

Abstract
The primary intent of a chemical probe is to establish the relationship between a molecular target, usually a protein whose function is modulated by the probe, and the biological consequences of that modulation. In order to fulfill this purpose, a chemical probe must be profiled for selectivity, mechanism of action, and cellular activity, as the cell is the minimal system in which ‘biology’ can be explored. This review provides a brief overview of progress toward chemical probes for methyl lysine reader domains with a focus on recent progress targeting chromodomains.
Introduction
Advances in understanding the regulation of chromatin accessibility via post-translational modifications (PTMs) of histones have rejuvenated drug discovery directed toward modulation of transcription as the opportunities for pharmacological intervention are significantly better than direct perturbation of transcription factors [1–3]. Chemical biology is poised to play a central role in advancing scientific knowledge and assessing therapeutic opportunities in chromatin regulation. Specifically, cell penetrant, high-quality chemical probes that influence chromatin state are of great significance [4,5]. The advantages of a small molecule driven approach to modulating chromatin biology are numerous: temporal resolution; mechanistic flexibility (targeting a specific activity of a protein as opposed to ablating them all via DNA editing and RNA-interference techniques)[6]; ease of delivery; and most significantly, a small molecule tool has the potential to provide an immediate transition to a drug discovery effort, possibly cutting years off the time between target validation and therapeutic intervention [7,8].
While the enzymes that perform PTMs on histones are an important and precedented class of druggable targets [1,9], the biological consequences of many PTMs result from their recruitment of regulatory machinery via protein-protein interactions (PPI) directly facilitated by the PTM [10]. The binding domains involved in PTM recognition on chromatin are referred to as “readers”. We and others have been focused on exploration of the chemical biology of readers of methyl-lysine (Kme) as this PTM plays a central role in chromatin regulation and more than 200 Kme reader domains within several protein families occur within the human proteome, making this a large and relatively unexplored target-class for probe discovery [11–21].
Probe Validation
Characterization of selectivity and cellular target engagement are both essential and challenging aspects of probe validation [7,16]. In the case of the enzymes that regulate chromatin state, a knockdown of the target by siRNA, shRNA or gene editing directly perturbs a PTM that can be readily monitored at either a global level or at a specific gene locus [1,9]. For Kme readers genetic manipulations tend to result in biochemical or phenotypic outcomes that are less easily attributed to specific biochemical changes at the level of chromatin. Additionally, since most Kme readers occur in the context of multi-domain and hence multi-functional proteins, there is no a priori basis to expect that pharmacologic antagonism of the Kme reader function will be equivalent to the removal of the whole protein in which it is embedded.[6] For this reason, initial assessments of chromatin reader antagonism have frequently relied upon the effect of the probe on the localization or mobility of a tagged version of its reader target expressed in a cell of interest. This approach has been applied to bromodomains [22,23] and Kme readers in our own work [19]. While changes in target localization gives a readout that is both proximal to chromatin and logically attributable to the likely mechanism of action of the ligand, this phenomenology is difficult to relate to any specific biological function of the endogenous reader and does not directly establish a molecular pathway connection to phenotypic effects [15]. New technologies to assess cellular target engagement could have a significant impact on validation of Kme reader probes [24].
Selectivity assessment is perhaps the most important aspect of chemical probe characterization, and unfortunately, one that is often lacking in the literature [4,7,25]. While single-target specificity is not an absolute requirement, sufficient profiling data to confidently attribute in vivo effects to the in vitro profile of a probe are essential. We have attempted to address this for Kme readers (in collaboration with the Bedford lab at MD Anderson) by evaluating the binding of biotinylated versions of Kme reader probes to a nitro-cellulose membrane upon which hundreds of potential chromatin-associated effector domains have been spotted [26]. Binding is then observed with a streptavidin-dye conjugate and positive results are followed up via quantitative measurements in solution by isothermal titration calorimetry (ITC) [13,18]. In addition to assessing selectivity versus Kme reader proteins, probes must be profiled versus the enzyme families that modify lysine (PKMTs, lysine demethylases), as activity here would be likely to confound interpretation of both chromatin biochemical readouts and phenotypic outcomes. There is also a chemical logic for screening against these targets since Kme reader probes may mimic the substrates of these enzymes. Profiling versus general pharmacology panels is also performed in order to create a more complete assessment of potential off-target activities. While this data cannot rule out contributions from unexamined or unknown protein off-targets to a probe’s activity, it does support the case for specificity when cellular target-engagement has also been proven.[7] Additionally, in the absence of comprehensive profiling data against all possible cellular targets, the use of a close structural analogue as an inactive control compound that lacks biochemical target activity is critical in order to establish a correlation between on-target in vitro activity and cellular effects [25].
Chemical Strategies
Kme binding sites are generally made up of an aromatic cage involving 3 to 4 aromatic amino acids, and often an acidic residue to hydrogen bond to the Kme cation in the case of mono- and dimethyllysine (Kme1,2) recognition, or simply to balance the charge in the case of Kme3 [27,28]. The Patel lab introduced a useful division of Kme readers into “cavity insertion” versus “surface groove” binders [29], and subsequent work toward chemical probes has been informed by this ontology and confirmed its relevance to ligand design. Table 1 illustrates chemical strategies and principles applied thus far to the discovery of Kme reader antagonists.
Table 1.
Chemical strategies and principles applied to the discovery of antagonists for cavity insertion methyllysine binders and surface groove methyllysine binders.
| Cavity Insertion Binders | Surface Groove Binders | |
|---|---|---|
| Hit Discovery & Screening Strategies | Screen focused small molecule or fragment libraries Employ target class cross screening Utilize structure-based design for hit optimization |
Apply structure-based design Screen peptide or peptoid libraries |
| Design Principles | Exploit cation-π and H-bonding interactions in aromatic cage Utilize conformational constrained alkyl amines |
Use available Kme peptide SAR Target binding sites adjacent to aromatic cage Introduce unnatural amino acids, Kme mimics, and non-peptidic features |
| Major Challenge | Fragment-like size may result in low affinity | Low cell permeability may decrease overall utility |
| Example | L3MBTL1 + H4K20me2 (pdb 2PQW)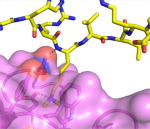
|
CBX7 + H3K27me3 (pdb 4X3K)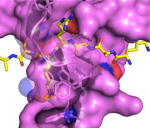
|
Our initial work focused on ligands for the MBT domains which utilize a cavity insertion recognition mode and led to the successful design of the first chemical probe for Kme readers, UNC1215 [19]. However, the relatively high affinity of UNC1215 for L3MBTL3 (Kd = 120 nM) depends upon a unique 2:2 dimer-binding mode that may not be accessible or relevant to other Kme reader targets. Based on our current experience, L3MBTL3 is the only MBT domain that elicits this recognition mode [30], although we continue to work toward Kme reader ligand engagement via dimerization, particularly in cases where this binding mechanism may be relevant in vivo, such as 53BP1 [14]. While we and others have reported weak Kme ligands discovered by high throughput screening (HTS) of diversity libraries [31–35], no hits from HTS have so far been optimized to potent chemical probes and our own experience suggests that many HTS hits are false positives that fail to confirm in biophysical assays. Kme cavity insertion binders have been largely targeted via traditional small molecule strategies (select inhibitors shown in Table 2) and this work has been summarized previously [15,36], while more recent progress toward Kme reader probes has resulted from ligand discovery efforts towards surface groove binding Kme readers and will be reviewed herein.
Table 2.
Select small molecule, non-peptidomimetic inhibitors of methyllysine reader proteins that engage the aromatic cage where methyllysine binds.
| Inhibitor | Structure | Potency (Assay) | Target(s) | Peptide Binding Mode | Ref. |
|---|---|---|---|---|---|
| UNC926 |
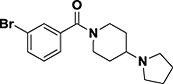
|
L3MBTL1 IC50 = 3.9 μM L3MBTL1 IC50 = 3.9 μM (Alphascreen) |
L3MBTL1, L3MBTL3 (MBT domains) | Cavity insertion | 21 |
| UNC1215* |
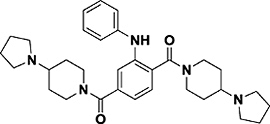
|
Kd = 0.12 μM (ITC) | L3MBTL3 (MBT domain) | Cavity insertion | 19 |
| Compound 56 |
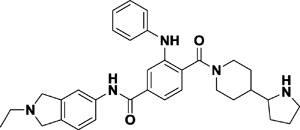
|
Kd = 0.35 μM (ITC) | L3MBTL3 (MBT domain) | Cavity insertion | 13 |
| CF16 |
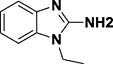
|
Kd = 7.3 mM (NMR) | Pygo2 (PHD finger) | Surface groove | 34 |
| MS37452 |
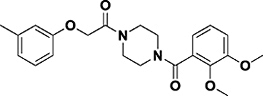
|
Kd = 29 μM (NMR) | CBX7 (Chromodomain) | Surface groove | 31 |
designates a high-quality chemical probe.
Peptidomimetic Chromodomain Inhibitors
Cavity insertion binding events between Kme readers and their cognate substrates are predominantly driven by localized interactions between the methyl-ammonium and a deep protein cleft, making them candidates for intervention with drug-like small molecules, whereas surface groove interactions cover a much larger area and rely on contacts with a number of adjacent residues in addition to Kme [29]. Consequently, in recent efforts targeting chromodomains, which are characterized by an induced fit, surface groove interaction with methylated histone substrates, we and others have employed peptide mimetic discovery strategies and scaffolds for the successful development of chromodomain inhibitors [17,18,37]. These efforts have resulted in inhibitors of the polycomb (Pc) chromodomains, well known for their participation in Polycomb repressive complex 1 (PRC1) [38]. PRC1 is one of the major chromatin regulatory complexes involved in repression of gene transcription, and as such, plays a central role in differentiation and development [39,40]. The CBX chromodomains facilitate PRC1-mediated transcriptional repression by targeting the complex to the H3K27me3 mark. Consequently, PRC1 chromodomain chemical probes could be employed to explore the therapeutic potential of CBX chromodomains, investigate pharmacologic synergy with inhibitors of the H3K27me3 methyltransferase, EZH2 [41], and understand the precise roles of CBX target genes in different cancer types.
There are five human proteins that belong to the polycomb chromodomain family: CBX2, -4, -6, -7, and -8; all of which share a high level of sequence identity within their chromodomains, suggesting that development of selective inhibitors could be a challenge. Ligand design must also take into account the shallow aromatic cage that binds Kme3 and the continuous β-sheet-like interaction that includes a sequence of hydrogen bonds with the histone backbone; therefore development of peptidomimetic ligands has been pursued, despite the anticipated difficulties in cell permeability.
Hof and co-workers reported the first inhibitors of a chromodomain, CBX7, derived from a five-residue peptide sequence (FALKme3S) from the protein SETDB1, which was reported to have slightly higher in vitro affinity for CBX7 than the H3K27me3 sequence itself [17,42]. They performed a systematic study in which each amino acid of the parent peptide was substituted with both natural and unnatural residues, and the affinity of these and subsequently derived ligands for CBX7 was determined by fluorescence polarization and ITC. Iterative ligand design was further guided by X-ray crystallography and 2D NMR structural studies of ligands bound to CBX7. Upon evaluation of the tolerance of each site to modification, select compounds bearing multiple well-tolerated substitutions were prepared. This ultimately led to compound 64 (Figure 1a), which binds CBX7 with a Kd of approximately 200 nM and bears a number of modifications relative to the FALKme3S sequence. The most significant potency enhancement results from a benzoic amide at the N-terminus. The affinity of compound 64 for three other chromodomains was determined; resulting in 10-fold selectivity over CBX8, equal potency for CBX4, and no measurable affinity for the HP1 family chromodomain, CBX1.
Figure 1. Peptidomimetic chromodomain ligands.
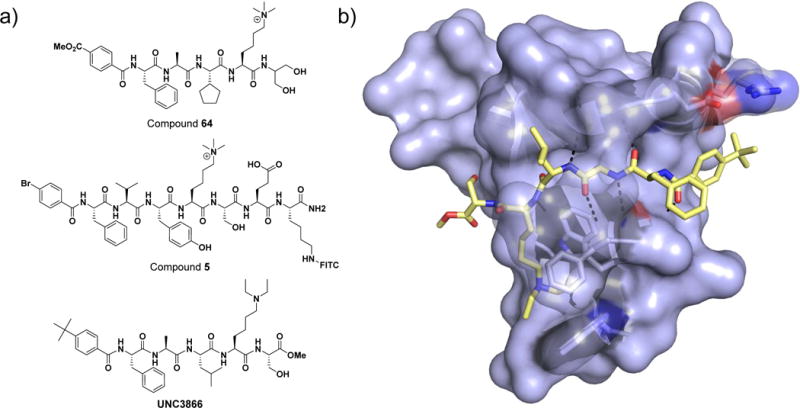
a) Chemical structures of peptidic chromodomain inhibitors. b) Co-crystal structure of chemical probe UNC3866 bound to CBX7 (pdb 5EPJ). The protein surface is shown in light blue and UNC3866 in yellow. Hydrogen bonds between the protein and UNC3866 are shown by a black dotted line.
More recently, the Hof group has elegantly demonstrated the ability of peptidomimetics to achieve selectivity within the family of polycomb CBX chromodomains. Structural knowledge of the features broadly required for CBX binding led to modification of the residue two amino acids from the Kme3 site (−2 position) which resulted in enhanced CBX selectivity [37]. Fluorescently labeled ligands were profiled against CBX2, -4, -6, -7, -8, and the HP1 paralog CBX1 via direct fluorescent polarization (FP) titrations. Inclusion of an isopropyl group in compound 5 at the (−2) position resulted in 90-, 20-, 18-, 6-, and 7-fold selectivity for CBX6 over CBX1, -2, -4, -7, and -8, respectively, accompanied by only a 3-fold loss in potency for CBX6 (Kd = 0.9 μM) relative to the analogous alanine containing compound (Figure 1a). This reveals that subtle modifications to this ligand scaffold are able to greatly diminish CBX7 binding and clearly demonstrates that occupying binding sites other than the Kme3-binding aromatic cage can modulate potency and selectivity.
Concurrent to the efforts of the Hof group, we initiated a program to discover high-quality chemical probes of the polycomb CBX chromodomains based on their known disease relevance, guided largely by molecular dynamics simulations, cocrystallization studies, and structure-activity relationships. This led to the design and characterization of UNC3866 (Figure 1), which binds the chromodomains of CBX4 and CBX7 most potently (Kd ~100nM), and is 6- to 18-fold selective as compared to the other polycomb CBXs [18]. Knowledge of the activity profile of a molecule is essential in order to associate its cellular effects with the modulation of a specific molecular target(s); therefore, UNC3866 was evaluated against >250 other protein targets including Kme readers, bromodomains (acetyllysine reader proteins), protein methyltransferases and demethylases, and GPCRs (G protein coupled receptors), among others (Figure 2). This revealed that in addition to the known polycomb targets, UNC3866 binds three members of the CDY family of chromodomains (CDY1, CDYL1b, and CDYL2). Importantly, UNC3866 was also shown to engage the intact PRC1 complex, antagonize PRC1 chromodomains in cells, exhibit a high level of stability in cells, and inhibit PC3 cell proliferation (a known CBX7 phenotype), supporting further investigation of PRC1 chromodomain antagonists as oncology therapeutics. The replacement of the quaternary amine of the native peptide ligand with an unnatural tertiary amine mimetic was a key achievement in the development of a cellularly active ligand, despite its relatively low permeability, and signifies that a quaternary amine is not essential for inhibition of Kme3 reader proteins. The design and concomitant use of an inactive negative control compound, differing from UNC3866 by addition of a single methyl group, further supported the characterization of UNC3866 as the first high-quality chemical probe for a chromodomain. Taken together, our progress toward cellularly active, peptidic chromodomain antagonists and the Hof’s group demonstration of selectivity tuning within the PRC1 chromodomains bodes well for further breakthroughs in this area.
Figure 2. Selectivity profile of UNC3866.
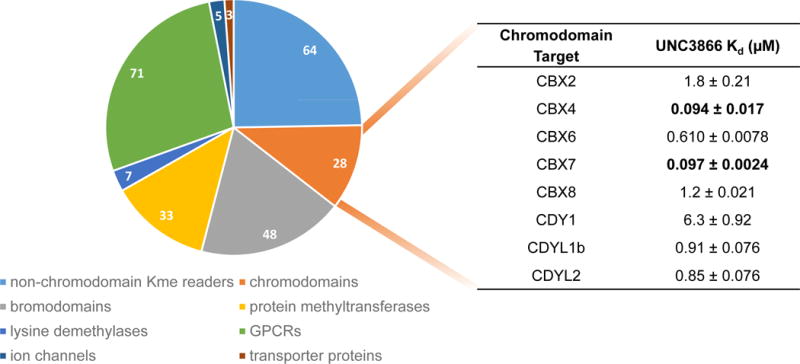
UNC3866 antagonizes the Kme reading function of the Polycomb CBX and the CDY families of chromodomains and is highly selective over >250 other protein targets evaluated. The affinity of UNC3866 for its chromodomain targets was determined by ITC.
Conclusions
The pursuit of well validated chemical probes for Kme reader proteins represents an emerging area of chemical biology that will create new understanding of chromatin regulation and prove useful in validation of Kme readers implicated in disease. As this target-class is well represented in databases of proteins genetically altered in cancer and neurological diseases [43], the opportunity for translational impact is promising. While peptidic probes seem most likely to deliver high-affinity and selectivity for groove-binding Kme readers, such as chromodomains, progress toward more drug-like compounds for these domains could be spurred on by target validation studies with peptide-like chemical probes. [44,45]
Highlights.
Methyl lysine reader domains are a major class of chromatin regulatory modules that are being explored for therapeutic relevance.
Chemical probes are molecules that are suitable for target validation studies in cells based on their potency, demonstrated target engagement, and selectivity.
Recent progress toward methyl lysine reader domain chemical probes has resulted in potent, cellularly active and selective ligands for the chromodomains of the polycomb repressive complex 1.
Acknowledgments
The authors thank Jake Stuckey for assistance in generating the structural images in the figures above. The research described here was supported by the National Institute of General Medical Sciences, US National Institutes of Health (grants RC1GM090732 and R01GM100919), the Carolina Partnership and the University Cancer Research Fund. The authors also wish to recognize the collaborative contributions of all co-authors in our cited references and especially the Structural Genomics Consortium.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1•.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11:384–400. doi: 10.1038/nrd3674. This is an excellent review that covers the different epigenetic protein families and their links with disease, their basic molecular mechanisms of action, and progress toward the pharmacological modulation of each. [DOI] [PubMed] [Google Scholar]
- 2.Ptashne M. Epigenetics: core misconcept. Proc Natl Acad Sci U S A. 2013;110:7101–7103. doi: 10.1073/pnas.1305399110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fierz B, Muir TW. Chromatin as an expansive canvas for chemical biology. Nat Chem Biol. 2012:417–427. doi: 10.1038/nchembio.938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4•.Frye SV. The art of the chemical probe. Nat Chem Biol. 2010;6:159–161. doi: 10.1038/nchembio.296. This commentary proposes a set of principles to guide probe qualification. [DOI] [PubMed] [Google Scholar]
- 5•.Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, Bountra C, Brennan PE, Brown PJ, Bunnage ME, et al. The promise and peril of chemical probes. Nat Chem Biol. 2015;11:536–541. doi: 10.1038/nchembio.1867. This commentary outlines the requirements of high-quality chemical probes and a new resource made available by chemical biologists to increase the proper use of the best available probe compounds by the scientific community. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiss WA, Taylor SS, Shokat KM. Recognizing and exploiting differences between RNAi and small-molecule inhibitors. Nat Chem Biol. 2007;3:739–744. doi: 10.1038/nchembio1207-739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bunnage ME, Chekler ELP, Jones LH. Target validation using chemical probes. Nature Chemical Biology. 2013;9:195–199. doi: 10.1038/nchembio.1197. [DOI] [PubMed] [Google Scholar]
- 8.Chung C-w, Tough DF. Bromodomains: a new target class for small molecule drug discovery. Drug Discovery Today: Therapeutic Strategies. 2012;9:e111–e120. [Google Scholar]
- 9.Copeland RA, Olhava EJ, Scott MP. Targeting epigenetic enzymes for drug discovery. Current Opinion in Chemical Biology. 2010;14:505–510. doi: 10.1016/j.cbpa.2010.06.174. [DOI] [PubMed] [Google Scholar]
- 10.Musselman CA, Lalonde ME, Cote J, Kutateladze TG. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol. 2012;19:1218–1227. doi: 10.1038/nsmb.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao C, Herold JM, Kireev D, Wigle T, Norris JL, Frye S. Biophysical probes reveal a “compromise” nature of the methyl-lysine binding pocket in L3MBTL1. J Am Chem Soc. 2011;133:5357–5362. doi: 10.1021/ja110432e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herold JM, Wigle TJ, Norris JL, Lam R, Korboukh VK, Gao C, Ingerman LA, Kireev DB, Senisterra G, Vedadi M, et al. Small-molecule ligands of methyl-lysine binding proteins. J Med Chem. 2011;54:2504–2511. doi: 10.1021/jm200045v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.James LI, Korboukh VK, Krichevsky L, Baughman BM, Herold JM, Norris JL, Jin J, Kireev DB, Janzen WP, Arrowsmith CH, et al. Small-Molecule Ligands of Methyl-Lysine Binding Proteins: Optimization of Selectivity for L3MBTL3. J Med Chem. 2013;56:7358–7371. doi: 10.1021/jm400919p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perfetti MT, Baughman BM, Dickson BM, Mu Y, Cui G, Mader P, Dong A, Norris JL, Rothbart SB, Strahl BD, et al. Identification of a fragment-like small molecule ligand for the methyl-lysine binding protein, 53BP1. ACS Chem Biol. 2015;10:1072–1081. doi: 10.1021/cb500956g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frye SV, James LI. Small-Molecule Modulation of Methyl-Lysine-Mediated Interactions. In: Zhou M-M, editor. Histone Recognition. Springer International Publishing; 2015. pp. 243–271. [Google Scholar]
- 16.Frye SV. Unlocking the potential of chemical probes for methyl-lysine reader proteins. Future Med Chem. 2015;7:1831–1833. doi: 10.4155/fmc.15.119. [DOI] [PubMed] [Google Scholar]
- 17••.Simhadri C, Daze KD, Douglas SF, Quon TT, Dev A, Gignac MC, Peng F, Heller M, Boulanger MJ, Wulff JE, et al. Chromodomain antagonists that target the polycomb-group methyllysine reader protein chromobox homolog 7 (CBX7) J Med Chem. 2014;57:2874–2883. doi: 10.1021/jm401487x. This paper describes peptidic compounds which represent the first inhibitors of any chromodomain. [DOI] [PubMed] [Google Scholar]
- 18••.Stuckey JI, Dickson BM, Cheng N, Liu Y, Norris JL, Cholensky SH, Tempel W, Qin S, Huber KG, Sagum C, et al. A cellular chemical probe targeting the chromodomains of Polycomb repressive complex 1. Nat Chem Biol. 2016;12:180–187. doi: 10.1038/nchembio.2007. This paper reports the first cellularly active chemical probe for a chromodomain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19••.James LI, Barsyte-Lovejoy D, Zhong N, Krichevsky L, Korboukh VK, Herold JM, MacNevin CJ, Norris JL, Sagum CA, Tempel W, et al. Discovery of a chemical probe for the L3MBTL3 methyllysine reader domain. Nat Chem Biol. 2013;9:184–191. doi: 10.1038/nchembio.1157. This paper describes the first chemical probe for a methyllysine reader domain, L3MBTL3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Camerino MA, Zhong N, Dong A, Dickson BM, James LI, Baughman BM, Norris JL, Kireev DB, Janzen WP, Arrowsmith CH, et al. The structure-activity relationships of L3MBTL3 inhibitors: flexibility of the dimer interface. Medchemcomm. 2013;4:1501–1507. doi: 10.1039/C3MD00197K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herold JM, James LI, Korboukh VK, Gao C, Coil KE, Bua DJ, Norris JL, Kireev DB, Brown PJ, Jin J, et al. Structure-activity relationships of methyl-lysine reader antagonists. MedChemComm. 2012;3:45–51. [Google Scholar]
- 22.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundbäck T, Nordlund P, Molina DM. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protocols. 2014;9:2100–2122. doi: 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- 25.Workman P, Collins I. Probing the probes: fitness factors for small molecule tools. Chem Biol. 2010;17:561–577. doi: 10.1016/j.chembiol.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, Zhang Y, Bedford MT, Tudor MBT. chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006;7:397–403. doi: 10.1038/sj.embor.7400625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kamps JJ, Huang J, Poater J, Xu C, Pieters BJ, Dong A, Min J, Sherman W, Beuming T, Matthias Bickelhaupt F, et al. Chemical basis for the recognition of trimethyllysine by epigenetic reader proteins. Nat Commun. 2015;6:8911. doi: 10.1038/ncomms9911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Fischle W, Wang W, Duncan EM, Liang L, Murakami-Ishibe S, Allis CD, Patel DJ. Structural basis for lower lysine methylation state-specific readout by MBT repeats of L3MBTL1 and an engineered PHD finger. Mol Cell. 2007;28:677–691. doi: 10.1016/j.molcel.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baughman BM, Pattenden SG, Norris JL, James LI, Frye SV. The L3MBTL3 Methyl-Lysine Reader Domain Functions As a Dimer. ACS Chem Biol. 2016;11:722–728. doi: 10.1021/acschembio.5b00632. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Ren C, Morohashi K, Plotnikov Alexander N, Jakoncic J, Smith Steven G, Li J, Zeng L, Rodriguez Y, Stojanoff V, Walsh M, et al. Small-Molecule Modulators of Methyl-Lysine Binding for the CBX7 Chromodomain. Chemistry & Biology. 2015;22:161–168. doi: 10.1016/j.chembiol.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wigle TJ, Herold JM, Senisterra GA, Vedadi M, Kireev DB, Arrowsmith CH, Frye SV, Janzen WP. Screening for Inhibitors of Low-Affinity Epigenetic Peptide-Protein Interactions: An AlphaScreenTM-Based Assay for Antagonists of Methyl-Lysine Binding Proteins. J Biomol Screen. 2010;15:62–71. doi: 10.1177/1087057109352902. [DOI] [PubMed] [Google Scholar]
- 33.Wagner EK, Nath N, Flemming R, Feltenberger JB, Denu JM. Identification and characterization of small molecule inhibitors of a plant homeodomain finger. Biochemistry. 2012;51:8293–8306. doi: 10.1021/bi3009278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller TC, Rutherford TJ, Birchall K, Chugh J, Fiedler M, Bienz M. Competitive binding of a benzimidazole to the histone-binding pocket of the Pygo PHD finger. ACS Chem Biol. 2014;9:2864–2874. doi: 10.1021/cb500585s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ren C, Smith SG, Yap K, Li S, Li J, Mezei M, Rodriguez Y, Vincek A, Aguilo F, Walsh MJ, et al. Structure-Guided Discovery of Selective Antagonists for the Chromodomain of Polycomb Repressive Protein CBX7. ACS Medicinal Chemistry Letters. 2016 doi: 10.1021/acsmedchemlett.6b00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36•.Milosevich N, Hof F. Chemical Inhibitors of Epigenetic Methyllysine Reader Proteins. Biochemistry. 2016;55:1570–1583. doi: 10.1021/acs.biochem.5b01073. This is an recent, comprehensive review of the inhibitors that have been developed for the Kme reader target class. [DOI] [PubMed] [Google Scholar]
- 37••.Milosevich N, Gignac MC, McFarlane J, Simhadri C, Horvath S, Daze KD, Croft CS, Dheri A, Quon TT, Douglas SF, et al. Selective Inhibition of CBX6: A Methyllysine Reader Protein in the Polycomb Family. ACS Med Chem Lett. 2016;7:139–144. doi: 10.1021/acsmedchemlett.5b00378. This paper demonstrates the ability to achieve selective inhibition of a single member of the family of Polycomb CBX Kme readers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aranda S, Mas G, Di Croce L. Regulation of gene transcription by Polycomb proteins. Sci Adv. 2015;1:e1500737. doi: 10.1126/sciadv.1500737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muller J, Verrijzer P. Biochemical mechanisms of gene regulation by polycomb group protein complexes. Curr Opin Genet Dev. 2009;19:150–158. doi: 10.1016/j.gde.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 40.Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6:846–856. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- 41.Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, MacNevin CJ, Liu F, Gao C, Huang X-P, Kuznetsova E, et al. An Orally Bioavailable Chemical Probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chemical Biology. 2013;8:1324–1334. doi: 10.1021/cb400133j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaustov L, Ouyang H, Amaya M, Lemak A, Nady N, Duan S, Wasney GA, Li Z, Vedadi M, Schapira M, et al. Recognition and specificity determinants of the human cbx chromodomains. J Biol Chem. 2011;286:521–529. doi: 10.1074/jbc.M110.191411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dawson MA, Kouzarides T, Huntly BJ. Targeting epigenetic readers in cancer. N Engl J Med. 2012;367:647–657. doi: 10.1056/NEJMra1112635. [DOI] [PubMed] [Google Scholar]
- 44.Wójcik P, Berlicki Ł. Peptide-based inhibitors of protein–protein interactions. Bioorganic & Medicinal Chemistry Letters. 2016;26:707–713. doi: 10.1016/j.bmcl.2015.12.084. [DOI] [PubMed] [Google Scholar]
- 45.Craik DJ, Fairlie DP, Liras S, Price D. The Future of Peptide-based Drugs. Chemical Biology & Drug Design. 2013;81:136–147. doi: 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]