

Abstract
Chronic inflammatory demyelinating polyneuropathy (CIDP) is the most common chronic autoimmune neuropathy. While both cell‐mediated and humoral mechanisms contribute to its pathogenesis, the rapid clinical response to plasmapheresis implicates a circulating factor responsible for peripheral nerve injury. We report that treatment‐naïve patients with CIDP show increased serum and CSF levels of the anaphylatoxin C5a and the soluble terminal complement complex (sTCC). Systemic terminal complement activation correlates with clinical disease severity as determined by the Inflammatory Neuropathy Cause and Treatment (INCAT) disability scale. These data indicate that complement activation contributes to peripheral nerve injury and suggest that complement inhibition should be explored for its potential therapeutic merit in CIDP.
Introduction
Chronic inflammatory demyelinating polyneuropathy (CIDP) is the most prevalent chronic autoimmune neuropathy and the most common autoimmune‐mediated disease of the peripheral nervous system.1, 2 Both cell‐mediated and humoral mechanisms contribute to its pathogenesis and the rapid clinical response to plasmapheresis therapy implicates a circulating factor responsible for peripheral nerve tissue injury in patients with CIDP.1, 2 Consistent with this hypothesis, complement‐fixing IgG and IgM deposits are found on the myelin sheath in sural nerve biopsies from patients with CIDP.3, 4 Furthermore, sera derived from patients who responded well to plasma exchange were shown to induce demyelination and reduction of conduction velocity if adoptively transferred to nonhuman primates or intraneurally injected into rat sciatic nerves.5, 6 Most patients with CIDP lack detectable titers of antibodies specific for major compact myelin proteins which suggests that serum constituents other than myelin‐directed antibodies, such as noncompact myelin‐specific antibodies, cytokines, or components of the complement cascade might contribute to peripheral nerve injury.1, 2, 7
Complement activation has long be thought to constitute a potential pathogenic mechanism in CIDP since the complement component C3d was found deposited on the outer surface of Schwann cells and the compact myelin in biopsies from CIDP patients.3, 4 Its proinflammatory function is reflected by the ability of terminal complement components such as C5a to recruit myeloid cells such as macrophages to sites of inflammation through complement receptors and in inducing tissue injury through formation of the terminal complement complex (TCC), that is, membrane attack complex.
Koski and colleagues reported that serum TCC levels are increased in patients with Guillain–Barré syndrome (GBS), decline with clinical improvement and become undetectable 1 month after onset of symptoms.8 They also reported elevated TCC serum levels in six out of seven patients diagnosed with chronic recurrent polyneuritis8. In patients with GBS, membrane bound TCC components were found to be deposited on the abaxonal Schwann cell surface8, 9. Hartung et al. reported elevated C3a und C5a CSF levels in patients with GBS if compared to patients with noninflammatory neurological diseases.10 We could previously show that the presence of serum IgG antibodies enriched for IgG‐Fc glycovariants that efficiently activate the complement cascade is associated with disease activity in patients with CIDP.11 Here, we hypothesized that complement activation is increased in CIDP patients and associated with disease severity.
Patients and Methods
Patients
Samples and clinical data were collected between 2010 and 2015 at the Department of Neurology, University of Marburg, Germany. All patients with CIDP fulfilled the European Federation of Neurological Societies/Peripheral Nerve Society (EFNS/PNS) diagnostic criteria 12 and were newly diagnosed with an interval between onset of symptoms and blood/CSF draw of <3 months.
Patients showed symmetrical, proximal and distal sensory and motor deficits and were therefore classified as idiopathic/typical CIDP. Control samples were collected from 29 patients with noninflammatory neurological disease conditions (tension‐type or hypertension‐induced headache, movement disorders, stenosis of the lumbal spinal canal, noninflammatory polyneuropathies). Sera from patients with systemic lupus erythematosus (SLE) were collected in 2016 at the Department of Rheumatology and Clinical Immunology, Charité Universitätsmedizin Berlin, Germany. All patients met the updated and revised criteria proposed by the American College of Rheumatology (ACR) for the diagnosis of SLE13 and received immunomodulatory therapy at the time of blood draw (Table 1). After blood draw, serum was separated by centrifugation at 4°C and immediately frozen at −80°C within 30 min of venipuncture. CSF collected from both patients with CIDP and controls was also frozen within 30 min of collection and stored at −80°C. To assess treatment response, the modified INCAT score 14, 15 was used routinely before and 4 weeks after IVIG‐treatment (Gamunex‐C (Grifols) and Privigen (CSL Behring); 2 g/kg body weight over 5 consecutive days). The study was approved by the local Institutional Review Boards and all subjects provided informed consent.
Table 1.
Demographic an clinical characteristics of CIDP patients and their controls
| CIDP n = 21 | Controls n = 29 | SLE n = 25a | |
|---|---|---|---|
| Age (Years ± SD; range) | 64 ± 13 (38–80) | 49 ± 19 (21–81) | 43 ± 14 (23–71) |
| Male to female ratio | 1.3 | 1.3 | 0.09 |
| Fulfilling modified AAN criteria, n; % | 21; 100 | NA | NA |
| Fulfilling EFNS/PNS criteria, n; % | 21; 100 | NA | NA |
| Clinical course | NA | NA | |
| CP, n; % | 19; 90.5 | – | NA |
| Monophasicb, n; % | 2; 9.5 | – | NA |
| CIDP subtype | NA | NA | |
| CIDP‐I, n; % | 19; 90.5 | – | NA |
| CIDP‐MGUS, n; % | 2; 9.5 | – | NA |
| Treatment responsec, n; % | 16; 80 | NA | NA |
AAN, American Academy of Neurology; CP, chronic progressive; CIDP, chronic inflammatory demyelinating polyneuropathy CIDP‐I, idiopathic CIDP; CIDP‐MGUS, CIDP with monoclonal gammopathy of uncertain significance; EFNS, European Federation of Neurological Societies; NA, not applicable; PNS, Peripheral Nerve Society; SD, standard deviation; SLE, systemic lupus erythematosus.
All patients with SLE received immunomodulatory/immunosuppressive treatment such as belimumab (n = 9/25), a monoclonal antibody targeting the soluble human B lymphocyte stimulator protein BLyS, low‐dose glucocorticosteroids (n = 21/25), azathioprine (n = 21/25), low‐dose methotrexate (n = 3/25), mycophenolate mofetil (n = 3/25), and cyclosporine (n = 1/25).
But no Guillain–Barré syndrome.
Defined as ≥1 point decrease in the modified Rankin’ scale. No pretreatment data was available for one patient.
ELISA
C5a and TCC levels in serum and CSF samples were quantified by ELISA (Tecomedical AG, Sissach, Switzerland) according to the manufacturer's recommendations. CSF indices for C5a and sTCC were calculated as follows: (C5a or sTCCCSF/C5a or sTCCserum)/(albuminCSF/albuminserum).
Statistics
Protein expression levels in serum and CSF samples were compared using the nonparametric Mann–Whitney U test. Correlation analyses were performed using the Spearman's rank correlation coefficient. A P < 0.05 was considered significant. All graphs and statistics were done using GraphPad Prism 5 (GraphPad Software, Inc.; La Jolla, CA).
Results
A total of 21 newly diagnosed and treatment‐naïve patients with CIDP fulfilling the EFNS/PNS diagnostic criteria12 and 29 controls with noninflammatory neurological disease conditions were included in the analysis (Table 1). All patients underwent neurological examination at the time of diagnosis, serum and CSF were collected before induction of immunomodulatory therapy with intravenous immunoglobulin (2 g/kg body weight over 5 consecutive days) and patients were re‐examined after 4 weeks. Complement activation can be triggered by various stimuli including immune complexes, extracellular matrix proteins and apoptotic/necrotic cells17. Complement activating pathways converge to the generation of the anaphylatoxin C5a and the TCC, generated by the assembly of C5b through C9. The C5b‐9 that fails to assemble in the membrane forms a soluble, lytically inert complex, termed sTCC, and complement‐mediated cell lysis was shown to closely correlate with the generation of sTCC levels in vitro.16 Here, terminal complement activation was quantified in serum and CSF samples by ELISAs specific for C5a and sTCC. Compared to controls, patients with CIDP showed significantly higher levels of C5a and sTCC in both serum and CSF (Fig. 1). C5a serum levels higher than 95.6 ng/mL were exclusively observed in patients with CIDP. We additionally analyzed C5a and sTCC serum levels in patients with SLE receiving immunomodulatory therapy (Table 1). Compared to patients with CIDP, patients with SLE showed lower levels of C5a (mean: 43.18 ng/mL, SD: 22.68; P < 0.0001) and sTCC (mean: 2782 ng/mL, SD: 1533; P < 0.0001). Increased levels of C5a and sTCC in CSF samples of CIDP patients correlated with CSF albumin concentrations (sTCC: r = 0.82, P = 0.0001; C5a: r = 0.77, P < 0.0001) and CSF indices for C5a and TCC were lower in patients with CIDP compared to controls (P < 0.0001 for C5a and P < 0.012 for sTCC) suggesting a systemic increase of terminal complement activation as opposed to local production within the CSF compartment. Serum levels of albumin, chosen as control protein, were unchanged in patients with CIDP (Fig. 1). Thus, systemic terminal complement activation as reflected by C5a and sTCC and levels is increased in treatment‐naïve patients with CIDP.
Figure 1.
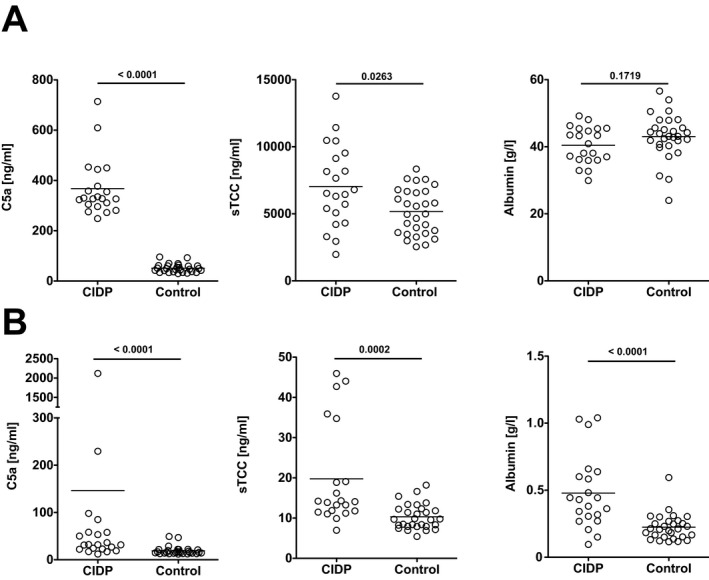
Increased terminal complement activation in serum and CSF of CIDP patients. Shown are (A) serum and (B) CSF levels of C5a, sTCC and albumin in treatment‐naïve patients with CIDP compared to patients with noninflammatory neurological diseases (controls). Statistics were performed by Mann–Whitney U test. CIDP, Chronic inflammatory demyelinating polyneuropathy; sTCC, soluble terminal complement complex.
We next investigated whether increased terminal complement activation is associated with or anticipates clinical disease severity in patients with CIDP. Disease severity was monitored using the INCAT (Inflammatory Neuropathy Cause and Treatment) disability score 14, 15 during the diagnostic workup and 4 weeks after initiation of IVIG therapy. INCAT disability scores were reduced in 16/21 (80%) patients 4 weeks after IVIG therapy induction. Both C5a and sTCC levels tended to be higher in patients with higher disability at the time of diagnosis (Fig. 2A) and significantly correlated with INCAT scores determined at 4 week follow‐up visits (Fig. 2B). No significant correlation could be observed for clinical disease severity and albumin concentrations (Fig. 2). CSF levels of C5a and TCC at the initial presentation did not significantly correlate with INCAT disability scores at any time point (data not shown). Thus, systemic terminal complement activation is associated with residual disease severity 4 weeks after effective IVIG therapy in patients with CIDP.
Figure 2.
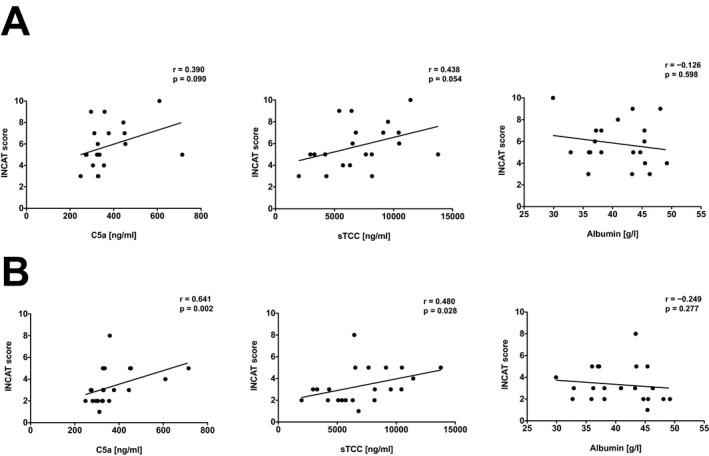
Systemic terminal complement activation correlates with disease severity in patients with CIDP. Shown are Spearman's rank correlation analyses of serum C5a, sTCC and albumin levels with disease severity (defined by INCAT score) of (A) newly diagnosed, previously untreated patients with CIDP and (B) 4 weeks after immunomodulatory therapy with IVIG (2 g/kg body weight over 5 consecutive days). Each dot reflects an individual patient. Displayed are linear regression curves. Statistics were performed by Spearman test. CIDP, Chronic inflammatory demyelinating polyneuropathy; INCAT, Inflammatory Neuropathy Cause and Treatment; sTCC, soluble terminal complement complex.
Discussion
Our study demonstrates that systemic terminal complement activation as defined by serum C5a and TCC levels is increased and associated with disease severity in patients with CIDP. Sera from CIDP patients which, upon adoptive transfer, induced conduction block and demyelination in rat sciatic nerves show IgG and C3d binding to myelin components on normal peripheral nerve tissue, indicating that their demyelinating capacity is antibody‐mediated and complement‐dependent.6 Our data suggest that systemic and local terminal complement activation is a characteristic feature of inflammatory demyelinating polyneuropathies and support a role of complement activation in the pathogenesis of CIDP.
Deficiencies of early components of the classical complement activation pathway such as C1q, C2, and C4 are strongly associated with the development of SLE, presumably due to impaired complement‐mediated clearance of dying cells,17 and low concentrations of complement components are observed in a majority of patients with active and severe SLE. In contrast, terminal complement activation products such as C5a and sTCC, although reported to correlate with clinical disease activity in individual patients, are either normal or only slightly elevated in sera from patients with established SLE.18, 19 The finding that C5a and sTCC levels are substantially higher in patients with CIDP compared to SLE suggest differential mechanisms and functions of terminal complement activation in these diseases, although treatment‐related effects cannot be excluded since all SLE patients included in our study received immunomodulatory therapy.
Terminal complement activation can be inhibited therapeutically, for example, by eculizumab, a humanized monoclonal antibody which blocks the cleavage from C5 to C5a and C5b, thereby preventing the generation of C5a and TCC formation. Eculizumab is approved for the treatment of paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome, was shown to be therapeutically effective in a murine model of acute antibody‐mediated neuropathy20 and is currently tested for its safety and efficacy in patients with GBS. In an open‐label clinical trial conducted in 13 patients with multifocal motor neuropathy (MMN), eculizumab treatment did not show objective clinical benefits, although improvement of some secondary outcomes such as patient‐rated subjective scores and selected clinical and electrophysiological measurements were reported.21 Our data strongly indicate that inhibition of terminal complement activation should be explored for its potential therapeutic merit in patients with CIDP.
There are limitations to our study. First, the relatively small number of patients requires validation in larger independent cohorts. Second, we did not monitor serum C5a and TCC levels over time in order to investigate whether terminal complement activation remains increased during disease progression and under immunomodulatory therapy since transient reduction of complement proteins and/or complement activation occur after plasmapheresis,22 during corticosteroid therapy23 as well as following IVIG treatment,24. A small open‐label clinical trial on eculizumab in patients with MMN who concomitantly received IVIG reported that most patients continued to require IVIG despite full inhibition of terminal complement function 21 suggesting that the therapeutic efficacy of IVIG is mediated by immunomodulatory mechanisms other than complement inhibition. Furthermore, our study was restricted to patients with ideopathic or typical CIDP characterized by symmetrical, proximal and distal sensory and motor deficits which comprise only 50–60% of patients with CIDP.1, 2, 7 Our findings, however, provide incentive to conduct larger prospective investigations to examine terminal complement activation as a potential surrogate marker for clinical disease activity, progression, and response to immunomodulatory therapies in patients with major phenotypic variants of CIDP. It remains to be evaluated whether inhibition of terminal complement activation can be harnessed to improve the clinical outcome in patients with CIDP.
Author Contribution
Conception and design of the study: I.Q., J.D.L.; acquisition and analysis of data: I.Q., C.W.K., F.H., B.T., J.D.L.; drafting of the manuscript and figures: I.Q., C.W.K., F.H., B.T., J.D.L.
Conflict of Interest
The authors declare that there are no financial or other relationships that might lead to a perceived conflict of interest.
Acknowledgments
We thank our patients for their continuous cooperation. Isaak Quast was supported by a DOC scholarship provided by the Austrian Academy of Sciences (ÖAW). Christian W. Keller was supported by a Forschungskredit doctoral fellowship (FK‐14‐021) provided by the University of Zurich.
References
- 1. Koller H, Kieseier BC, Jander S, Hartung HP. Chronic inflammatory demyelinating polyneuropathy. N Engl J Med 2005;352:1343–1356. [DOI] [PubMed] [Google Scholar]
- 2. Dalakas MC. Advances in the diagnosis, pathogenesis and treatment of CIDP. Nat Rev Neurol 2011;7:507–517. [DOI] [PubMed] [Google Scholar]
- 3. Dalakas MC, Engel WK. Immunoglobulin and complement deposits in nerves of patients with chronic relapsing polyneuropathy. Arch Neurol 1980;37:637–640. [DOI] [PubMed] [Google Scholar]
- 4. Hays AP, Lee SS, Latov N. Immune reactive C3d on the surface of myelin sheaths in neuropathy. J Neuroimmunol 1988;18:231–244. [DOI] [PubMed] [Google Scholar]
- 5. Heininger K, Liebert UG, Toyka KV, et al. Chronic inflammatory polyneuropathy. Reduction of nerve conduction velocities in monkeys by systemic passive transfer of immunoglobulin G. J Neurol Sci 1984;66:1–14. [DOI] [PubMed] [Google Scholar]
- 6. Yan WX, Taylor J, Andrias‐Kauba S, Pollard JD. Passive transfer of demyelination by serum or IgG from chronic inflammatory demyelinating polyneuropathy patients. Ann Neurol 2000;47:765–775. [PubMed] [Google Scholar]
- 7. Mathey EK, Park SB, Hughes RA, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatry 2015;86:973–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koski CL, Sanders ME, Swoveland PT, et al. Activation of terminal components of complement in patients with Guillain‐Barre syndrome and other demyelinating neuropathies. J Clin Invest 1987;80:1492–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hafer‐Macko CE, Sheikh KA, Li CY, et al. Immune attack on the Schwann cell surface in acute inflammatory demyelinating polyneuropathy. Ann Neurol 1996;39:625–635. [DOI] [PubMed] [Google Scholar]
- 10. Hartung HP, Schwenke C, Bitter‐Suermann D, Toyka KV. Guillain‐Barre syndrome: activated complement components C3a and C5a in CSF. Neurology 1987;37:1006–1009. [DOI] [PubMed] [Google Scholar]
- 11. Quast I, Keller CW, Maurer MA, et al. Sialylation of IgG Fc domain impairs complement‐dependent cytotoxicity. J Clin Invest 2015;125:4160–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Joint Task Force of the E, and the PNS . European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society–First Revision. J Peripher Nerv Syst 2010;15:1–9. [DOI] [PubMed] [Google Scholar]
- 13. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997;40:1725. [DOI] [PubMed] [Google Scholar]
- 14. Merkies IS, Schmitz PI, van der Meche FG, et al. Inflammatory Neuropathy C, and Treatment g . Clinimetric evaluation of a new overall disability scale in immune mediated polyneuropathies. J Neurol Neurosurg Psychiatry. 2002;72:596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hughes RA, Donofrio P, Bril V, et al. Intravenous immune globulin (10% caprylate‐chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): a randomised placebo‐controlled trial. Lancet Neurol 2008;7:136–144. [DOI] [PubMed] [Google Scholar]
- 16. Maillet F, Fremeaux‐Bacchi V, Uhring‐Lambert B, Kazatchkine MD. Assessment of complement activation in clinical samples. Comparison of immunochemical and functional measurements of complement components with quantitation of activation fragments. J Immunol Methods 1992;156:171–178. [DOI] [PubMed] [Google Scholar]
- 17. Sjoberg AP, Trouw LA, Blom AM. Complement activation and inhibition: a delicate balance. Trends Immunol 2009;30:83–90. [DOI] [PubMed] [Google Scholar]
- 18. Belmont HM, Hopkins P, Edelson HS, et al. Complement activation during systemic lupus erythematosus. C3a and C5a anaphylatoxins circulate during exacerbations of disease. Arthritis Rheum 1986;29:1085–1089. [DOI] [PubMed] [Google Scholar]
- 19. Mollnes TE, Haga HJ, Brun JG, et al. Complement activation in patients with systemic lupus erythematosus without nephritis. Rheumatology (Oxford) 1999;38:933–940. [DOI] [PubMed] [Google Scholar]
- 20. Halstead SK, Zitman FM, Humphreys PD, et al. Eculizumab prevents anti‐ganglioside antibody‐mediated neuropathy in a murine model. Brain 2008;131(Pt 5):1197–1208. [DOI] [PubMed] [Google Scholar]
- 21. Fitzpatrick AM, Mann CA, Barry S, et al. An open label clinical trial of complement inhibition in multifocal motor neuropathy. J Peripher Nerv Syst 2011;16:84–91. [DOI] [PubMed] [Google Scholar]
- 22. Wood L, Jacobs P. The effect of serial therapeutic plasmapheresis on platelet count, coagulation factors, plasma immunoglobulin, and complement levels. J Clin Apher 1986;3:124–128. [DOI] [PubMed] [Google Scholar]
- 23. Brandslund I, Peters ND, Ejstrup L. Steroids reduce complement activation in rheumatoid arthritis. Int J Tissue React 1985;7:161–165. [PubMed] [Google Scholar]
- 24. Basta M, Dalakas MC. High‐dose intravenous immunoglobulin exerts its beneficial effect in patients with dermatomyositis by blocking endomysial deposition of activated complement fragments. J Clin Invest 1994;94:1729–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]