

Abstract
An individual’s genetic makeup plays an important role in determining susceptibility to cognitive aging. Identifying the specific genes that contribute to cognitive aging may aid in early diagnosis of at-risk patients, as well as identify novel therapeutics targets to treat or prevent development of symptoms. Challenges to identifying these specific genes in human studies include complex genetics, difficulty in controlling environmental factors, and limited access to human brain tissue. Here, we identify Hp1bp3 as a novel modulator of cognitive aging using a genetically diverse population of mice, and confirm that HP1BP3 protein levels are significantly reduced in the hippocampi of cognitively impaired elderly humans relative to cognitively intact controls. Deletion of functional Hp1bp3 in mice recapitulates memory deficits characteristic of aged impaired mice and humans, further supporting the idea that Hp1bp3 and associated molecular networks are modulators of cognitive aging. Overall, our results suggest Hp1bp3 may serve as a potential target against cognitive aging and demonstrate the utility of genetically diverse animal models for the study of complex human disease.
Keywords: systems genetics, cognitive aging, fear conditioning, BXD, gene set enrichment analysis
1. Introduction
Aging is associated with a decline in cognitive performance that begins around midlife (i.e. 45–50 yrs, but the onset and severity varies greatly among individuals (Davies, et al., 2015; Singh-Manoux, et al., 2012). Although variation in cognitive performance at midlife is highly heritable [i.e. 62–78% of variance is attributable to genetic factors, (McClearn, et al., 1997; Plomin and Deary, 2015)], a large portion of this heritability remains unexplained by currently identified gene variants (Johnson, et al., 2015). Several studies have identified associations between apolipoprotein E, brain derived neurotrophic factor, and catechol-O-methyl transferase with either cognitive ability or rate of cognitive decline in older people (Harris and Deary, 2011; Laukka, et al., 2013; Payton, 2009; Tapia-Arancibia, et al., 2008; Wisdom, et al., 2011). However, even when polymorphisms in these genes are significantly associated with cognitive phenotypes, effect sizes are typically small (Harris and Deary, 2011), indicating that additional genes contribute significantly to the regulation of cognitive decline in human populations. The identification of specific genes that modify the development and progression of cognitive decline may aid in early diagnosis of at-risk patients, as well as identify novel targets for the development of therapeutics to prevent or delay the onset of disease.
Challenges to identifying DNA variants that modify cognitive aging in humans include substantial genetic heterogeneity, difficulty in controlling environmental factors, and limited molecular data from disease-relevant human brain tissue. In order to circumvent some of these challenges, murine genetic reference panels (GRPs) have been designed to model some of the genetic and phenotypic complexity of human populations (International HapMap, et al., 2010; Peirce, et al., 2004; Williams, et al., 2001), allowing a researcher to exploit phenotypic heterogeneity across a population while controlling for environmental factors (Williams and Auwerx, 2015). One such model, a well-characterized GRP known as the BXDs (Peirce, et al., 2004; Taylor, 1978; Taylor, et al., 1999), was derived by crossing two common inbred strains, C57BL/6J (B6) and DBA/2J (D2). The BXDs have been successfully used to identify genomic regions important for determining learning and memory capabilities early in life (Wehner, et al., 1997), but have not yet been used to study cognitive aging. To identify genes and molecular pathways regulating memory capabilities during aging, here we perform a forward systems genetic analysis on an aged cohort of strains from the BXD GRP.
2. Methods
2.1 Animals
Male and female mice were group housed (2–5 per cage) and maintained in colony housing (12-hour light/dark cycle) with ad libitum access to food and water. All mouse experiments were conducted in accordance with the University of Tennessee Health Science Center Animal Care and Use Committee, the Institutional Committee on Animal Care and Use at the Hebrew University of Jerusalem, as well as the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Middle-aged mice (15 ± 0.3 mo, 2–8 mice per strain) from 21 BXD recombinant inbred strains were phenotyped. Although experiments in rodents using a single inbred strain are often carried out using 7–12 replicates (Kaczorowski, et al., 2012; Kaczorowski and Disterhoft, 2009; Kaczorowski, et al., 2011), mapping studies using the BXD panel gain much more power by increasing numbers of unique genotypes rather than replicates per strain (Belknap, 1998). This is because at each locus, roughly half of the lines inherit B/B genotypes and the other half D/D genotypes (see Figure 1B in Andreux et al., 2012). The BXDs were derived by inbreeding the F2 progeny of a C57BL/6J (B6) and DBA/2J (D2) intercross to create a panel that models some of the genetic complexity of human populations (Chesler, et al., 2005; International HapMap, et al., 2010). The parental strains, B6 and D2, differ in a variety of traits, including memory function (Balogh, et al., 2002), amyloid precursor protein processing (Lehman, et al., 2003), adult hippocampal neurogenesis (Kempermann, et al., 2006), and hippocampal excitability (Oksman, et al., 2005), which confers wide phenotypic variability to the resulting BXD strains. Each BXD line has been inbred for >20 generations and their genomes are stable, which allows for replication studies across time and laboratories (Peirce, et al., 2004). In addition to collected ‘health records’ at GeneNetwork.org, both genotype and transcriptome data for ~40 tissues and cells, including hippocampus, have been generated for many of the strains, which we combined with full-sequence genome data for both B6 and D2 to facilitate genetic mapping to identify individual genetic variants that correlate with traits of interest (Chesler, et al., 2005; Houtkooper, et al., 2013; Wang, et al., 2016). Hp1bp3tm1a(EUCOMM)Wtsi mice (n = 6/group) were acquired from the European Conditional Mouse Mutagenesis program and are described elsewhere (Garfinkel, et al., 2015a).
2.2 Contextual Fear Conditioning
All mice were habituated to transport for at least three days prior to behavioral tests. On the fourth day, mice were trained on a standard contextual fear conditioning paradigm (Neuner, et al., 2014). Briefly, contextual fear conditioning consisted of a 180 s baseline period followed by 4 mild foot shocks (1 s, 0.9 mA, inter-shock interval 115 ± 20 s). The average activity burst following each shock was determined for each by manual timing and used to quantify the post-shock reactivity for each mouse to ensure no difference in pain sensitivity. Twenty-four hours later, hippocampus-dependent contextual fear memory (Chen, et al., 1996) was tested in a 10 min session. Behavioral freezing, an index of conditioned fear, was measured using Freeze Frame software (Coulbourn Instruments, PA) at the University of Tennessee or EthoVision software (Noldus Information Technology, Netherlands) at the Hebrew University of Jerusalem. Our lab has previously demonstrated that measures of freezing obtained via video monitoring software correlates well with hand-scored measures of freezing (Kaczorowski, et al., 2012). Experimenters were blind to the genotype during behavioral tests and data analysis.
2.3 Calculation of Memory Index for Adult BXD Strains
BXD strains aged 8–9 weeks had previously been subjected to contextual fear conditioning by Philip and colleagues (2010). The dataset used in our study (including males and females) is publically available on GeneNetwork.org as BXD Published Phenotypes Record ID 11908. The memory index used in Trait 11908 is a measure of activity (beam breaks) throughout CFM testing. This measure of activity has been previously shown to correlate well with measures of hand-scored freezing behavior (Valentinuzzi, et al., 1998). As the dynamic range of human-observed freezing behavior is 0–100% (Anagnostaras, et al., 2000), and an activity score of zero is equivalent to 100% freezing, we subtracted these activity measures from 100 to obtain a measure of inactivity that parallels the CFM index (% freezing) calculated for middle-aged strains. Specifically, the mean time each adult BXD strain spent active during CFM testing was subtracted from 100 to obtain a measure of inactivity. The genotype of each strain at the Hp1bp3 locus was then obtained from GeneNetwork.org and used for genotype-by-memory analyses.
2.4 T-maze
The T-maze test was performed as described previously (Deacon and Rawlins, 2006). Mice were given 10 minutes to habituate to the testing room. The T-maze floor was coated with fresh woodchips before each round of trials. Mice were placed into the T-maze and allowed to choose one of two “goal” arms. The mouse was then confined to that arm for 30 seconds using a central partition. The mice were removed from the maze and immediately placed back at the starting point, the partition removed, and the mouse allowed to choose between the two goal arms again (Deacon and Rawlins, 2006). An “alternation” occurred when the mouse entered the opposite arm than that to which it was just confined. The criterion point used was whole animal in goal arm, including tail tip. A total of six trials were performed, with animals tested no more than twice a day. The alternation scores across all six trials were averaged to obtain an average alternation score for each mouse.
2.5 QTL identification and candidate gene selection
After CFM tests for 21 BXD strains was completed, the mean time spent freezing was averaged for each strain. Strain means were entered in GeneNetwork.org and are publically available as BXD Published Phenotypes Record ID: 18395. Hippocampal transcript data from aged BXD strains available on GeneNetwork.org as UTHSC BXD Aged Hippocampus Affy MoGene1.0ST (May15) RMA Gene Level, were used to identify cis (locally)-regulated genes (Andreux, et al., 2012; Chesler, et al., 2005) from within intervals of interest. The same transcript data was used to identify genes whose expression correlated with memory function in aged animals. Only probes that did not overlap single nucleotide polymorphisms were used in order to avoid hybridization artifacts that may cause apparent differences in expression due to technical rather than biological variance. When multiple SNP-free gene level probes were available (e.g. Hp1bp3), the mean expression of exon-targeting probes was used. In cases where SNP-free gene level probes were not available, exon-level hippocampal transcript data from aged BXD strains was used (UTHSC BXD Aged Hippocampus Affy Mouse Gene1.0ST (Sep12) RMA Exon Level, dataset accession ID GN392). If exon level probes were used, the probe with highest average expression across all strains was selected. All probes were verified by BLAT (UCSC Genome Browser). Correlation analyses for initial candidate gene prioritization used Spearman’s rho and were adjusted for multiple comparisons using Bonferroni correction (95% confidence interval).
2.6 Western Blots
Western blots were performed as previously described (Hatfield, et al., 2015; Neuner, et al., 2014). Frozen human hippocampal samples (n=3/group) collected postmortem were obtained from the University of Kentucky Sanders-Brown Center on Aging and stored at −80°C until use. Briefly, hippocampal lysates were prepared from frozen tissue, protein concentration was determined using a Nanodrop2000 Spectrophotometer (ThermoScientific), and 20 μg of total protein was loaded and separated on a 10% SDS-PAGE gel. Proteins were transferred using the Bio-Rad TurboTransfer system and blocked for 30 min at room temperature. Primary antibodies for HP1BP3 and GAPDH (ProteinTech #24556-1-AP and Fitzgerald #10R-G109a, respectively) were incubated overnight and detected by anti-mouse and anti-rabbit fluorescent conjugated antibodies. Visualization was performed using an Odyssey image scanner and blots were quantified using the Odyssey software version 5.0 (LiCOR). Results were replicated in 2 independent Western blots. Observed double band staining is typical expression pattern for HP1BP3 (Garfinkel, et al., 2015b) and overlaps with positive control HP1BP3 overexpression lysate from human 293T cells (Abnova #H00050809-T02), which was used as a positive control. Loading-dye only lanes served as negative control.
2.7 Gene Set Enrichment Analysis
Gene Set Enrichment Analysis was performed as described previously using version 2.2.0 (Mootha, et al., 2003; Subramanian, et al., 2005). Briefly, all nominally significant correlates of hippocampal Hp1bp3 were extracted from whole-genome hippocampal transcript data from BXD strains age-matched to those used for contextual fear conditioning. Correlates were ranked based on correlation coefficient and this preranked list was used to calculate an enrichment score for gene sets obtained from the Molecular Signatures Database (MSigDB) version 5.0. Enrichment scores are obtained by calculating a cumulative (“running-sum”) statistic, which is increased when a gene is present in the set being tested and decreased when a gene is not. The maximum deviation of this cumulative score from zero is the enrichment score. Then, normalized enrichment scores (NES) were obtained to account for the size of the gene set being tested. The proportion of false positives were controlled using established methods (Mootha, et al., 2003; Subramanian, et al., 2005). Based on these criteria, any gene set with a false discovery rate (FDR) of ≤ 0.25 was accepted as significantly enriched in our dataset.
2.8 Statistical analysis
Statistical analysis was performed using SPSS software (IBM) and Microsoft Excel. Analyses included independent t-tests, repeated measures, one-way and two-way ANOVA, and Pearson correlation with confidence level set at p < 0.05. All analyses were corrected for multiple comparisons and described in the Results section. Unless otherwise stated, data values reported here are given as mean ± SEM. The order of behavioral tests and mice allocated to each condition were randomized. The experimenter was blind to group allocation for all behavioral studies, and analysis of raw data was conducted blind to experimental groups.
3. Results
3.1 Genetic mapping identifies an interval on chromosome 4 associated with memory status at midlife
To identify genes involved in the regulation of cognitive aging, we analyzed hippocampus-dependent memory function across a cohort of middle-aged mice (15 ± 0.3 m) that model the genetic and phenotypic variation of human populations (the BXD genetic reference panel, Fig 1a). Specifically, mice were trained on standard contextual fear conditioning (Kaczorowski and Disterhoft, 2009). During training on day 1, mice received 4 scrambled foot shocks in the conditioning chamber over a 10-minute training session. Twenty-four hours later, mice were returned to the original conditioning chamber and the percentage of time each mouse spent freezing over the 10-minute test was measured as an index of contextual fear memory (CFM). CFM is highly variable across this aged BXD family (21 strains tested, n = 2–8 mice/strain, Fig 1b) and heritability estimates that compare the observed genetic (between-strain) variance to technical (within-strain) variance (heritability, h2 ≈ 0.7) demonstrate that much of this variability is attributable to genetic factors. Although strain-specific differences in acquisition were also observed, the variance in acquisition was not sufficient to explain the differences in CFM across the panel.
Figure 1.

Identification of Hp1bp3 as top candidate modulator of cognitive aging (a) The BXD panel was derived by inbreeding the F2 progeny of a C57BL/6J (B6) and DBA/2J (D2) intercross. (b) Contextual fear memory (CFM) varies widely across 21 BXD strains (n = 2–8/strain, mean age = 15 ± 0.3 mo). Memory index was quantified by percent time spent freezing during 10 min test. (c) Genetic interval mapping revealed a significant CFM QTL on mouse chromosome 4 (Chr4: 137.5–140.5 Mb). Pink horizontal line: Genome-wide statistical significance (p = 0.05), Green additive effect line: D parental allele increases trait values, red: B allele increases trait values. Yellow tick marks: presence of genome sequence variant. (d) Hp1bp3 expression is highly correlated with CFM across BXD strains (Pearson r = 0.6, p < 0.05), with strains inheriting the D allele having lower levels of hippocampal Hp1bp3 and poorer CFM.
Subsequent interval mapping using the average memory index for each strain highlighted a region of chromosome 4 (Chr 4, 137.7–140.5 Mb) that was significantly associated with variation in CFM in middle-aged mice (Fig 1c). This quantitative trait locus (QTL) had not associated with memory function in studies using younger adult sex-matched BXDs (Philip, et al., 2010), suggesting the QTL contains variants that contribute to variation in cognitive aging as opposed to general memory function.
3.2 Prioritization of positional candidates identifies Hp1bp3 as putative regulator of cognitive aging across BXD panel
In order to identify genes located in the QTL that may be causally involved in regulating CFM abilities at midlife, positional gene candidates were prioritized based on: 1) annotated sequence differences segregating among the BXD strains, 2) local control of gene expression as determined by expression QTL analysis using hippocampal transcriptome data from aged BXD strains, and 3) significant correlation between hippocampal gene expression and CFM. The gene heterochromatin protein 1 binding protein 3 (Hp1bp3) emerged as the single best positional candidate (Table 1).
Table 1.
Locally (cis) regulated positional candidates.
| Gene Symbol | Description | SNP Count | Trait ID in Gene Network | Location | Max LRS | Correlation with CFM |
|---|---|---|---|---|---|---|
| Hp1bp3 | Heterochromatin protein 1 binding protein 3 | 106 | 10338180*, 10344447*, 10342675* | Chr 4: 137.7 | Chr4: 139.3 | r = 0.7, p < 0.05 |
| Eif4g3 | Eukaryotic translation initiation factor 4 gamma 3 | 763 | 10509463* | Chr4: 137.5 | Chr4: 140.2 | r = 0.5, N.S. |
| Capzb | Capping protein (actin filament) muscle Z-line, beta | 448 | 10509620, 10509628* | Chr4: 138.7 | Chr4: 138.2 | r = 0.4, N.S. |
| Iffo2 | Intermediate filament family orphan 2 | 225 | 10509777, 10509781* | Chr4: 139.1 | Chr4: 139.3 | r = −0.4, N.S. |
| Padi2 | Peptidyl arginine deiminase, type II | 132 | 10509838, 10509846* | Chr4: 140.5 | Chr4: 140.5 | r = 0.4, N.S. |
| Kif17 | Kinesin family member 17 | 119 | 10509526, 10509532* | Chr4: 137.8 | Chr4: 138.2 | r = −0.1, N.S. |
| Pla2g2f | Phospholipase A2, group IIF | 40 | 10517646, 10517654* | Chr4: 138.3 | Chr4: 140.2 | r = 0.2, N.S. |
| Mrto4 | MRT4, mRNA turnover 4 homolog (S. Cerevisiae) | 32 | 10517706, 10517711* | Chr4: 138.9 | Chr4: 139.9 | r = −0.5, N.S. |
| Nbl1 | Neuroblastoma, suppression of tumorigenicity 1 | 10 | 10517677, 10517681* | Chr4: 138.6 | Chr4: 138.2 | r = −0.5, N.S. |
| Camk2n1 | Calcium/calmodulin-dependent protein kinase II inhibitor 1 | 4 | 10509568, no SNP-free probe | Chr4: 138.0 | Chr4: 140.5 | -- |
Genes located under the significant CFM QTL were first prioritized for presence of annotated sequence variants between parental strains B6 and D2. Whole-genome transcript data from aged BXD strains was then used to perform expression QTL mapping and those genes whose expression had a peak LRS at the location of the gene (cis, or locally, regulated genes) were given higher priority. These genes were tested for correlation with the CFM trait using SNP-free probes from gene-level analyses (or exon-level analyses where necessary, see Methods section 2.5). Spearman’s rho correlations are reported.
Denotes SNP-free probe
N.S.; not significant
Hp1bp3 contains multiple missense variants in coding regions, numerous non-coding variants, and insertions/deletions predicted to impact protein function, transcriptional regulation and/or splicing (McLaren, et al., 2010); Supplementary Table 1). Hippocampal Hp1bp3 expression was significantly correlated with CFM status, with those BXD strains inheriting the D parental allele exhibiting lower levels of Hp1bp3 transcript and worse CFM performance relative to the B allele (Fig 1d). While Hp1bp3 genotype had no effect on memory status in younger adult mice (age of 8–9 weeks, Fig 2a, secondary analysis of data from Philip and colleagues, 2010), strains inheriting the D allele exhibited significantly impaired CFM at midlife (Fig 2b). Since variation in the CFM index at midlife was not due to genetic differences in baseline anxiety or post-shock pain sensitivity confounds (Fig 3), these data suggest a genotype-by-age interaction in which reductions in Hp1bp3 expression correspond specifically to aging-related cognitive impairment (Figs 1–3). In support, the genotype at Hp1bp3 can account for as much as 52% of the strain variance in CFM performance at midlife. However, calculations of genetic variance explained by a QTL are biased upward by an average of 30% due to a combination of epistatic, genotype-by-environment interactions, and small sample size (n < 1000) (Beavis, 1998; Wellenreuther and Hansson, 2016; Wurschum and Kraft, 2014). Therefore, we estimate that the actual biological heritable variation in CFM at midlife explained by Hp1bp3 genotype is more likely in the range of 20–25%.
Figure 2.

Effect of Hp1bp3 genotype on CFM status is age-dependent. (a) Memory index for adult BXDs (age = 8–9 weeks) was calculated using CFM data generated by Philip and colleagues (2010) and is publically available on GeneNetwork.org (Trait ID 110908, see Methods section 2.3). Parental origin of Hp1bp3 allele did not significantly affect memory status in adulthood (D allele memory index = 81.0 ± 1.3, B =84.3 ± 1.3, t(54) = −1.86, p = 0.068). (b) Middle-aged (age = 15.0 ± 0.3 months) BXD strains inheriting the D parental allele performed significantly worse on CFM tests than strains inheriting the B allele (D = 4.8 ± 1.0, B = 41.5 ± 20.7, t(14) = −4.6, p < 0.001). Two-way ANOVA revealed a significant interaction between genotype and age [F(1,66) = 33.5, p < 0.001], suggesting that Hp1bp3 genotype influences memory status in an age-dependent manner.
Figure 3.

Differences in CFM were not attributable to baseline freezing or post-shock reactivity. There were no significant effects of strain on either (a) baseline freezing [F(20,50) = 1.009, p = 0.47] or (b) length of post-shock activity burst [F(20,49) = 1.668, p = 0.07], indicating no strain-specific differences in baseline fear/anxiety or pain sensitivity, respectively, across 21 BXD strains tested.
3.3 Functional validation in a knock-out mouse model confirms novel role for Hp1bp3 in cognitive function
To assess the functional consequence of loss of Hp1bp3 on hippocampus-dependent learning and memory, we employed a reverse genetics approach. During CFM training, Hp1bp3 knock-out (KO) and wild-type (WT) mice (Garfinkel, et al., 2015a) showed no differences in baseline activity (Fig 4a) or shock reactivity (Fig 4b), and KO mice exhibited comparable acquisition of conditioned fear, indicating they successfully learned the context-shock association (Fig 4a). Nevertheless, KO mice exhibited long-term CFM deficits when tested 24 hours later (Fig 4c). Notably, the effect of Hp1bp3 KO on CFM was similar to the effect of genotype at the Hp1bp3 locus. As the KO mice were generated on a B6 background, the WT mice carry the B version of the Hp1bp3 allele, and as expected, perform comparable to B6 mice from the BXD GRP. However, KO mice performed more similarly to BXDs harboring the D allele, which exhibited a 20–40% reduction in conditioned freezing during CFM tests compared to strains with the B allele (Fig 2b), and suggests the D allele functions similarly to a loss-of-function mutation.
Figure 4.
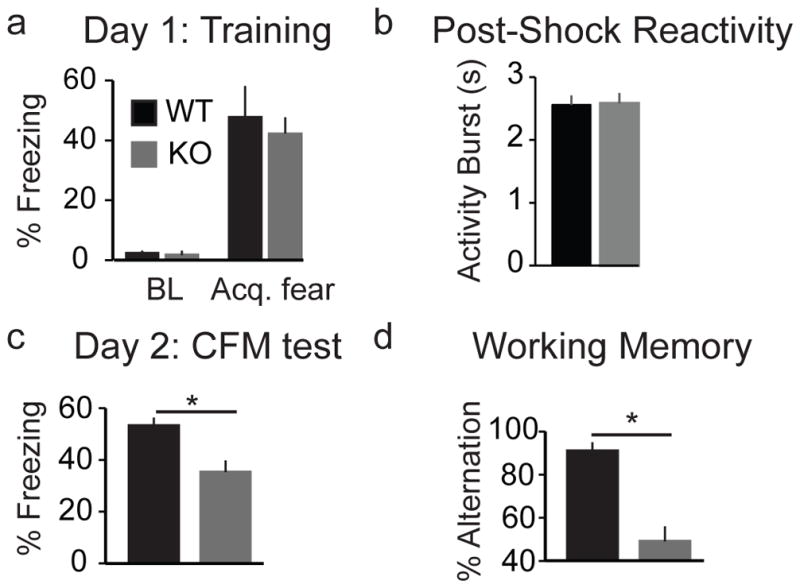
Hp1bp3 knockout (KO) mice exhibit impairment on hippocampus-dependent long-term and working memory tasks. (a) Comparable baseline (BL) freezing and acquisition of conditioned fear (Acq. fear) by the final trial in wild-type (WT) and Hp1bp3 knock-out (KO) mice (n = 6/group) during contextual fear conditioning. (b) There were no differences in post-shock reactivity during fear conditioning training between WT and Hp1bp3 KO mice, indicating no differences in pain sensitivity. Videos were manually analyzed to determine the average length of activity burst for each mouse. WT = 2.6 ± 0.2 s, KO = 2.6 ± 0.2 s; t(1,10)= −0.11, p = 0.91. (c) KO mice were significantly impaired on CFM [t(10) = 3.10, p < 0.001)]. (d) KO mice also exhibited spatial working memory deficits [t(12) = 5.095, p < 0.001].
To determine if loss of Hp1bp3 mimics aging-related effects on additional hippocampus-dependent cognitive domains (Zornetzer, et al., 1982), working memory was assessed using the T-maze test of spontaneous alternation (Deacon and Rawlins, 2006). KO mice were significantly impaired, and like aged mice (Lalonde, 2002), performed at chance levels (Fig 4d). Taken together, these results demonstrate, for the first time, that Hp1bp3 is necessary for successful hippocampus-dependent memory function spanning multiple cognitive domains - and suggest that therapeutic interventions to restore levels of Hp1bp3 may improve cognitive function.
3.4 Hp1bp3 is associated with memory function in human cognitive aging
To test the hypothesis that Hp1bp3 is also involved in human cognitive aging and to evaluate the translational relevance of our murine results, we next tested whether hippocampal Hp1bp3 expression correlated with cognitive performance in elderly humans (Table 2). Caveats to working with human brain samples include variable post-mortem intervals and undefined brain pathology (Bennett, et al., 2014; Hargis, 2016). Therefore, in order to limit potential confounds we analyzed hippocampal tissue from humans with ‘normal’ cognitive aging (i.e. no distinct Alzheimer’s disease pathology), which is not typically associated with gross neurodegeneration (Burke and Barnes, 2006; Korbo, et al., 2004). Quantitative Western blots for HP1BP3 were performed on postmortem hippocampal tissue lysates prepared from cognitively intact (MMSE = 29.3 ± 0.3, age = 78 ± 9.7 years) and cognitively impaired humans (MMSE = 22.6 ± 3.0, age = 84 ± 4.4 years). HP1BP3 levels were significantly lower in humans diagnosed with cognitive impairment compared to age-matched controls (Fig 5). Double band staining is typical of HP1BP3 expression, indicative of multiple splice variants present in brain tissue (Garfinkel, et al., 2015b). These results suggest either lower or reduced expression of Hp1bp3 may underlie cognitive deficits observed in human populations, and highlight the value and potential relevance of aged cohorts of diverse strains of mice for identifying molecular correlates of cognitive decline in humans.
Table 2.
Demographics for elderly humans used for postmortem hippocampal tissue analysis.
| Patient | Age | Sex | MMSE | Group | Post-Mortem Interval (h) |
|---|---|---|---|---|---|
| H1 | 79 | Female | 29 | Intact | 1.75 |
| H2 | 93 | Female | 30 | Intact | 2.25 |
| H3 | 81 | Male | 29 | Intact | 2.83 |
| H4 | 86 | Female | 27 | Impaired | 2.48 |
| H5 | 90 | Female | 24 | Impaired | 2.5 |
| H6 | 59 | Male | 17 | Impaired | 5.85 |
MMSE; Mini-Mental State Exam score (maximum score indicating normal cognition = 30)
Figure 5.

HP1BP3 protein is correlated with memory status in aging humans (a) Western blot shows hippocampal HP1BP3 protein is reduced in cognitively impaired elderly humans (n = 3, MMSE = 22.6 ± 3.0, age = 84 ± 4 years) relative to cognitively intact controls (n = 3, MMSE = 29.3 ± 0.3, age = 78 ± 10 years). (b) Quantification of hippocampal HP1BP3 protein (Cog. Intact = 1.02 ± 0.21 adjusted density, Impaired = 0.60 ± 0.04, t(1,4) = −3.344, p = 0.03).
3.5 Genetic correlation analyses elucidate functional roles for Hp1bp3 in cognitive aging
There is evidence that groups of highly correlated genes are likely to play a similar biological function and/or act as part of the same biological pathway (Eisen, et al., 1998; Zhang and Horvath, 2005). To identify mechanisms through which Hp1bp3 may act to regulate cognitive abilities at midlife, genes whose hippocampal expression significantly correlated with that of Hp1bp3 were identified using whole-genome hippocampal transcript data from middle-aged BXD strains. All nominally significant correlates (uncorrected p ≤ 0.05, 2074 genes) were sorted into a ranked list, with the most highly positively correlated genes at the top (Supplementary Table 2). This list was then used for Gene Set Enrichment Analysis (GSEA, Broad Institute; (Subramanian, et al., 2005) and tested against gene sets publicly available from Broad Institute’s Molecular Signatures Database (MSigDB), including Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (Kanehisa and Goto, 2000), Reactome pathways (Matthews, et al., 2009), and Gene Ontology (GO) classification (Harris, et al., 2004), using a false discovery rate of ≤ 0.25 to identify significantly enriched gene sets per standard methods (Subramanian, et al., 2005).
Functions of Hp1bp3 that have been identified include regulation of chromatin structure (Dutta, et al., 2014), gene expression (Garfinkel, et al., 2015b), cell cycle progression (Dutta, et al., 2014), and insulin signaling (Garfinkel, et al., 2015a), several of which were supported by our analyses. For example, positive correlates of Hp1bp3 were significantly enriched both in nuclear localization (Figure 6a) and insulin signaling (Fig 6b). Additionally, we found Hp1bp3 gene correlates enriched for terms related to plasma membrane localization and for functions related to neuronal function and excitability, including voltage-gated channel activity, ion channel activity, and G-protein coupled receptor activity (Fig 6c). Together, these results support the hypothesis that Hp1bp3 may critically influence expression of ion channels and receptors located in the plasma membrane that mediate neurogenesis, neuronal excitability, and plasticity, mechanisms that are disrupted in hippocampal neurons of aged rodent models with cognitive deficits (Burke and Barnes, 2006; Kaczorowski and Disterhoft, 2009; Mattson and Magnus, 2006). Finally, Hp1bp3 gene correlates also show significant overlap with genes differentially expressed between Alzheimer’s disease patients and controls (Blalock, et al., 2004); Fig 6d), suggesting the role of Hp1bp3 in cognitive aging may extend into the regulation of cognitive deficits observed in AD.
Figure 6.

Gene Set Enrichment Analysis (GSEA). All nominally significant hippocampal correlates (uncorrected p ≤ 0.05, n = 2074, see Supplementary Table 2) of Hp1bp3 were extracted from whole-genome transcriptome data from BXD strains age-matched to those used for contextual fear conditioning. The genes were sorted based on correlation coefficient, with the most highly positively correlated genes at the top of the list. Graphs illustrate calculation of enrichment score, or deviation from random distribution throughout the preranked list. Each vertical black line below the graph represents a gene set member and its position in the preranked list (red=genes positively correlated with Hp1bp3, blue=genes negatively correlated with Hp1bp3). A FDR cut-off of q ≤ 0.25 was used to identify significantly enriched sets according to established methods. (a, top) Positive correlates were significantly enriched for localization to the nucleus. (a, bottom) Genes localized to the nucleus were also present in significantly enriched functional gene sets including RNA splicing, nuclear transport, chromosome organization, cell cycle, and transcription cofactor activity. Normalized enrichment score (NES) represents enrichment score after adjustment to account for size of the given gene set. (b) Positive correlates were significantly enriched for the KEGG pathway corresponding to insulin signaling. (c, top) Negative correlates of Hp1bp3 were significantly enriched for localization to the plasma membrane. (c, bottom) Genes localized to the plasma membrane were also present in significantly enriched functional gene sets including ion channel activity, substrate-specific channel activity, transmembrane transporter activity, voltage-gated channel activity, and G-protein coupled receptor activity. (d) Correlates of Hp1bp3 also show significant overlap with a set of genes determined by Blalock and colleagues (2004) to be significantly differentially expressed between Alzheimer’s disease patients and corresponding controls.
4. Discussion
The goal of this study was to utilize a genetically diverse panel of mice in order to identify genetic factors involved in the regulation of cognitive aging that may have gone undetected in either complex human studies or murine studies utilizing only a single genetic background. Aging is a leading risk factor for age-associated dementias such as Alzheimer’s disease, and our work and others suggest that genetic factors and mechanisms underlying biological processes during midlife play a key role in determining an individual’s susceptibility or resilience to transitioning between healthy brain aging and pathological brain aging (Douaud, et al., 2013; Jack, et al., 2013; Kaczorowski, et al., 2011; Keller, 2006; Miller, et al., 2008). Thus, there is a critical need to understand gene variants that play a role in determining memory capabilities at this transition point (midlife). To directly address this need, and to overcome some of the barriers inherent to human studies, we turned to a well-characterized murine GRP, which allows for the exploitation of phenotypic heterogeneity across a population while exerting precise control of environmental conditions.
4.1 Identification and validation of Hp1bp3 as top positional candidate modulating cognitive aging
By combining forward and reverse murine genetic approaches and by joint analysis of mouse and human cohorts, we progressed from the identification of a significant QTL containing variants regulating CFM at midlife to the demonstration that our top positional candidate, Hp1bp3, is important for hippocampus-dependent long-term and spatial working memory. Knockout of Hp1bp3 has been associated with viability and growth abnormalities (Garfinkel, et al., 2015b) that may affect behavior. Therefore, we examined the behavior of Hp1bp3 KO mice on three tasks that may confound contextual fear memory testing (e.g. baseline exploratory activity, post-shock reactivity, and acquisition of contextual fear). We found no differences between the Hp1bp3 KO and WT mice on these measures, suggesting that the effects of Hp1bp3 KO reported herein are not due to gross behavioral abnormalities. Other positional candidates identified in the CFM QTL, such as Pink1 and Kif17 (Table 1), have been linked to cognitive deficits (Roberson, et al., 2008) and neurodegeneration (Moisoi, et al., 2014) and contain variants that could impact gene function or expression. Here, we focused on genetic correlates of CFM expressed in the hippocampus due to the hippocampal-dependent nature of contextual fear conditioning (Chen, et al., 1996) and the fact that the hippocampus is one of the first structures affected in aging (Burke and Barnes, 2006; Gant, et al., 2006). However, given that the formation and recall of CFM involves a distributed network of brain regions (Tovote, et al., 2015), it is possible that altered gene expression in other regions (e.g. prefrontal cortex) may also contribute to variation in cognitive aging. As variation in Hp1bp3 genotype was estimated to account for ~20–25% of the heritable variation in CFM at midlife, it is possible that additional genes, acting alone or in combination with Hp1bp3, also influence some of the observed variation in cognitive decline and further investigation into these candidates is warranted.
4.2 Hp1bp3 likely plays conserved role in humans
Reduced expression of Hp1bp3 is observed in both cognitively impaired aged mice and humans, suggesting that decreased expression of Hp1bp3 contributes to cognitive aging in both species. In support of this idea, Hp1bp3 is among the top 100 genes upregulated in response to metformin hydrochloride (Lamb, et al., 2006), a drug known to enhance memory in mice (Wang, et al., 2012), which was recently approved for clinical trial to prevent or reduce effects of aging, including cognitive decline (Targeting Aging with Metformin, TAME study). In addition, Hp1bp3 is highly conserved in mammals, with 93% similarity between the human and murine primary sequences (Garfinkel, et al., 2015b) indicating Hp1bp3 likely plays an important functional role in both species. Deletion of the region of the human genome syntenic to our QTL results in a condition known as Deletion 1p36 syndrome (Battaglia, et al., 2008). Although specific phenotypes vary according to size and location of deletion breakpoint (Gajecka, et al., 2007), many with this syndrome exhibit cognitive deficits and mental retardation (Battaglia, et al., 2008), supporting the idea that genes in this area are necessary for cognitive function in both humans and animals.
4.3 Gene set enrichment analyses highlight putative functional roles for Hp1bp3
Hp1bp3 has been shown to be evolutionarily and structurally related to the linker histone H1 family, members of which confer higher-order organization to chromatin by binding to the surface of nucleosomes (Garfinkel, et al., 2015b). A number of studies suggest this family of proteins play a highly specific role in gene expression, possibly due to their role in chromatin organization (Garfinkel, et al., 2015b). It is thought that Hp1bp3 contributes to the inter-conversion of heterochromatin and euchromatin (Dutta, et al., 2014), thereby activating or silencing specific genes as needed. As long-term memory and cognition depend on de novo gene expression in response to a learning and/or training event (Cavallaro, et al., 2002), it is possible that Hp1bp3 is regulating the transcription of specific genes necessary for successful cognitive function. Genes significantly positively correlated with Hp1bp3 were enriched for localization in the nucleus, with functions including transcription and RNA processing. Genes negatively correlated with Hp1bp3 include genes with known links to cognition and neuron function, such as channel and transporter activity (Fig 6c). In addition, genes effected by Hp1bp3 knockdown in cell lines are known to have important neuronal and/or memory functions in vivo, including the regulation of neuronal excitability (Averaimo, et al., 2014; Gulledge, et al., 2013; Richards, et al., 2007), Ca2+ homeostasis (Jia, et al., 2015), synaptic plasticity (Lee, et al., 2012) and inhibitory neurotransmission (Marsden, et al., 2007) in the hippocampus (e.g. Ca2+ ATPase, Na+/K+ ATPase, CLCC1 and CLIC channels, and GABARAP). However, our GSEA results do not differentiate whether changes in nuclear or plasma membrane proteins occur first, so it is possible that compensatory changes in transcription and RNA processing are occurring due to aging-induced alterations in plasma membrane ion channels and receptors. A targeted in vivo knockdown of Hp1bp3 and subsequent gene expression analysis will help to clarify the role of Hp1bp3 in gene expression under physiological conditions.
5. Conclusion
Here, we demonstrate for the first time that Hp1bp3 is a key modulator of cognitive aging. In addition, although the BXD family has previously been used to study cognition in young mice (Philip, et al., 2010; Wehner, et al., 1997), but Hp1bp3 had not emerged as a key regulator of CFM, our results suggest that variation in Hp1bp3 genotype may influence memory in an age-dependent manner. As biological processes regulating memory function at midlife may play a critical role in the development of AD dementia (Douaud, et al., 2013; Jack, et al., 2013; Kaczorowski, et al., 2011; Keller, 2006; Miller, et al., 2008), and Hp1bp3 gene correlates also show significant overlap with genes differentially expressed between AD patients and controls (Blalock, et al., 2004); Fig 6d), we speculate that treatments that restore Hp1bp3 expression and/or function may improve cognition in patients with normal cognitive decline as well as AD dementia. This prediction is further supported by evidence that metformin hydrochloride, a drug known to increase Hp1bp3 expression (Lamb, et al., 2006), has been shown to enhance memory in mice (Wang, et al., 2012) and is approved for an anti-aging clinical trial in humans (Targeting Aging with Metformin, TAME study). Overall, our results suggest Hp1bp3 and related networks may serve as potential targets against cognitive aging and demonstrate the utility of genetically diverse mouse models for the study of complex human disease.
Supplementary Material
Genetic variants in Hp1bp3 across the BXDs.
All sequence polymorphisms and structural variants (insertions/deletions) between the reference sequence (B6) and parental strain D2 according to the Sanger Institute’s Mouse Genomes Project. Those variants predicted to have ‘moderate’ impact on transcript structure, splicing, and/or protein function according to the Ensembl Variant Effect Predictor (McLaren, et al., 2010) are highlighted in red.
GSEA input.
All genes significantly correlated (uncorrected p ≤ 0.05) with Hp1bp3 in the hippocampus of aged BXD strains. The genes were ranked according to correlation coefficient and used for Gene Set Enrichment Analysis according to established methods (Mootha, et al., 2003; Subramanian, et al., 2005)
Highlights.
Contextual fear memory abilities at midlife are highly heritable
Genetic mapping identified area of mouse chromosome 4 associated with memory decline
Heterochromatin protein 1 binding protein 3 emerged as top positional candidate
Genetic deletion of Hp1bp3 recapitulates memory deficits characteristic of aging
HP1BP3 protein levels are significantly reduced in hippocampus of cognitively impaired humans
Acknowledgments
Support was provided by the National Institute on Aging (F31 AG050357 to SMN, R00 AG039511 to CCK, R01 043930 to RWW), as well as the American Federation for Aging Research (PD15013 to LAW, RAG14141 to CCK). The UTHSC BXD colony is supported by the Center for Integrative and Translational Genomics, and GeneNetwork is supported by U01 AA016662 and U01 AA013499. BPG and JO were supported by The United States-Israel Binational Science Foundation (Grant 2009326 to JO). The authors would also like to thank the University of Kentucky Sanders-Brown Center on Aging for providing patient samples generously supported by NIA grant P30-AG0-28383.
Abbreviations
- CFM
contextual fear memory
- GRP
genetic reference panel
- QTL
quantitative trait locus
- Hp1bp3
heterochromatin 1 binding protein
- GSEA
gene set enrichment analysis
Footnotes
Author Contributions: SMN and CCK conceived of the experiments. SMN, LAW, BPG, BMIJ, AC, JO, LL, GK, KMSO, RWW and CCK designed and performed the experiments. SMN, LAW, BPG, BMIJ, LL, RWO, MKM and CCK analyzed the data. SMN, AC, JO, LL, GK, RWW, and CCK contributed reagents/materials/analysis tools. SMN, KMSO, and CCK wrote the manuscript. All authors reviewed and contributed intellectually to the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anagnostaras SG, Josselyn SA, Frankland PW, Silva AJ. Computer-assisted behavioral assessment of Pavlovian fear conditioning in mice. Learning & memory. 2000;7(1):58–72. doi: 10.1101/lm.7.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreux PA, Williams EG, Koutnikova H, Houtkooper RH, Champy MF, Henry H, Schoonjans K, Williams RW, Auwerx J. Systems genetics of metabolism: the use of the BXD murine reference panel for multiscalar integration of traits. Cell. 2012;150(6):1287–99. doi: 10.1016/j.cell.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Averaimo S, Gritti M, Barini E, Gasparini L, Mazzanti M. CLIC1 functional expression is required for cAMP-induced neurite elongation in post-natal mouse retinal ganglion cells. Journal of neurochemistry. 2014;131(4):444–56. doi: 10.1111/jnc.12832. [DOI] [PubMed] [Google Scholar]
- Balogh SA, Radcliffe RA, Logue SF, Wehner JM. Contextual and cued fear conditioning in C57BL/6J and DBA/2J mice: context discrimination and the effects of retention interval. Behavioral neuroscience. 2002;116(6):947–57. doi: 10.1037//0735-7044.116.6.947. [DOI] [PubMed] [Google Scholar]
- Battaglia A, Hoyme HE, Dallapiccola B, Zackai E, Hudgins L, McDonald-McGinn D, Bahi-Buisson N, Romano C, Williams CA, Brailey LL, Zuberi SM, Carey JC. Further delineation of deletion 1p36 syndrome in 60 patients: a recognizable phenotype and common cause of developmental delay and mental retardation. Pediatrics. 2008;121(2):404–10. doi: 10.1542/peds.2007-0929. [DOI] [PubMed] [Google Scholar]
- Beavis WD. QTL analyses: power, precision, and accuracy. In: Patterson AH, editor. Molecular Dissection of Complex Traits. CRC Press; New York: 1998. pp. 145–62. [Google Scholar]
- Belknap JK. Effect of within-strain sample size on QTL detection and mapping using recombinant inbred mouse strains. Behavior genetics. 1998;28(1):29–38. doi: 10.1023/a:1021404714631. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Yu L, De Jager PL. Building a pipeline to discover and validate novel therapeutic targets and lead compounds for Alzheimer’s disease. Biochemical pharmacology. 2014;88(4):617–30. doi: 10.1016/j.bcp.2014.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(7):2173–8. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke SN, Barnes CA. Neural plasticity in the ageing brain. Nature reviews Neuroscience. 2006;7(1):30–40. doi: 10.1038/nrn1809. [DOI] [PubMed] [Google Scholar]
- Cavallaro S, D’Agata V, Manickam P, Dufour F, Alkon DL. Memory-specific temporal profiles of gene expression in the hippocampus. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(25):16279–84. doi: 10.1073/pnas.242597199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Kim JJ, Thompson RF, Tonegawa S. Hippocampal lesions impair contextual fear conditioning in two strains of mice. Behavioral neuroscience. 1996;110(5):1177–80. doi: 10.1037//0735-7044.110.5.1177. [DOI] [PubMed] [Google Scholar]
- Chesler EJ, Lu L, Shou S, Qu Y, Gu J, Wang J, Hsu HC, Mountz JD, Baldwin NE, Langston MA, Threadgill DW, Manly KF, Williams RW. Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nature genetics. 2005;37(3):233–42. doi: 10.1038/ng1518. [DOI] [PubMed] [Google Scholar]
- Davies G, Armstrong N, Bis JC, Bressler J, Chouraki V, Giddaluru S, Hofer E, Ibrahim-Verbaas CA, Kirin M, Lahti J, van der Lee SJ, Le Hellard S, Liu T, Marioni RE, Oldmeadow C, Postmus I, Smith AV, Smith JA, Thalamuthu A, Thomson R, Vitart V, Wang J, Yu L, Zgaga L, Zhao W, Boxall R, Harris SE, Hill WD, Liewald DC, Luciano M, Adams H, Ames D, Amin N, Amouyel P, Assareh AA, Au R, Becker JT, Beiser A, Berr C, Bertram L, Boerwinkle E, Buckley BM, Campbell H, Corley J, De Jager PL, Dufouil C, Eriksson JG, Espeseth T, Faul JD, Ford I, Generation S, Gottesman RF, Griswold ME, Gudnason V, Harris TB, Heiss G, Hofman A, Holliday EG, Huffman J, Kardia SL, Kochan N, Knopman DS, Kwok JB, Lambert JC, Lee T, Li G, Li SC, Loitfelder M, Lopez OL, Lundervold AJ, Lundqvist A, Mather KA, Mirza SS, Nyberg L, Oostra BA, Palotie A, Papenberg G, Pattie A, Petrovic K, Polasek O, Psaty BM, Redmond P, Reppermund S, Rotter JI, Schmidt H, Schuur M, Schofield PW, Scott RJ, Steen VM, Stott DJ, van Swieten JC, Taylor KD, Trollor J, Trompet S, Uitterlinden AG, Weinstein G, Widen E, Windham BG, Jukema JW, Wright AF, Wright MJ, Yang Q, Amieva H, Attia JR, Bennett DA, Brodaty H, de Craen AJ, Hayward C, Ikram MA, Lindenberger U, Nilsson LG, Porteous DJ, Raikkonen K, Reinvang I, Rudan I, Sachdev PS, Schmidt R, Schofield PR, Srikanth V, Starr JM, Turner ST, Weir DR, Wilson JF, van Duijn C, Launer L, Fitzpatrick AL, Seshadri S, Mosley TH, Jr, Deary IJ. Genetic contributions to variation in general cognitive function: a meta-analysis of genome-wide association studies in the CHARGE consortium (N=53949) Molecular psychiatry. 2015;20(2):183–92. doi: 10.1038/mp.2014.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon RM, Rawlins JN. T-maze alternation in the rodent. Nature protocols. 2006;1(1):7–12. doi: 10.1038/nprot.2006.2. [DOI] [PubMed] [Google Scholar]
- Douaud G, Menke RA, Gass A, Monsch AU, Rao A, Whitcher B, Zamboni G, Matthews PM, Sollberger M, Smith S. Brain microstructure reveals early abnormalities more than two years prior to clinical progression from mild cognitive impairment to Alzheimer’s disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33(5):2147–55. doi: 10.1523/JNEUROSCI.4437-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta B, Ren Y, Hao P, Sim KH, Cheow E, Adav S, Tam JP, Sze SK. Profiling of the Chromatin-Associated Proteome Identifies HP1BP3 as a Novel Regulator of Cell Cycle Progression. Molecular & cellular proteomics: MCP. 2014 doi: 10.1074/mcp.M113.034975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(25):14863–8. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajecka M, Mackay KL, Shaffer LG. Monosomy 1p36 deletion syndrome. Am J Med Genet C Semin Med Genet. 2007;145C(4):346–56. doi: 10.1002/ajmg.c.30154. [DOI] [PubMed] [Google Scholar]
- Gant JC, Sama MM, Landfield PW, Thibault O. Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+-induced Ca2+ release. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26(13):3482–90. doi: 10.1523/JNEUROSCI.4171-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garfinkel BP, Arad S, Le PT, Bustin M, Rosen CJ, Gabet Y, Orly J. Proportionate Dwarfism in Mice Lacking Heterochromatin Protein 1 Binding Protein 3 (HP1BP3) Is Associated With Alterations in the Endocrine IGF-1 Pathway. Endocrinology. 2015a;156(12):4558–70. doi: 10.1210/en.2015-1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garfinkel BP, Melamed-Book N, Anuka E, Bustin M, Orly J. HP1BP3 is a novel histone H1 related protein with essential roles in viability and growth. Nucleic acids research. 2015b;43(4):2074–90. doi: 10.1093/nar/gkv089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Dasari S, Onoue K, Stephens EK, Hasse JM, Avesar D. A sodium-pump-mediated afterhyperpolarization in pyramidal neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33(32):13025–41. doi: 10.1523/JNEUROSCI.0220-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargis KWB, EM Transcriptional signatures of brain aging and Alzheimer’s disease: What are our rodent models telling us? Behavioural brain research. 2016 doi: 10.1016/j.bbr.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, Richter J, Rubin GM, Blake JA, Bult C, Dolan M, Drabkin H, Eppig JT, Hill DP, Ni L, Ringwald M, Balakrishnan R, Cherry JM, Christie KR, Costanzo MC, Dwight SS, Engel S, Fisk DG, Hirschman JE, Hong EL, Nash RS, Sethuraman A, Theesfeld CL, Botstein D, Dolinski K, Feierbach B, Berardini T, Mundodi S, Rhee SY, Apweiler R, Barrell D, Camon E, Dimmer E, Lee V, Chisholm R, Gaudet P, Kibbe W, Kishore R, Schwarz EM, Sternberg P, Gwinn M, Hannick L, Wortman J, Berriman M, Wood V, de la Cruz N, Tonellato P, Jaiswal P, Seigfried T, White R, Gene Ontology C. The Gene Ontology (GO) database and informatics resource. Nucleic acids research. 2004;32(Database issue):D258–61. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SE, Deary IJ. The genetics of cognitive ability and cognitive ageing in healthy older people. Trends in cognitive sciences. 2011;15(9):388–94. doi: 10.1016/j.tics.2011.07.004. [DOI] [PubMed] [Google Scholar]
- Hatfield I, Harvey I, Yates ER, Redd JR, Reiter LT, Bridges D. The role of TORC1 in muscle development in Drosophila. Scientific reports. 2015;5:9676. doi: 10.1038/srep09676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497(7450):451–7. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap C. Altshuler DM, Gibbs RA, Peltonen L, Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F, Peltonen L, Dermitzakis E, Bonnen PE, Altshuler DM, Gibbs RA, de Bakker PI, Deloukas P, Gabriel SB, Gwilliam R, Hunt S, Inouye M, Jia X, Palotie A, Parkin M, Whittaker P, Yu F, Chang K, Hawes A, Lewis LR, Ren Y, Wheeler D, Gibbs RA, Muzny DM, Barnes C, Darvishi K, Hurles M, Korn JM, Kristiansson K, Lee C, McCarrol SA, Nemesh J, Dermitzakis E, Keinan A, Montgomery SB, Pollack S, Price AL, Soranzo N, Bonnen PE, Gibbs RA, Gonzaga-Jauregui C, Keinan A, Price AL, Yu F, Anttila V, Brodeur W, Daly MJ, Leslie S, McVean G, Moutsianas L, Nguyen H, Schaffner SF, Zhang Q, Ghori MJ, McGinnis R, McLaren W, Pollack S, Price AL, Schaffner SF, Takeuchi F, Grossman SR, Shlyakhter I, Hostetter EB, Sabeti PC, Adebamowo CA, Foster MW, Gordon DR, Licinio J, Manca MC, Marshall PA, Matsuda I, Ngare D, Wang VO, Reddy D, Rotimi CN, Royal CD, Sharp RR, Zeng C, Brooks LD, McEwen JE. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467(7311):52–8. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, Lesnick TG, Pankratz VS, Donohue MC, Trojanowski JQ. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12(2):207–16. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Jucius TJ, Cook SA, Ackerman SL. Loss of Clcc1 Results in ER Stress, Misfolded Protein Accumulation, and Neurodegeneration. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2015;35(7):3001–9. doi: 10.1523/JNEUROSCI.3678-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MR, Shkura K, Langley SR, Delahaye-Duriez A, Srivastava P, Hill WD, Rackham OJ, Davies G, Harris SE, Moreno-Moral A, Rotival M, Speed D, Petrovski S, Katz A, Hayward C, Porteous DJ, Smith BH, Padmanabhan S, Hocking LJ, Starr JM, Liewald DC, Visconti A, Falchi M, Bottolo L, Rossetti T, Danis B, Mazzuferi M, Foerch P, Grote A, Helmstaedter C, Becker AJ, Kaminski RM, Deary IJ, Petretto E. Systems genetics identifies a convergent gene network for cognition and neurodevelopmental disease. Nature neuroscience. 2015 doi: 10.1038/nn.4205. [DOI] [PubMed] [Google Scholar]
- Kaczorowski CC, Davis SJ, Moyer JR., Jr Aging redistributes medial prefrontal neuronal excitability and impedes extinction of trace fear conditioning. Neurobiology of aging. 2012;33(8):1744–57. doi: 10.1016/j.neurobiolaging.2011.03.020. [DOI] [PubMed] [Google Scholar]
- Kaczorowski CC, Disterhoft JF. Memory deficits are associated with impaired ability to modulate neuronal excitability in middle-aged mice. Learning & memory. 2009;16(6):362–6. doi: 10.1101/lm.1365609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczorowski CC, Sametsky E, Shah S, Vassar R, Disterhoft JF. Mechanisms underlying basal and learning-related intrinsic excitability in a mouse model of Alzheimer’s disease. Neurobiology of aging. 2011;32(8):1452–65. doi: 10.1016/j.neurobiolaging.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JN. Age-related neuropathology, cognitive decline, and Alzheimer’s disease. Ageing research reviews. 2006;5(1):1–13. doi: 10.1016/j.arr.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Chesler EJ, Lu L, Williams RW, Gage FH. Natural variation and genetic covariance in adult hippocampal neurogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(3):780–5. doi: 10.1073/pnas.0510291103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbo L, Amrein I, Lipp HP, Wolfer D, Regeur L, Oster S, Pakkenberg B. No evidence for loss of hippocampal neurons in non-Alzheimer dementia patients. Acta neurologica Scandinavica. 2004;109(2):132–9. doi: 10.1034/j.1600-0404.2003.00182.x. [DOI] [PubMed] [Google Scholar]
- Lalonde R. The neurobiological basis of spontaneous alternation. Neuroscience and biobehavioral reviews. 2002;26(1):91–104. doi: 10.1016/s0149-7634(01)00041-0. [DOI] [PubMed] [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–35. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- Laukka EJ, Lovden M, Herlitz A, Karlsson S, Ferencz B, Pantzar A, Keller L, Graff C, Fratiglioni L, Backman L. Genetic effects on old-age cognitive functioning: a population-based study. Psychology and aging. 2013;28(1):262–74. doi: 10.1037/a0030829. [DOI] [PubMed] [Google Scholar]
- Lee SH, Kim KR, Ryu SY, Son S, Hong HS, Mook-Jung I, Lee SH, Ho WK. Impaired short-term plasticity in mossy fiber synapses caused by mitochondrial dysfunction of dentate granule cells is the earliest synaptic deficit in a mouse model of Alzheimer’s disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32(17):5953–63. doi: 10.1523/JNEUROSCI.0465-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman EJ, Kulnane LS, Gao Y, Petriello MC, Pimpis KM, Younkin L, Dolios G, Wang R, Younkin SG, Lamb BT. Genetic background regulates beta-amyloid precursor protein processing and beta-amyloid deposition in the mouse. Human molecular genetics. 2003;12(22):2949–56. doi: 10.1093/hmg/ddg322. [DOI] [PubMed] [Google Scholar]
- Marsden KC, Beattie JB, Friedenthal J, Carroll RC. NMDA receptor activation potentiates inhibitory transmission through GABA receptor-associated protein-dependent exocytosis of GABA(A) receptors. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27(52):14326–37. doi: 10.1523/JNEUROSCI.4433-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews L, Gopinath G, Gillespie M, Caudy M, Croft D, de Bono B, Garapati P, Hemish J, Hermjakob H, Jassal B, Kanapin A, Lewis S, Mahajan S, May B, Schmidt E, Vastrik I, Wu G, Birney E, Stein L, D’Eustachio P. Reactome knowledgebase of human biological pathways and processes. Nucleic acids research. 2009;37(Database issue):D619–22. doi: 10.1093/nar/gkn863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Magnus T. Ageing and neuronal vulnerability. Nature reviews Neuroscience. 2006;7(4):278–94. doi: 10.1038/nrn1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClearn GE, Johansson B, Berg S, Pedersen NL, Ahern F, Petrill SA, Plomin R. Substantial genetic influence on cognitive abilities in twins 80 or more years old. Science. 1997;276(5318):1560–3. doi: 10.1126/science.276.5318.1560. [DOI] [PubMed] [Google Scholar]
- McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26(16):2069–70. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Oldham MC, Geschwind DH. A systems level analysis of transcriptional changes in Alzheimer’s disease and normal aging. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28(6):1410–20. doi: 10.1523/JNEUROSCI.4098-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moisoi N, Fedele V, Edwards J, Martins LM. Loss of PINK1 enhances neurodegeneration in a mouse model of Parkinson’s disease triggered by mitochondrial stress. Neuropharmacology. 2014;77:350–7. doi: 10.1016/j.neuropharm.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature genetics. 2003;34(3):267–73. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Neuner SM, Wilmott LA, Hope KA, Hoffmann B, Chong JA, Abramowitz J, Birnbaumer L, O’Connell K, Tryba AK, Greene AS, Chan CS, Kaczorowski CC. TRPC3 channels critically regulate hippocampal excitability and contextual fear memory. Behavioural brain research. 2014 doi: 10.1016/j.bbr.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksman GY, Li K, Rose G, Skrebitsky VG, Fedorov NB. Increase in slow afterhyperpolarization led to learning delay in DBA mice. Bulletin of experimental biology and medicine. 2005;140(3):274–7. doi: 10.1007/s10517-005-0465-1. [DOI] [PubMed] [Google Scholar]
- Payton A. The impact of genetic research on our understanding of normal cognitive ageing: 1995 to 2009. Neuropsychol Rev. 2009;19(4):451–77. doi: 10.1007/s11065-009-9116-z. [DOI] [PubMed] [Google Scholar]
- Peirce JL, Lu L, Gu J, Silver LM, Williams RW. A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC genetics. 2004;5:7. doi: 10.1186/1471-2156-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip VM, Duvvuru S, Gomero B, Ansah TA, Blaha CD, Cook MN, Hamre KM, Lariviere WR, Matthews DB, Mittleman G, Goldowitz D, Chesler EJ. High-throughput behavioral phenotyping in the expanded panel of BXD recombinant inbred strains. Genes, brain, and behavior. 2010;9(2):129–59. doi: 10.1111/j.1601-183X.2009.00540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomin R, Deary IJ. Genetics and intelligence differences: five special findings. Molecular psychiatry. 2015;20(1):98–108. doi: 10.1038/mp.2014.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards KS, Bommert K, Szabo G, Miles R. Differential expression of Na+/K+-ATPase alpha-subunits in mouse hippocampal interneurones and pyramidal cells. The Journal of physiology. 2007;585(Pt 2):491–505. doi: 10.1113/jphysiol.2007.144733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson R, Toso L, Abebe D, Spong CY. Altered expression of KIF17, a kinesin motor protein associated with NR2B trafficking, may mediate learning deficits in a Down syndrome mouse model. American journal of obstetrics and gynecology. 2008;198(3):313, e1–4. doi: 10.1016/j.ajog.2008.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh-Manoux A, Kivimaki M, Glymour MM, Elbaz A, Berr C, Ebmeier KP, Ferrie JE, Dugravot A. Timing of onset of cognitive decline: results from Whitehall II prospective cohort study. Bmj. 2012;344:d7622. doi: 10.1136/bmj.d7622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia-Arancibia L, Aliaga E, Silhol M, Arancibia S. New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res Rev. 2008;59(1):201–20. doi: 10.1016/j.brainresrev.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Taylor BA. Recombinant inbred strains: use in genetic mapping. In: HM III, editor. Origins of Inbred Mice. Academic Press; New York: 1978. pp. 423–38. [Google Scholar]
- Taylor BA, Wnek C, Kotlus BS, Roemer N, MacTaggart T, Phillips SJ. Genotyping new BXD recombinant inbred mouse strains and comparison of BXD and consensus maps. Mammalian genome: official journal of the International Mammalian Genome Society. 1999;10(4):335–48. doi: 10.1007/s003359900998. [DOI] [PubMed] [Google Scholar]
- Tovote P, Fadok JP, Luthi A. Neuronal circuits for fear and anxiety. Nature reviews Neuroscience. 2015;16(6):317–31. doi: 10.1038/nrn3945. [DOI] [PubMed] [Google Scholar]
- Valentinuzzi VS, Kolker DE, Vitaterna MH, Shimomura K, Whiteley A, Low-Zeddies S, Turek FW, Ferrari EA, Paylor R, Takahashi JS. Automated measurement of mouse freezing behavior and its use for quantitative trait locus analysis of contextual fear conditioning in (BALB/cJ x C57BL/6J)F2 mice. Learning & memory. 1998;5(4–5):391–403. [PMC free article] [PubMed] [Google Scholar]
- Wang J, Gallagher D, DeVito LM, Cancino GI, Tsui D, He L, Keller GM, Frankland PW, Kaplan DR, Miller FD. Metformin activates an atypical PKC-CBP pathway to promote neurogenesis and enhance spatial memory formation. Cell Stem Cell. 2012;11(1):23–35. doi: 10.1016/j.stem.2012.03.016. [DOI] [PubMed] [Google Scholar]
- Wang X, Pandey AK, Mulligan MK, Williams EG, Mozhui K, Li Z, Jovaisaite V, Quarles LD, Xiao Z, Huang J, Capra JA, Chen Z, Taylor WL, Bastarache L, Niu X, Pollard KS, Ciobanu DC, Reznik AO, Tishkov AV, Zhulin IB, Peng J, Nelson SF, Denny JC, Auwerx J, Lu L, Williams RW. Joint mouse-human phenome-wide association to test gene function and disease risk. Nat Commun. 2016;7:10464. doi: 10.1038/ncomms10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehner JM, Radcliffe RA, Rosmann ST, Christensen SC, Rasmussen DL, Fulker DW, Wiles M. Quantitative trait locus analysis of contextual fear conditioning in mice. Nature genetics. 1997;17(3):331–4. doi: 10.1038/ng1197-331. [DOI] [PubMed] [Google Scholar]
- Wellenreuther M, Hansson B. Detecting Polygenic Evolution: Problems, Pitfalls, and Promises. Trends in genetics: TIG. 2016;32(3):155–64. doi: 10.1016/j.tig.2015.12.004. [DOI] [PubMed] [Google Scholar]
- Williams EG, Auwerx J. The Convergence of Systems and Reductionist Approaches in Complex Trait Analysis. Cell. 2015;162(1):23–32. doi: 10.1016/j.cell.2015.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RW, Gu J, Qi S, Lu L. The genetic structure of recombinant inbred mice: high-resolution consensus maps for complex trait analysis. Genome biology. 2001;2(11):RESEARCH0046. doi: 10.1186/gb-2001-2-11-research0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisdom NM, Callahan JL, Hawkins KA. The effects of apolipoprotein E on non-impaired cognitive functioning: a meta-analysis. Neurobiology of aging. 2011;32(1):63–74. doi: 10.1016/j.neurobiolaging.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Wurschum T, Kraft T. Cross-validation in association mapping and its relevance for the estimation of QTL parameters of complex traits. Heredity. 2014;112(4):463–8. doi: 10.1038/hdy.2013.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Statistical applications in genetics and molecular biology. 2005;4 doi: 10.2202/1544-6115.1128. Article 17. [DOI] [PubMed] [Google Scholar]
- Zornetzer SF, Thompson R, Rogers J. Rapid forgetting in aged rats. Behavioral and neural biology. 1982;36(1):49–60. doi: 10.1016/s0163-1047(82)90234-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genetic variants in Hp1bp3 across the BXDs.
All sequence polymorphisms and structural variants (insertions/deletions) between the reference sequence (B6) and parental strain D2 according to the Sanger Institute’s Mouse Genomes Project. Those variants predicted to have ‘moderate’ impact on transcript structure, splicing, and/or protein function according to the Ensembl Variant Effect Predictor (McLaren, et al., 2010) are highlighted in red.
GSEA input.
All genes significantly correlated (uncorrected p ≤ 0.05) with Hp1bp3 in the hippocampus of aged BXD strains. The genes were ranked according to correlation coefficient and used for Gene Set Enrichment Analysis according to established methods (Mootha, et al., 2003; Subramanian, et al., 2005)