with its Greek etymology, the term “mesoscopic” refers to something with a scope in the middle, without specifying “the middle of what” is being addressed. In our case, the model connects the organizational levels of molecular biology and biochemistry on the one hand and of physiology and psychiatry on the other. Within this intermediate realm, our mesoscopic model has an intended low degree of conceptual resolution, which allows us to reduce complexity by retaining resolution only in the most necessary components. Expressed differently, to include all microscopic details in a comprehensive macroscopic model would presently not be possible. At our level of analysis, it is necessary to step back and take a broader perspective in order to retain a comprehensible framework, which is necessarily coarse-grained, at least in some aspects. Other types of coarse-grained models, such as the famous Hodgkin-Huxley model of the initiation and propagation of action potentials in neurons, which ignores many aspects of neuronal functionality, demonstrate the benefits of mesoscopic approaches for understanding and advanced further research.
A particular strength of any computational model is the ease with which various scenarios can be explored. For example, in one typical simulation we perturbed individual neurotransmitter projections from the PFC. The model easily captured how these perturbations migrated through the connectivity graph in Figure 2, governed by the interaction matrix and its differential equations. The model then took this information and revealed the ultimate, dynamic effects on neurotransmitters, which were visualized with different mobiles. For the case of PFC, two mobiles were developed: one for glutamatergic neurons and the other for GABAergic neurons. In addition, the levels of each neurotransmitter system were summed over all relevant brain regions, and this process resulted in an overall mobile for the whole brain. The results of this type of simulation suggest that GABAergic neurons in PFC are not likely responsible for working memory deficit. Secondly, the overall mobile exhibits imbalances in response to many different changes, which suggests that this particular mobile is not discerning enough as a model for working memory deficit in schizophrenia. Instead, our simulation results suggest that the mobile for glutamatergic neurons in PFC responds much more specifically to genetic mutations and pharmacological perturbations on working memory in the context of schizophrenia. It might therefore be a reasonable model for studying cognitive deficits.
Other simulation results showed that chronic administration of ketamine to the whole brain can cause a variety of imbalances within the mobile. Specifically, DA and NE outweigh ACh, GABA, and Glu (Fig. 3B). By contrast, application of an antagonist of the D2 receptor in PFC has the opposite effect (Fig. 4B). Various other perturbations, as displayed in Table 2, resulted in different combinations of imbalances within the mobile. For instance, application of an NE antagonist on the DA neurons in VTA leads to an imbalance where ACh/Glu/GABA are down, while DA and NE are lifted up. At the same time, the balance between DA and NE tilts toward a higher position of DA. Some brain regions, such as GPi and GPe, appear to have negligible influence on working memory performance. An important overall conclusion is that it appears to be likely that multiple failure routes can lead to working memory deficit. If this conclusion is indeed correct, distinct treatments might have to be developed for different root causes of such deficits.
A related, yet different advantage of the mobile is that it can be applied as a heuristic tool also for other perturbations, such as alcohol dependence, which we have shown in a simplified analysis elsewhere [11]. This applicability shows that the model structure represents some general principles of neurotransmitter interactions, whereas a more detailed model, due to today’s modeling capabilities, has more limited generalizability.
A crucial challenge for any modeling task is to find the most appropriate level of conceptual resolution so that the model is neither too simple regarding the body of empirical knowledge nor that it is so complex that it precludes understanding. Here, the interaction diagram in Figure 2 is complicated, but certainly not as detailed as reality. For instance, the hippocampus has been associated with working memory deficit in schizophrenia [105], but is not included in this study. Furthermore, sub-regions within each brain region may be responsible for different tasks of working memory. As an example, the left posterior parietal cortex is activated during verbal working memory tasks, while spatial tasks tend to recruit areas in the right hemisphere [106]. In addition, each neurotransmitter system might contain multiple receptor subtypes, which could have very different actions, as it is the case for D1 and D2. From a mathematical point of view, extensions of the model to more brain regions and to different receptor subtypes are no issue in principle. However, such extensions require large amounts of additional data, which are presently unavailable.
Selecting an intermediate level of conceptual resolution for modeling has the substantial advantage that it facilitates interdisciplinary communication regarding the neurochemical dynamics in schizophrenia. Effective tools for communication are of obvious importance for clinical psychiatrists and psychopharmacologists, as neuropsychology, neurophysiology, brain imaging, experimental neurobiology, neurochemistry, histology, genetics, and computational science use very different, sophisticated methodologies that are intellectually challenging. Without doubt, all these disciplines contribute greatly to our knowledge about schizophrenia, but it is difficult, even for the expert, to obtain an integrative view of the disease from these disparate fields. The interaction-mobile model is obviously coarse, but it nevertheless relates an immediate and intuitive impression of how the neurochemical state of the brain or of a brain region is altered by disease and treatment.
This study provides heuristic and systemic access to the biochemical interactions among neurotransmitter systems and their physiological or behavioral effects in the context of working memory deficit in schizophrenia. As the results in Table 1 demonstrate, the model reflects available data well and may be particularly useful as a tool for the education of medical students and the lay public. The models and the method may assist with the prediction of novel targets for pharmacotherapeutic drug design for working memory deficit. It is also clear that this modeling approach might be applied similarly to other symptoms of schizophrenia.
Highlights.
A heuristic model combining biochemical and physiological levels in schizophrenia is proposed
The model accounts for numerous interactions among neurotransmitters in various brain regions
This study suggests that, mathematically, glutamatergic neuron in PFC may be a good indicator of neurotransmitter imbalances related to working memory deficits
Some brain regions appear to have negligible influence on working memory performance
The model can serve as an intuitive tool for communicating clinical and biological observations
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (P01-ES016731, GWM, PI; R01HL095479) and an endowment from the Georgia Research Alliance (EOV, PI). Any opinions, findings, conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the sponsoring institutions.
Abbreviations
- 5,7-DHT
5,7-Dihydroxytryptamine
- PCP
Phencyclidine
- ACh
acetylcholine
- DA
dopamine
- DRN
dorsal raphe nucleus
- DLPFC
dorsolateral prefrontal cortex
- GABA
gamma-aminobutyric acid
- GPe
global pallidus external
- GPi
global pallidus internal
- Glu
glutamate
- LDT
laterodorsal tegmental nucleus
- LC
locus coeruleus
- mPFC
medial prefrontal cortex
- NE
norepinephrine
- ODEs
ordinary differential equations
- PFC
prefrontal cortex
- 5-HT
serotonin
- SNpc
substantia nigra pars compacta
- STN
subthalamic nucleus
- VTA
ventral tegmental area
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Z.Q., F.T., and E.O.V. designed the study; Z.Q. and G.P.Y. performed the computational research and analyzed the results; Z.Q. and E.O.V. wrote the paper; F.T., O.P., and A.A.G. provided expertise and feedback. All authors critically reviewed the content and approved the final version for publication.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.van Os J, Kapur S. Schizophrenia. Lancet. 2009;374:635–645. doi: 10.1016/S0140-6736(09)60995-8. [DOI] [PubMed] [Google Scholar]
- 2.APA. Diagnostic and Statistical Manual of Mental Disorders (DSM-5) American Psychiatric Association; 2013. [DOI] [PubMed] [Google Scholar]
- 3.Barch DM, Sheffield JM. Cognitive impairments in psychotic disorders: common mechanisms and measurement. World psychiatry: official journal of the World Psychiatric Association. 2014;13:224–232. doi: 10.1002/wps.20145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carlsson A. The neurochemical circuitry of schizophrenia. Pharmacopsychiatry. 2006;39(Suppl 1):S10–14. doi: 10.1055/s-2006-931483. [DOI] [PubMed] [Google Scholar]
- 5.Leicht G, Kirsch V, Giegling I, Karch S, Hantschk I, Moller HJ, Pogarell O, Hegerl U, Rujescu D, Mulert C. Reduced early auditory evoked gamma-band response in patients with schizophrenia. Biological psychiatry. 2010;67:224–231. doi: 10.1016/j.biopsych.2009.07.033. [DOI] [PubMed] [Google Scholar]
- 6.Tseng KY, O’Donnell P. Dopamine-glutamate interactions controlling prefrontal cortical pyramidal cell excitability involve multiple signaling mechanisms. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2004;24:5131–5139. doi: 10.1523/JNEUROSCI.1021-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuen EY, Yan Z. Dopamine D4 receptors regulate AMPA receptor trafficking and glutamatergic transmission in GABAergic interneurons of prefrontal cortex. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:550–562. doi: 10.1523/JNEUROSCI.5050-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tretter F, Winterer G, Gebicke-Haerter PJ, Mendoza ER. Systems Biology in Psychiatric Research: From High-Throughput Data to Mathematical Modeling, Wiley-Blackwell. 2010 [Google Scholar]
- 9.Fritze J. Biologische Psychiatrie, Fischer, Stuttgart. 1989 [Google Scholar]
- 10.Bender W, Albus M, Moller HJ, Tretter F. Towards systemic theories in biological psychiatry. Pharmacopsychiatry. 2006;39(Suppl 1):S4–9. doi: 10.1055/s-2006-931482. [DOI] [PubMed] [Google Scholar]
- 11.Qi Z, Tretter F, Voit EO. A heuristic model of alcohol dependence. PloS one. 2014;9:e92221. doi: 10.1371/journal.pone.0092221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qi Z, Fieni D, Tretter F, Voit EO. The neurochemical mobile with non-linear interaction matrix: an exploratory computational model. Pharmacopsychiatry. 2013;46(Suppl 1):S53–63. doi: 10.1055/s-0033-1337920. [DOI] [PubMed] [Google Scholar]
- 13.Carlsson A. The current status of the dopamine hypothesis of schizophrenia. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 1988;1:179–186. doi: 10.1016/0893-133x(88)90012-7. [DOI] [PubMed] [Google Scholar]
- 14.Moghaddam B. Bringing order to the glutamate chaos in schizophrenia. Neuron. 2003;40:881–884. doi: 10.1016/s0896-6273(03)00757-8. [DOI] [PubMed] [Google Scholar]
- 15.Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia, Nature reviews. Neuroscience. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- 16.Aghajanian GK, Marek GJ. Serotonin model of schizophrenia: emerging role of glutamate mechanisms. Brain research Brain research reviews. 2000;31:302–312. doi: 10.1016/s0165-0173(99)00046-6. [DOI] [PubMed] [Google Scholar]
- 17.Pogarell O, Koch W, Karch S, Dehning S, Muller N, Tatsch K, Poepperl G, Moller HJ. Dopaminergic neurotransmission in patients with schizophrenia in relation to positive and negative symptoms. Pharmacopsychiatry. 2012;45(Suppl 1):S36–41. doi: 10.1055/s-0032-1306313. [DOI] [PubMed] [Google Scholar]
- 18.Deserno L, Huys QJ, Boehme R, Buchert R, Heinze HJ, Grace AA, Dolan RJ, Heinz A, Schlagenhauf F. Ventral striatal dopamine reflects behavioral and neural signatures of model-based control during sequential decision making. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:1595–1600. doi: 10.1073/pnas.1417219112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zahrt J, Taylor JR, Mathew RG, Arnsten AF. Supranormal stimulation of D1 dopamine receptors in the rodent prefrontal cortex impairs spatial working memory performance. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1997;17:8528–8535. doi: 10.1523/JNEUROSCI.17-21-08528.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rao SG, Williams GV, Goldman-Rakic PS. Destruction and creation of spatial tuning by disinhibition: GABA(A) blockade of prefrontal cortical neurons engaged by working memory. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2000;20:485–494. doi: 10.1523/JNEUROSCI.20-01-00485.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arnsten AF, Paspalas CD, Gamo NJ, Yang Y, Wang M. Dynamic Network Connectivity: A new form of neuroplasticity. Trends in cognitive sciences. 2010;14:365–375. doi: 10.1016/j.tics.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robbins TW, Arnsten AF. The neuropsychopharmacology of fronto-executive function: monoaminergic modulation. Annual review of neuroscience. 2009;32:267–287. doi: 10.1146/annurev.neuro.051508.135535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castner SA, Vosler PS, Goldman-Rakic PS. Amphetamine sensitization impairs cognition and reduces dopamine turnover in primate prefrontal cortex. Biological psychiatry. 2005;57:743–751. doi: 10.1016/j.biopsych.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 24.Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1997;17:2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldman-Rakic PS. The physiological approach: functional architecture of working memory and disordered cognition in schizophrenia. Biological psychiatry. 1999;46:650–661. doi: 10.1016/s0006-3223(99)00130-4. [DOI] [PubMed] [Google Scholar]
- 26.Lett TA, Voineskos AN, Kennedy JL, Levine B, Daskalakis ZJ. Treating working memory deficits in schizophrenia: a review of the neurobiology. Biological psychiatry. 2014;75:361–370. doi: 10.1016/j.biopsych.2013.07.026. [DOI] [PubMed] [Google Scholar]
- 27.Cai HL, Zhu RH, Li HD, Zhang XH, Hu L, Yang W, Ye HS. Elevated plasma gamma-aminobutyrate/glutamate ratio and responses to risperidone antipsychotic treatment in schizophrenia. Progress in neuro-psychopharmacology & biological psychiatry. 2010;34:1273–1278. doi: 10.1016/j.pnpbp.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 28.Luciana M, Collins PF, Depue RA. Opposing roles for dopamine and serotonin in the modulation of human spatial working memory functions. Cerebral cortex. 1998;8:218–226. doi: 10.1093/cercor/8.3.218. [DOI] [PubMed] [Google Scholar]
- 29.Slaney TR, Mabrouk OS, Porter-Stransky KA, Aragona BJ, Kennedy RT. Chemical gradients within brain extracellular space measured using low flow push-pull perfusion sampling in vivo. ACS chemical neuroscience. 2013;4:321–329. doi: 10.1021/cn300158p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tretter F, Albus M. “Computational Neuropsychiatry” of Working Memory Disorders in Schizophrenia: The Network Connectivity in Prefrontal Cortex - Data and Models. Pharmacopsychiatry. 2007;40:S2–S16. [Google Scholar]
- 31.Durstewitz D, Seamans JK, Sejnowski TJ. Neurocomputational models of working memory, Nature neuroscience. 2000;3(Suppl):1184–1191. doi: 10.1038/81460. [DOI] [PubMed] [Google Scholar]
- 32.Deco G, Ponce-Alvarez A, Hagmann P, Romani GL, Mantini D, Corbetta M. How local excitation-inhibition ratio impacts the whole brain dynamics. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014;34:7886–7898. doi: 10.1523/JNEUROSCI.5068-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang XJ, Tegner J, Constantinidis C, Goldman-Rakic PS. Division of labor among distinct subtypes of inhibitory neurons in a cortical microcircuit of working memory. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:1368–1373. doi: 10.1073/pnas.0305337101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang XJ. Neurophysiological and computational principles of cortical rhythms in cognition. Physiological reviews. 2010;90:1195–1268. doi: 10.1152/physrev.00035.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geerts H, Roberts P, Spiros A. Assessing the synergy between cholinomimetics and memantine as augmentation therapy in cognitive impairment in schizophrenia. A virtual human patient trial using quantitative systems pharmacology. Frontiers in pharmacology. 2015;6:198. doi: 10.3389/fphar.2015.00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qi Z, Miller GM, Voit EO. Mathematical Models in Schizophrenia. In: Ritsner MS, editor. Handbook of Schizophrenia Spectrum Disorders. Springer Verlag; New York: 2011. [Google Scholar]
- 37.Qi Z, Miller GW, Voit EO. Computational modeling of synaptic neurotransmission as a tool for assessing dopamine hypotheses of schizophrenia. Pharmacopsychiatry. 2010;43(Suppl 1):S50–60. doi: 10.1055/s-0030-1248317. [DOI] [PubMed] [Google Scholar]
- 38.Qi Z, Miller GW, Voit EO. Computational analysis of determinants of dopamine (DA) dysfunction in DA nerve terminals. Synapse. 2009;63:1133–1142. doi: 10.1002/syn.20686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Voit EO, Qi Z, Miller GW. Steps of modeling complex biological systems. Pharmacopsychiatry. 2008;41(Suppl 1):S78–84. doi: 10.1055/s-2008-1080911. [DOI] [PubMed] [Google Scholar]
- 40.Qi Z, Miller GW, Voit EO. A mathematical model of presynaptic dopamine homeostasis: implications for schizophrenia. Pharmacopsychiatry. 2008;41(Suppl 1):S89–98. doi: 10.1055/s-2008-1080936. [DOI] [PubMed] [Google Scholar]
- 41.Qi Z, Miller GW, Voit EO. Computational systems analysis of dopamine metabolism. PloS one. 2008;3:e2444. doi: 10.1371/journal.pone.0002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.an der Heiden U. Schizophrenia as a dynamical disease. Pharmacopsychiatry. 2006;39(Suppl 1):S36–42. doi: 10.1055/s-2006-931487. [DOI] [PubMed] [Google Scholar]
- 43.Wang XJ. Toward a prefrontal microcircuit model for cognitive deficits in schizophrenia. Pharmacopsychiatry. 2006;39(Suppl 1):S80–87. doi: 10.1055/s-2006-931501. [DOI] [PubMed] [Google Scholar]
- 44.Sabria J, Torres D, Pasto M, Peralba JM, Allali-Hassani A, Pares X. Release of neurotransmitters from rat brain nerve terminals after chronic ethanol ingestion: differential effects in cortex and hippocampus. Addiction biology. 2003;8:287–294. doi: 10.1080/13556210310001602194. [DOI] [PubMed] [Google Scholar]
- 45.Uramura K, Maejima Y, Shimomura K, Santoso P, Katsuda S, Kobayashi D, Jodo E, Kodaira M, Otgon-Uul Z, Yang Y, Sakuma K, Takigawa M, Hazama A, Yada T. Chronic phencyclidine treatment induces long-lasting glutamatergic activation of VTA dopamine neurons. Neuroscience letters. 2014;564:72–77. doi: 10.1016/j.neulet.2014.01.032. [DOI] [PubMed] [Google Scholar]
- 46.Chatterjee M, Verma R, Ganguly S, Palit G. Neurochemical and molecular characterization of ketamine-induced experimental psychosis model in mice. Neuropharmacology. 2012;63:1161–1171. doi: 10.1016/j.neuropharm.2012.05.041. [DOI] [PubMed] [Google Scholar]
- 47.Zugno AI, Matos MP, Canever L, Fraga DB, De Luca RD, Ghedim FV, Deroza PF, de Oliveira MB, Pacheco FD, Valvassori SS, Volpato AM, Budni J, Quevedo J. Evaluation of acetylcholinesterase activity and behavioural alterations induced by ketamine in an animal model of schizophrenia. Acta neuropsychiatrica. 2014;26:43–50. doi: 10.1017/neu.2013.31. [DOI] [PubMed] [Google Scholar]
- 48.Lindefors N, Barati S, O’Connor WT. Differential effects of single and repeated ketamine administration on dopamine, serotonin and GABA transmission in rat medial prefrontal cortex. Brain research. 1997;759:205–212. doi: 10.1016/s0006-8993(97)00255-2. [DOI] [PubMed] [Google Scholar]
- 49.Pietila K, Ahtee L. Chronic nicotine administration in the drinking water affects the striatal dopamine in mice. Pharmacology, biochemistry, and behavior. 2000;66:95–103. doi: 10.1016/s0091-3057(00)00235-5. [DOI] [PubMed] [Google Scholar]
- 50.Falasca S, Ranc V, Petruzziello F, Khani A, Kretz R, Zhang X, Rainer G. Altered neurochemical levels in the rat brain following chronic nicotine treatment. Journal of chemical neuroanatomy. 2014;59–60:29–35. doi: 10.1016/j.jchemneu.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 51.Pawlak R, Takada Y, Takahashi H, Urano T, Ihara H, Nagai N, Takada A. Differential effects of nicotine against stress-induced changes in dopaminergic system in rat striatum and hippocampus. European journal of pharmacology. 2000;387:171–177. doi: 10.1016/s0014-2999(99)00778-5. [DOI] [PubMed] [Google Scholar]
- 52.Guiard BP, El Mansari M, Merali Z, Blier P. Functional interactions between dopamine, serotonin and norepinephrine neurons: an in-vivo electrophysiological study in rats with monoaminergic lesions. The international journal of neuropsychopharmacology/official scientific journal of the Collegium Internationale Neuropsychopharmacologicum. 2008;11:625–639. doi: 10.1017/S1461145707008383. [DOI] [PubMed] [Google Scholar]
- 53.Tanda G, Frau R, Di Chiara G. Chronic desipramine and fluoxetine differentially affect extracellular dopamine in the rat prefrontal cortex. Psychopharmacology. 1996;127:83–87. doi: 10.1007/BF02805978. [DOI] [PubMed] [Google Scholar]
- 54.Seager MA, Barth VN, Phebus LA, Rasmussen K. Chronic coadministration of olanzapine and fluoxetine activates locus coeruleus neurons in rats: implications for bipolar disorder. Psychopharmacology. 2005;181:126–133. doi: 10.1007/s00213-005-2198-2. [DOI] [PubMed] [Google Scholar]
- 55.Gronier BS, Rasmussen K. Electrophysiological effects of acute and chronic olanzapine and fluoxetine in the rat prefrontal cortex. Neuroscience letters. 2003;349:196–200. doi: 10.1016/s0304-3940(03)00851-6. [DOI] [PubMed] [Google Scholar]
- 56.Waider J, Proft F, Langlhofer G, Asan E, Lesch KP, Gutknecht L. GABA concentration and GABAergic neuron populations in limbic areas are differentially altered by brain serotonin deficiency in Tph2 knockout mice. Histochemistry and cell biology. 2013;139:267–281. doi: 10.1007/s00418-012-1029-x. [DOI] [PubMed] [Google Scholar]
- 57.Abdallah L, Bonasera SJ, Hopf FW, O’Dell L, Giorgetti M, Jongsma M, Carra S, Pierucci M, Di Giovanni G, Esposito E, Parsons LH, Bonci A, Tecott LH. Impact of serotonin 2C receptor null mutation on physiology and behavior associated with nigrostriatal dopamine pathway function. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:8156–8165. doi: 10.1523/JNEUROSCI.3905-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamamoto BK, Cooperman MA. Differential effects of chronic antipsychotic drug treatment on extracellular glutamate and dopamine concentrations. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1994;14:4159–4166. doi: 10.1523/JNEUROSCI.14-07-04159.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jaskiw GE, Kirkbride B, Bongiovanni R. In rats chronically treated with clozapine, tyrosine depletion attenuates the clozapine-induced in vivo increase in prefrontal cortex dopamine and norepinephrine levels. Psychopharmacology. 2006;185:416–422. doi: 10.1007/s00213-005-0283-1. [DOI] [PubMed] [Google Scholar]
- 60.Youngren KD, Moghaddam B, Bunney BS, Roth RH. Preferential activation of dopamine overflow in prefrontal cortex produced by chronic clozapine treatment. Neuroscience letters. 1994;165:41–44. doi: 10.1016/0304-3940(94)90704-8. [DOI] [PubMed] [Google Scholar]
- 61.Pietraszek M, Golembiowska K, Bijak M, Ossowska K, Wolfarth S. Differential effects of chronic haloperidol and clozapine administration on glutamatergic transmission in the fronto-parietal cortex in rats: microdialysis and electrophysiological studies. Naunyn-Schmiedeberg’s archives of pharmacology. 2002;366:417–424. doi: 10.1007/s00210-002-0619-x. [DOI] [PubMed] [Google Scholar]
- 62.Higashino K, Ago Y, Umehara M, Kita Y, Fujita K, Takuma K, Matsuda T. Effects of acute and chronic administration of venlafaxine and desipramine on extracellular monoamine levels in the mouse prefrontal cortex and striatum. European journal of pharmacology. 2014;729:86–93. doi: 10.1016/j.ejphar.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 63.Hernandez LF, Segovia G, Mora F. Chronic treatment with a dopamine uptake blocker changes dopamine and acetylcholine but not glutamate and GABA concentrations in prefrontal cortex, striatum and nucleus accumbens of the awake rat. Neurochemistry international. 2008;52:457–469. doi: 10.1016/j.neuint.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 64.Wang S, Zhang QJ, Liu J, Wu ZH, Ali U, Wang Y, Chen L, Gui ZH. The firing activity of pyramidal neurons in medial prefrontal cortex and their response to 5-hydroxytryptamine-1A receptor stimulation in a rat model of Parkinson’s disease. Neuroscience. 2009;162:1091–1100. doi: 10.1016/j.neuroscience.2009.04.069. [DOI] [PubMed] [Google Scholar]
- 65.Haddjeri N, Blier P, de Montigny C. Acute and long-term actions of the antidepressant drug mirtazapine on central 5-HT neurotransmission. Journal of affective disorders. 1998;51:255–266. doi: 10.1016/s0165-0327(98)00223-7. [DOI] [PubMed] [Google Scholar]
- 66.Haddjeri N, Blier P, de Montigny C. Effect of the alpha-2 adrenoceptor antagonist mirtazapine on the 5-hydroxytryptamine system in the rat brain. The Journal of pharmacology and experimental therapeutics. 1996;277:861–871. [PubMed] [Google Scholar]
- 67.Parr-Brownlie LC, Poloskey SL, Flanagan KK, Eisenhofer G, Bergstrom DA, Walters JR. Dopamine lesion-induced changes in subthalamic nucleus activity are not associated with alterations in firing rate or pattern in layer V neurons of the anterior cingulate cortex in anesthetized rats. The European journal of neuroscience. 2007;26:1925–1939. doi: 10.1111/j.1460-9568.2007.05814.x. [DOI] [PubMed] [Google Scholar]
- 68.Vila M, Perier C, Feger J, Yelnik J, Faucheux B, Ruberg M, Raisman-Vozari R, Agid Y, Hirsch EC. Evolution of changes in neuronal activity in the subthalamic nucleus of rats with unilateral lesion of the substantia nigra assessed by metabolic and electrophysiological measurements. The European journal of neuroscience. 2000;12:337–344. doi: 10.1046/j.1460-9568.2000.00901.x. [DOI] [PubMed] [Google Scholar]
- 69.Breit S, Bouali-Benazzouz R, Popa RC, Gasser T, Benabid AL, Benazzouz A. Effects of 6-hydroxydopamine-induced severe or partial lesion of the nigrostriatal pathway on the neuronal activity of pallido-subthalamic network in the rat. Experimental neurology. 2007;205:36–47. doi: 10.1016/j.expneurol.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 70.Janssen ML, Zwartjes DG, Tan SK, Vlamings R, Jahanshahi A, Heida T, Hoogland G, Steinbusch HW, Visser-Vandewalle V, Temel Y. Mild dopaminergic lesions are accompanied by robust changes in subthalamic nucleus activity. Neuroscience letters. 2012;508:101–105. doi: 10.1016/j.neulet.2011.12.027. [DOI] [PubMed] [Google Scholar]
- 71.Carlsson A, Waters N, Carlsson ML. Neurotransmitter interactions in schizophrenia–therapeutic implications. Biological psychiatry. 1999;46:1388–1395. doi: 10.1016/s0006-3223(99)00117-1. [DOI] [PubMed] [Google Scholar]
- 72.Carlsson A, Waters N, Carlsson ML. Neurotransmitter interactions in schizophrenia-therapeutic implications. European archives of psychiatry and clinical neuroscience. 1999;249(Suppl 4):37–43. doi: 10.1007/pl00014183. [DOI] [PubMed] [Google Scholar]
- 73.Hazy TE, Frank MJ, O’Reilly R C. Towards an executive without a homunculus: computational models of the prefrontal cortex/basal ganglia system, Philosophical transactions of the Royal Society of London. Series B, Biological sciences. 2007;362:1601–1613. doi: 10.1098/rstb.2007.2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Autry AE, Monteggia LM. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacological reviews. 2012;64:238–258. doi: 10.1124/pr.111.005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sumiyoshi T, Higuchi Y, Uehara T. Neural basis for the ability of atypical antipsychotic drugs to improve cognition in schizophrenia. Frontiers in behavioral neuroscience. 2013;7:140. doi: 10.3389/fnbeh.2013.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schroll H, Vitay J, Hamker FH. Working memory and response selection: a computational account of interactions among cortico-basalganglio-thalamic loops. Neural networks: the official journal of the International Neural Network Society. 2012;26:59–74. doi: 10.1016/j.neunet.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 77.Taylor JG, Taylor NR. Analysis of recurrent cortico-basal ganglia-thalamic loops for working memory. Biological cybernetics. 2000;82:415–432. doi: 10.1007/s004220050595. [DOI] [PubMed] [Google Scholar]
- 78.Zhou FM, Wilson CJ, Dani JA. Cholinergic interneuron characteristics and nicotinic properties in the striatum. Journal of neurobiology. 2002;53:590–605. doi: 10.1002/neu.10150. [DOI] [PubMed] [Google Scholar]
- 79.Williams GV, Goldman-Rakic PS. Modulation of memory fields by dopamine D1 receptors in prefrontal cortex. Nature. 1995;376:572–575. doi: 10.1038/376572a0. [DOI] [PubMed] [Google Scholar]
- 80.Kodama T, Hikosaka K, Watanabe M. Differential changes in glutamate concentration in the primate prefrontal cortex during spatial delayed alternation and sensory-guided tasks. Experimental brain research. 2002;145:133–141. doi: 10.1007/s00221-002-1084-y. [DOI] [PubMed] [Google Scholar]
- 81.Utter AA, Basso MA. The basal ganglia: an overview of circuits and function. Neuroscience and biobehavioral reviews. 2008;32:333–342. doi: 10.1016/j.neubiorev.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 82.Muly EC, 3rd, Szigeti K, Goldman-Rakic PS. D1 receptor in interneurons of macaque prefrontal cortex: distribution and subcellular localization. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1998;18:10553–10565. doi: 10.1523/JNEUROSCI.18-24-10553.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.DeLong MR, Wichmann T. Circuits and circuit disorders of the basal ganglia. Archives of neurology. 2007;64:20–24. doi: 10.1001/archneur.64.1.20. [DOI] [PubMed] [Google Scholar]
- 84.Jo YS, Lee J, Mizumori SJ. Effects of prefrontal cortical inactivation on neural activity in the ventral tegmental area. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:8159–8171. doi: 10.1523/JNEUROSCI.0118-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sari Y, Johnson VR, Weedman JM. Role of the serotonergic system in alcohol dependence: from animal models to clinics. Progress in molecular biology and translational science. 2011;98:401–443. doi: 10.1016/B978-0-12-385506-0.00010-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.de Bartolomeis A, Buonaguro EF, Iasevoli F. Serotonin-glutamate and serotonin-dopamine reciprocal interactions as putative molecular targets for novel antipsychotic treatments: from receptor heterodimers to postsynaptic scaffolding and effector proteins. Psychopharmacology. 2013;225:1–19. doi: 10.1007/s00213-012-2921-8. [DOI] [PubMed] [Google Scholar]
- 87.Di Matteo V, Di Giovanni G, Pierucci M, Esposito E. Serotonin control of central dopaminergic function: focus on in vivo microdialysis studies. Progress in brain research. 2008;172:7–44. doi: 10.1016/S0079-6123(08)00902-3. [DOI] [PubMed] [Google Scholar]
- 88.Nakamura K. The role of the dorsal raphe nucleus in reward-seeking behavior. Frontiers in integrative neuroscience. 2013;7:60. doi: 10.3389/fnint.2013.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gervais J, Rouillard C. Dorsal raphe stimulation differentially modulates dopaminergic neurons in the ventral tegmental area and substantia nigra. Synapse. 2000;35:281–291. doi: 10.1002/(SICI)1098-2396(20000315)35:4<281::AID-SYN6>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 90.Avery MC, Dutt N, Krichmar JL. A large-scale neural network model of the influence of neuromodulatory levels on working memory and behavior. Frontiers in computational neuroscience. 2013;7:133. doi: 10.3389/fncom.2013.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Roozendaal B, McGaugh JL. Memory modulation. Behavioral neuroscience. 2011;125:797–824. doi: 10.1037/a0026187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang Z, Cordeiro Matos S, Jego S, Adamantidis A, Seguela P. Norepinephrine drives persistent activity in prefrontal cortex via synergistic alpha1 and alpha2 adrenoceptors. PloS one. 2013;8:e66122. doi: 10.1371/journal.pone.0066122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kawaguchi Y, Shindou T. Noradrenergic excitation and inhibition of GABAergic cell types in rat frontal cortex. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1998;18:6963–6976. doi: 10.1523/JNEUROSCI.18-17-06963.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gibbs ME, Hutchinson DS, Summers RJ. Noradrenaline release in the locus coeruleus modulates memory formation and consolidation; roles for alpha- and beta-adrenergic receptors. Neuroscience. 2010;170:1209–1222. doi: 10.1016/j.neuroscience.2010.07.052. [DOI] [PubMed] [Google Scholar]
- 95.Lester DB, Rogers TD, Blaha CD. Acetylcholine-dopamine interactions in the pathophysiology and treatment of CNS disorders. CNS neuroscience & therapeutics. 2010;16:137–162. doi: 10.1111/j.1755-5949.2010.00142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brooks JM, Sarter M, Bruno JP. D2-like receptors in nucleus accumbens negatively modulate acetylcholine release in prefrontal cortex. Neuropharmacology. 2007;53:455–463. doi: 10.1016/j.neuropharm.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Raedler TJ, Bymaster FP, Tandon R, Copolov D, Dean B. Towards a muscarinic hypothesis of schizophrenia. Molecular psychiatry. 2007;12:232–246. doi: 10.1038/sj.mp.4001924. [DOI] [PubMed] [Google Scholar]
- 98.Voit EO. Computational analysis of biochemical systems: a practical guide for biochemists and molecular biologists. Cambridge University Press; Cambridge, U.K: 2000. [Google Scholar]
- 99.Voit EO. Biochemical Systems Theory: A Review. Int Scholarly Res Network. 2013:1–53. (ISRN – Biomathematics) [Google Scholar]
- 100.Torres NV, Voit EO. Pathway Analysis and Optimization in Metabolic Engineering. Cambridge University Press; Cambridge, U.K: 2002. [Google Scholar]
- 101.Savageau MA. Biochemical Systems Analysis: A Study of Function and Design in Molecular Biology. Addison-Wesley; Reading, MA: 1976. [Google Scholar]
- 102.Smith RC, Warner-Cohen J, Matute M, Butler E, Kelly E, Vaidhyanathaswamy S, Khan A. Effects of nicotine nasal spray on cognitive function in schizophrenia. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2006;31:637–643. doi: 10.1038/sj.npp.1300881. [DOI] [PubMed] [Google Scholar]
- 103.Depatie L, O’Driscoll GA, Holahan AL, Atkinson V, Thavundayil JX, Kin NN, Lal S. Nicotine and behavioral markers of risk for schizophrenia: a double-blind, placebo-controlled, cross-over study. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2002;27:1056–1070. doi: 10.1016/S0893-133X(02)00372-X. [DOI] [PubMed] [Google Scholar]
- 104.Ebert U, Oertel R, Wesnes KA, Kirch W. Effects of physostigmine on scopolamine–induced changes in quantitative electroencephalogram and cognitive performance. Human Psychopharmacology: Clinical and Experimental. 1998;13:199–210. [Google Scholar]
- 105.Yakel JL. Cholinergic receptors: functional role of nicotinic ACh receptors in brain circuits and disease. Pflugers Archiv: European journal of physiology. 2013;465:441–450. doi: 10.1007/s00424-012-1200-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Smith EE, Jonides J, Marshuetz C, Koeppe RA. Components of verbal working memory: evidence from neuroimaging. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:876–882. doi: 10.1073/pnas.95.3.876. [DOI] [PMC free article] [PubMed] [Google Scholar]