

Abstract
Employing a scaffold hopping approach, a series of allosteric HIV-1 integrase (IN) inhibitors (ALLINIs) have been synthesized based on an indole scaffold. These compounds incorporate the key elements utilized in quinoline-based ALLINIs for binding to the IN dimer interface at the principal LEDGF/p75 binding pocket. The most potent of these compounds displayed good activity in the LEDGF/p75 dependent integration assay (IC50 = 4.5 μM) and, as predicted based on the geometry of the five-versus six-membered ring, retained activity against the A128T IN mutant that confers resistance to many quinoline-based ALLINIs.
Keywords: HIV integrase inhibitor, ALLINI, Scaffold hopping
Graphical abstract
HIV-1 integrase (IN) plays a key role in viral replication by catalyzing the insertion of viral DNA into the host genome. The entire process is mediated by the well-ordered assembly of a stable synaptic complex (SSC) formed via ordered multimerization of HIV IN into a tetramer on the viral DNA.1 Based on the importance of HIV-1 IN for the viral infection, there has been significant interest in developing compounds which are capable of disrupting IN function. Much of the early work on IN inhibitors was focused on the discovery and optimization of active site inhibitors capable of coordination to divalent metal ions in the catalytic site.1,2 These compounds are best represented by the class of integrase strand transfer inhibitors (INSTIs) and include three FDA approved drugs, raltegravir, elvitegravir, and the second-generation INSTI dolutegravir.3-5 More recently, a number of allosteric HIV-1 IN inhibitors (ALLINIs), alternatively referred to as LEDGINs, non-catalytic site IN inhibitors (NCINIs), and IN-LEDGF allosteric inhibitors (INLAIs), have been reported.6-11 These compounds bind to the IN dimer at the principal LEDGF/p75 binding site and exhibit a multimodal mechanism of action.6,8,10,12-15 ALLINIs induce aberrant IN multimerization as well as inhibit IN binding to LEDGF/p75. These compounds possess two conserved binding elements, an aromatic ring which projects into a hydrophobic channel formed by helices at the interface of the two integrase subunits and a substituted acetic acid side chain responsible for forming a network of hydrogen bonds and electrostatic interactions with the E170, H171, and T174 residues.6,13,16 To date, these functional groups have been effectively appended to six-membered heterocyclic rings, including the quinoline core scaffold of BI-1001 (Figure 1).15 Although these compounds and many others have shown promising inhibitory activity, various resistance mutations, including the A128T IN mutation, have been observed for many of the reported quinoline-based ALLINIs.6,13
Figure 1.

A docking model overlay of BI-1001 (purple) and an indole analogue (white) in the LEDGF/p75 binding pocket to HIV-1 IN dimer interface. Individual IN subunits are shown in green and blue.
In an effort to more fully explore the structure-activity relationships of the ALLINIs and potentially attenuate resistance to known mutants, the central scaffold of these compounds was identified as a potential site for structural manipulation. Specifically, we wished to examine whether a scaffold hopping17 approach could be applied to this class through transfer of the key binding elements from a six-membered to a five-membered heterocyclic ring and whether this new ring system could effectively orient these groups in the LEDGF/p75 binding site. In analogy to the quinoline core found in many ALLINIs, the indole presents a similar bioisosteric benzannular nitrogen-containing framework, despite significant differences in electron density, relative geometry, and H-bonding capabilities. Synthetically, however, there are several potential benefits to employing an indole in the synthesis of ALLINI analogues, namely, the wide array of existing methods for the preparation of the core18 and the relative ease of functionalization around the periphery of this scaffold, both of which could be expected to further the development of new ALLINI analogues. With this in mind, a computational docking model of an indole based analogue with HIV-1 IN was examined using AutoDock 4.0 (Figure 1).19 This model predicted that indole-based analogues would maintain a binding mode similar to that of the prototypical quinoline-based ALLINI, BI-1001. Of note, as predicted in our preliminary structural analysis, the docking model indicates that the geometry of the smaller five-membered ring of the indole imparts a modest effect on the relative positioning of the core in the binding pocket. While the key acetic acid and aryl substituents are predicted to overlay well in both the quinoline and indole systems, a slight “tilt” of the indole aromatic ring away from the A128 residue relative to the quinoline ring system is observed, suggesting that the indole analogues may not interact with that site or be affected by the A128T mutation which confers marked resistance to quinoline-based ALLINI analogues.13 Encouraged by these results, we pursued the synthesis of a series of indole analogues to experimentally test these predictions.
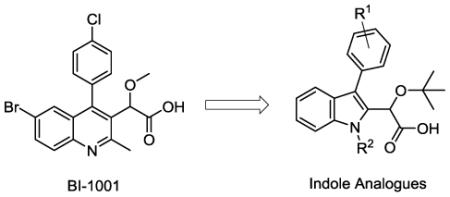
Key considerations in the development of these analogues were the efficiency of the route and the ability to easily introduce structural variation at the C3 substituent. Methods previously utilized to introduce functionality at this position in both the quinoline and pyridine systems have relied upon coupling reactions, typically a Suzuki coupling, to introduce the aryl substituents.9 A new strategy for the functionalization of the indole core could utilize alternative reactivity, ultimately facilitating the incorporation of other functional groups at that position. With this in mind, the inexpensive, commercially available reagent isatin was employed as a starting material for the synthesis (Scheme 1) and was quickly transformed using well-established methodology to the C3-substituted indole via nucleophilic addition of Grignard or aryllithium reagents and subsequent hydride reduction.20 Alkylation of the indole nitrogen was then carried out using either iodomethane (compounds 2a-d) or benzyl bromide (compound 2e). Installation of the critical acetic acid side chain was then accomplished through acylation of the electron rich indole ring system with oxalyl chloride followed by the addition of methanol to give ester 3.21 Selective reduction of the ketone with sodium borohydride cleanly provided alcohol 4. Alkylation using the resin bound acid, Amberlyst 15, successfully generated the desired t-butyl ether, although other conditions established for the introduction of the t-butyl ether functionality including the use of either t-BuOAc/perchloric acid or t-butylacetimidate have also been successfully applied to the indole system.13,22 Finally, saponification of the methyl ester furnished the sodium salt of the indole analogues 5a-e. Acidification of the salts to pH ≈ 4 provided the acids, but further lowering of the pH resulted in degradation of the compounds, presumably due to the potential reactivity of the indole ring in combination with the instability of the t-butyl group under these conditions. Due to the sensitivity of the indole system and the low yields obtained in the saponification and subsequent acidification, in several cases the acids were not protonated and the isolated carboxylates (sodium salts) were directly submitted for biological evaluation. The acidification was not considered critical in this case due to the use of neutral buffer conditions for the bioassay in which the molecule exists as the carboxylate anion. In total, five indoles (5a-e) were synthesized by employing this seven step sequence from isatin.
Scheme 1.
Method for preparation of indole analogues 5a-5e.
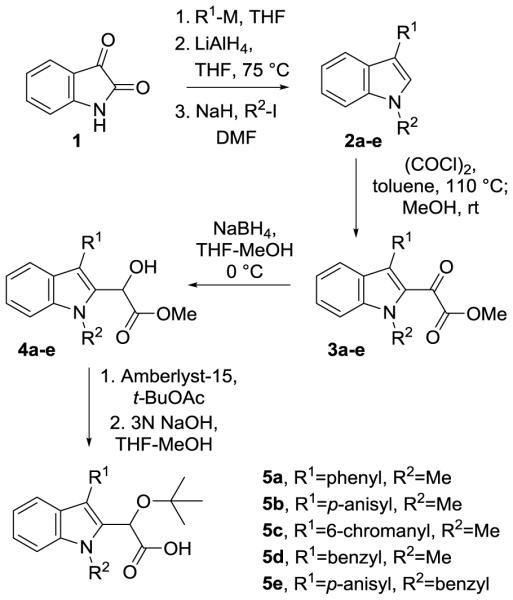
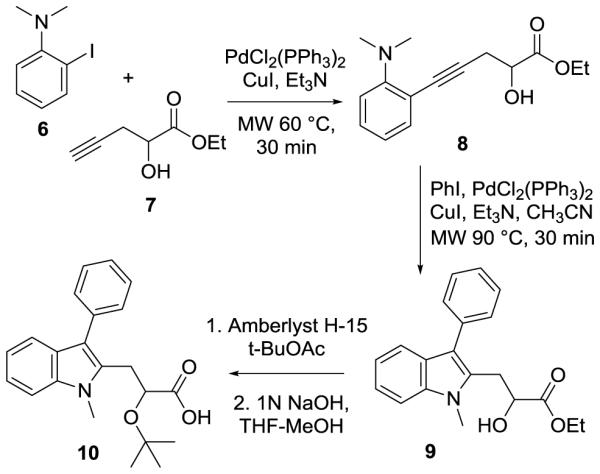
In addition to indoles 5a-e, two additional structural variants were synthesized to explore the synthetic flexibility and structure-activity relationships of this system. The first of these was the homologation of the acetic acid side chain to prepare compound 10 (Scheme 2). The extension of the chain was expected to provide more flexibility in the binding of the two key functional groups to IN and to affect the hydrogen bonding of the compound. Compound 10 was prepared through application of the chemistry of Larock.23 Upon coupling of alkyne 7 to iodoaniline 6, the indole was cyclized and the phenyl group appended to the C3 position under microwave conditions in the presence of palladium. The formation of the t-butyl ether and subsequent saponification were carried out in a manner analogous to the preparation of previous derivatives, generating a structural analogue of 5a. The second structural variant took advantage of functional group reversal (Scheme 3), a technique previously seen in indole containing compounds like indomethacin/clometacin and serotonin.24 For the preparation of compound 15 (Scheme 3), the anisole group was introduced directly onto 3-methylindole (11) via Ullmann coupling with iodoanisole (12). At that stage, the introduction of the acetic acid side chain once again could be accomplished as shown for the transformation of 2-5 to provide compound 15, which possesses the same functional groups as analogue 5b, in a total of five synthetic steps and with an overall yield of nearly 35% from inexpensive commercial reagents.
Scheme 2.
Synthesis of the homologated acid 10.
Scheme 3.
Functional inversion of indole leading to the preparation of 15.
The synthesized analogues were tested in a homogenous time-resolved fluorescence (HTRF)-based LEDGF/p75-dependent integration assay.13,15 The advantage of this assay is its ability to capture IN inhibitors with different modes of action. For example, compounds that impair IN catalytic activity, induce aberrant IN multimerization, or inhibit IN-LEDGF/p75 binding all can be identified using this approach. 13,15 Comparative analysis of the IC50 values (Table 1) for the indole-based compounds synthesized above have revealed 5c (entry 3) as the most promising lead compound with an IC50 value of 4.5 μM. Additional trends in this series of compounds, however, can also be observed to provide structure-activity relationships within this class. For example, entries 1-3 demonstrate the important role that substitution of the C3 aromatic ring plays on compound activity, with both the electron rich and slightly larger anisole and chromane rings resulting in increased activity over the unsubstituted phenyl ring. Surprisingly, the more flexible benzyl moiety was also reasonably well tolerated at the C3 position (5d, entry 4), giving similar results to the anisole in entry 2. Introduction of a benzyl group at the N1 position (5e, entry 5), however, was not well tolerated, resulting in a two-fold reduction in activity as compared to entry 2. The compounds containing the homologated acetic acid side chain (10, entry 6) and the inversion of the indole ring system (15, entry 7) failed to show any activity in the assay at concentrations up to 100 μM. Although the relative lack of activity for compound 15 is not clear, the extension of the acid chain in compound 10 likely affects the ability of the molecule to achieve efficient hydrogen bonding with the protein through the carboxylic acid moiety while simultaneously maintaining the proper orientation of the aryl ring in the binding site, a potential consequence of the increased length and conformational flexibility in this linker.
Table 1.
Respective IC50 values of indole-based compounds evaluated in LEDGF/p75-dependent integration assays.
| Entry | Compound |
IC50 for LEDGF/p75-
dependent integration (μM) |
|---|---|---|
| 1 | 5a | 44.1 ± 3.7 |
| 2 | 5b | 32.0 ± 2.5 |
| 3 | 5c | 4.5 ± 0.5 |
| 4 | 5d | 31.3 ± 0.9 |
| 5 | 5e | 59.1 ± 3.4 |
| 6 | 10 | >100 |
| 7 | 15 | >100 |
| 8 | BI-1001 13 | 1.3 ± 0.34 |
Based on the promising activity displayed by 5c in the LEDGF/p75 dependent integration assay, however, further mechanism of action studies were carried out using this compound. This work included additional HTRF-based assays that allow monitoring of LEDGF/p75-independent IN 3’-processing activity, IN binding to LEDGF/p75, and aberrant IN multimerization.15 The results of these assays, summarized in Table 2, indicate that 5c exhibits a multimodal mechanism of action. In particular, this compound not only effectively inhibited LEDGF/p75-dependent integration, but also LEDGF/p75-independent 3’-processing activity of IN. Furthermore, 5c induced aberrant IN multimerization, which could explain its ability to inhibit LEDGF/p75-independent 3’-processing activity of IN. In addition, 5c was able to inhibit IN-LEDGF/p75 binding, albeit at a somewhat higher concentration. As one may expect, this multimodal mechanism of action is reminiscent of the mechanism of action of quinoline-based ALLINI compounds such as BI-1001.13,15 Due to the similarity in activity to the quinoline-based ALLINIs, 5c was also screened against the single amino-acid A128T substitution in HIV-1 IN that confers marked resistance to quinoline-based inhibitors, including BI-1001.13 In sharp contrast to BI-1001, 5c promoted aberrant multimerization of both wild type and the A128T mutant INs with similar IC50 values (Figure 2). Likewise, 5c also showed similar inhibition of 3’-processing activity in both the wild type and A128T mutant with IC50 values of 3.2 and 0.9 μM, respectively.
Table 2.
Mechanistic investigation of compound 5c against WT and A128T IN (IC50 and EC50 values in uM).
| IC50 for LEDGF/p75 dependent Integration |
IC50 for LEDGF/p75 independent 3’-proc. activity |
EC50 for aberrant IN multimerization |
IC50 for IN- LEDGF/p75 Binding |
|
|---|---|---|---|---|
| WT | 4.5 ± 0.5 | 3.2 ± 0.5 | 16.1 ± 2.0 | 28.7 ± 3.2 |
| A128T | 0.9 ± 0.02 | 16.0 ± 1.4 |
Figure 2.
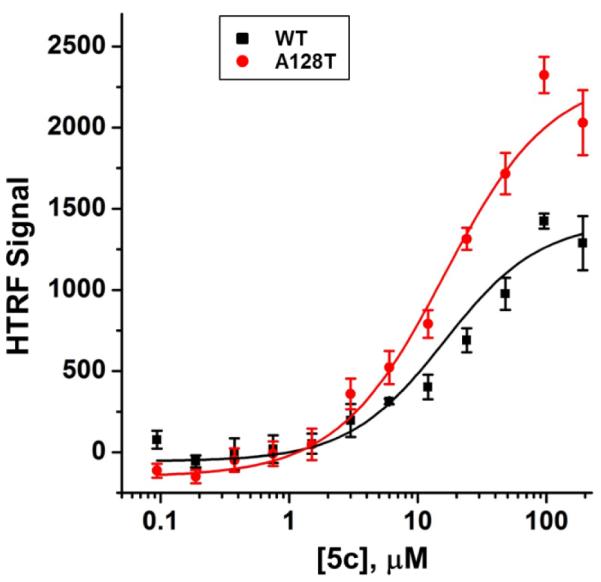
Dose response curves for 5c promoting aberrant multimerization of WT (black squares) and A128T (red circles) INs. Error bars indicate SD from three independent experiments. IC50 values for wild type and A128T INs were 16.1 ± 2.0 μM and 16.0 ± 1.4 μM, respectively.
To understand the structural basis for the mode of action of indole-based compounds the crystal structures of 5c bound to both the wild type and A128T IN CCDs have been solved. The x-ray crystallography studies have revealed that 5c binds at the IN dimer interface in the principal LEDGF/p75 binding pocket (Figure 3). Similar to its quinoline-based counterparts, 5c is anchored to the protein through the hydrogen bonding interactions between the carboxylic acid moiety and the backbone amides of Glu170 and His171 of subunit 2 with the t-butyl ether
Figure 3.

The structural orientation of compound 5c bound to the HIV1 Integrase CCD dimer in the presence and absence of the resistance A128T mutation. (A) The crystal structure of 5c in complex with CCD (PDB ID: 5KGW). The two CCD subunits are colored cyan and green with the electrostatic interaction and hydrogen bonds represented as black dashes. (B) Comparison of BI-1001 (PDB ID: 4DMN) and 5c binding to the CCD. (C) The binding of 5c is unchanged when comparing the overlay of 5KGW with the crystal structure of 5c bound to CCD containing the A128T substitution (PDB ID: 5KGX). The subunits of 5KGX are colored yellow and grey with 5c also colored in yellow.
oxygen atom providing additional hydrogen bonding to Thr174 (Figure 3A).13,16 At the same time, comparison of the binding sites of indole-based 5c and quinoline-based BI-1001 (Figure 3B) reveals important differences between these compounds. The quinoline ring system of BI-1001 extends toward the Ala128 residue in subunit 1 of IN. Accordingly, the replacement of Ala 128 with bulkier and polar Thr creates steric and electrostatic repulsions to BI-1001, and results in repositioning of BI-1001 away from subunit 1 as demonstrated in previous x-ray crystallographic studies.13 In contrast, very similar binding orientations of 5c have been observed in both the wild type and A128T CCD-CCD (Figure 3C) crystal structures. As predicted by the computational docking model, the five-membered indole ring structure of 5c mitigates the repulsion induced by the A128T mutation by tilting the aromatic ring away from the 128 residue of subunit 1, thus maintaining similar activities against both the WT and A128T proteins.
In conclusion, we have developed indole-based allosteric inhibitors of HIV-1 integrase. Similar to previously reported quinoline-based compounds, the indole-based compounds bind to the IN dimer interface at the LEDGF/p75 binding pocket and exhibit a multimodal mechanism of action. The five-membered indole ring, however, allows this class of compounds to avoid steric and electrostatic repulsions by the A128T substitution that leads to marked resistance to many quinoline-based ALLINIs. In addition, the synthetic versatility provided by the indole core provides numerous opportunities for functionalization and structural manipulation in the development of analogues. Therefore, despite the relatively lower potencies of this initial series of indole-based compounds as compared to the previously reported quinoline-based compounds, these indoles represent promising leads for the development of second generation of ALLINIs.
Supplementary Material
Acknowledgements
The authors wish to acknowledge support for this project from the National Institutes of Health grant (R21AI110270) and graduate support through the Egyptian Channel System to Y.M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Supplementary data associated with this article including experimental procedures and spectral data can be found, in the online version, at http://dx.doi.org/##.####/j.bmcl.2016.##.###.
References and notes
- 1.Lesbats P, Engelman AN, Cherepanov P. Chem. Rev. 2016 doi: 10.1021/acs.chemrev.6b00125. Article ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di Santo R. J. Med. Chem. 2014;57:539. doi: 10.1021/jm400674a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, Fiore F, Gardelli C, Paz OG, Hazuda DJ, Jones P, Kinzel O, Laufer R, Monteagudo E, Muraglia E, Nizi E, Orvieto F, Pace P, Pescatore G, Scarpelli R, Stillmock K, Witmer MV, Rowley M. J. Med. Chem. 2008;51:5843. doi: 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- 4.Shimura K, Kodama E, Sakagami Y, Matsuzaki Y, Watanabe W, Yamataka K, Watanabe Y, Ohata Y, Doi S, Sato M, Kano M, Ikeda S, Matsuoka M. J. Virol. 2008;82:764. doi: 10.1128/JVI.01534-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bailly F, Cotelle P. Expert Opin. Drug Discov. 2015;10:1243. doi: 10.1517/17460441.2015.1064896. [DOI] [PubMed] [Google Scholar]
- 6.Christ F, Voet A, Marchand A, Nicolet S, Desimmie BA, Marchand D, Bardiot D, Van der Veken NJ, Can Remoortel B, Strelkov SV, De Maeyer M, Chaltin P, Debyzer Z. Nat. Chem. Biol. 2010;6:442. doi: 10.1038/nchembio.370. [DOI] [PubMed] [Google Scholar]
- 7.Fader LD, Malenfant E, Parisien M, Carson R, Bilodeau F, Landry S, Pesant M, Brochu C, Morin S, Chabot C, Halmos T, Bousquet Y, Bailey MD, Kawai SH, Coulombe R, LaPlante S, Jakalian A, Bhardwaj PK, Wernic D, Schroeder P, Amad M, Edwards P, Garneau M, Duan J, Cordingley M, Bethell R, Mason SW, Bos M, Bonneau P, Poupart MA, Faucher AM, Simoneau B, Fenwick C, Yoakim C, Tsantrizos Y. ACS Med. Chem. Lett. 2014;5:422. doi: 10.1021/ml500002n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsiang M, Jones GS, Niedziela-Majka A, Kan E, Lansdon EB, Huang W, Hung M, Samuel D, Novikov N, Xu Y, Mitchell M, Guo H, Babaoglu K, Liu X, Geleziunas R, Sakowicz R. J. Biol. Chem. 2012;287:21189. doi: 10.1074/jbc.M112.347534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharma A, Slaughter A, Jena N, Feng L, Kessl JJ, Fadel HJ, Malani N, Male F, Wu L, Poeschla E, Bushman FD, Fuchs JR, Kvaratskhelia M. PLoS Pathog. 2014;10:e1004171. doi: 10.1371/journal.ppat.1004171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le Rouzic E, Bonnard D, Chasset S, Bruneau JM, Chevreuil F, Le Strat F, Nguyen J, Beauvoir R, Amadori C, Brias J, Vomscheid S, Eiler S, Levy N, Delelis O, Deprez E, Saib A, Zamborlini A, Emiliani S, Ruff M, Ledoussal B, Moreau F, Benarous R. Retrovirology. 2013;10:144. doi: 10.1186/1742-4690-10-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Bel N, va der Velden Y, Bonnard D, Le Rouzic E, Das AT, Benarous R, Berkhout B. PloS One. 2014;9:e103552. doi: 10.1371/journal.pone.0103552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jurado KA, Wang H, Slaughter A, Feng L, Kessl JJ, Koh Y, Wang W, Ballandras-Colas A, Patel PA, Fuchs JR, Kvaratskhelia M, Engelman A. Proc. Natl. Acad. Sci. U.S.A. 2013;110:8690. doi: 10.1073/pnas.1300703110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng L, Sharma A, Slaughter A, Jena N, Koh Y, Shkribai N, Larue RC, Patel PA, Mitsuya H, Kessl JJ, Engelman A, Fuchs JR, Kvaratskhelia M. J. Biol. Chem. 2013;288:15813. doi: 10.1074/jbc.M112.443390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupta K, Brady T, Dyer BM, Malani N, Hwang Y, Male F, Nolte RT, Wang L, Velthuisen E, Jeffrey J, Van Duyne GD, Bushman FD. J. Biol. Chem. 2014;289:20477. doi: 10.1074/jbc.M114.551119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kessl JJ, Jena N, Koh Y, Taskent-Sezgin H, Slaughter A, Feng L, de Silva S, Wu L, Le Grice SF, Engelman A, Fuchs JR, Kvaratskhelia M. J. Biol. Chem. 2012;287:16801. doi: 10.1074/jbc.M112.354373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Slaughter A, Jurado KA, Deng N, Feng L, Kessl JJ, Shkriabai N, Larue RC, Fadel HJ, Patel PA, Jena N, Fuchs JR, Poeschla E, Levy RM, Engelman A, Kvaratskhelia M. Retrovirology. 2014;11:100. doi: 10.1186/s12977-014-0100-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao H. Drug Disc. Today. 2007;12:149. doi: 10.1016/j.drudis.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Taber DF, Tirunahari PK. Tetrahedron. 2011;67:7195. doi: 10.1016/j.tet.2011.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. J. Comput. Chem. 2009;16:2785. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hewawasam P, Erway M, Moon SL, Knipe J, Weiner H, Boissard CG, Post-Munson DJ, Gao Q, Huang S, Gribkoff VK, Meanwell NA. J. Med. Chem. 2002;45:1487. doi: 10.1021/jm0101850. [DOI] [PubMed] [Google Scholar]
- 21.Jones AW, Bambang P, Bowyer PK, Mitchell PSR, Kumar N, Nugent SJ, Jolliffe KA, Black D. Tetrahedron. 2004;60:10779. [Google Scholar]
- 22.Fandrick KR, Li W, Zhang Y, Tang W, Gao J, Rodriguez S, Patel ND, Reeves DC, Wu JP, Sanyal S, Gonnella N, Qu B, Haddad N, Lorenz JC, Sidhu K, Wnag J, Ma S, Grinberg N, Lee H, Tsantrizos Y, Poupart MA, Busacca CA, Yee NK, Lu BZ, Senanayake CH. Angew. Chem. Int. Ed. Engl. 2015;54:7144. doi: 10.1002/anie.201501575. [DOI] [PubMed] [Google Scholar]
- 23.Chen Y, Markina NA, Larock RC. Tetrahedron. 2009;65:8908. doi: 10.1016/j.tet.2009.07.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bos M, Jenck F, Martin JR, Moreau JL, Sleight AJ, Wichmann J, Widmer U. J. Med. Chem. 1997;40:2762. doi: 10.1021/jm970030l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.