

Significance
CsrA proteins are repressors of translation that target the Shine–Dalgarno sequence of the ribosome-binding site. Small noncoding RNAs competitively sequester CsrA by offering multiple binding sites that mimic the Shine–Dalgarno. Antagonism of CsrA by small noncoding RNAs is a widely conserved mechanism of posttranscriptional regulation. Here we present the first crystal structure of protein FliW, which allosterically regulates CsrA in a highly specific manner. Our phylogenetic analysis reveals that the FliW–CsrA regulon is subject to coevolution and represents the ancestral state in flagellated bacteria.
Keywords: translation, CsrA, flagellum, FliW, crystallography
Abstract
Regulation of translation is critical for maintaining cellular protein levels, and thus protein homeostasis. The conserved RNA-binding protein CsrA (also called RsmA; for carbon storage regulator and regulator of secondary metabolism, respectively; hereafter called CsrA) represents a well-characterized example of regulation at the level of translation initiation in bacteria. Binding of a CsrA homodimer to the 5′UTR of an mRNA occludes the Shine–Dalgarno sequence, blocking ribosome access for translation. Small noncoding RNAs (sRNAs) can competitively antagonize CsrA activity by a well-understood mechanism. However, the regulation of CsrA by the protein FliW is just emerging. FliW antagonizes the CsrA-dependent repression of translation of the flagellar filament protein, flagellin. Crystal structures of the FliW monomer reveal a novel, minimal β-barrel-like fold. Structural analysis of the CsrA/FliW heterotetramer shows that FliW interacts with a C-terminal extension of CsrA. In contrast to the competitive regulation of CsrA by sRNAs, FliW allosterically antagonizes CsrA in a noncompetitive manner by excluding the 5′UTR from the CsrA–RNA binding site. Our phylogenetic analysis shows that the FliW-mediated regulation of CsrA regulation is the ancestral state in flagellated bacteria. We thus demonstrate fundamental mechanistic differences in the regulation of CsrA by sRNA in comparison with an ancient regulatory protein.
The ability of ribosomes to access mRNAs at the ribosome-binding site (RBS) is crucial for productive translation and presents an important element of translational control during bacterial gene expression. The Shine–Dalgarno sequence is part of the RBS and marks the starting point of translation, as it allows base pair interaction of the mRNA with the 3′-end of the 16S ribosomal RNA (1, 2). Translation is inhibited if an RBS is occluded either by base-paring RNAs or by regulatory proteins.
CsrA/RsmA proteins (for carbon storage regulator and regulator of secondary metabolism, respectively; hereafter called CsrA) are widely distributed regulators of bacterial gene expression that bind their target mRNAs at the RBS (3). Members of this family mediate the posttranscriptional regulation of a broad variety of bacterial genes and have pleiotropic effects on numerous processes, such as central carbon metabolism, secondary metabolite metabolism, motility, biofilm formation, and the expression of virulence factors in pathogenic bacteria (summarized in refs. 3–5).
CsrA proteins from different bacterial species form highly similar homodimers that are composed of a five-stranded, domain-swapped β-sheet with a barrel-like architecture (6–9). CsrA homodimers contain two RNA binding sites that establish optimal contacts with an A/UCANGGANGU/A sequence motif present in the 5′UTRs (8, 10). When bound by CsrA, the ANGGAN core folds into a loop stabilized by a 3-bp stem of the flanking nucleotides. In this clamp-like structure, the Shine–Dalgarno is sequestered and translation is repressed.
Small noncoding RNAs (sRNAs; e.g., CsrB/C or RsmX/Y/Z) antagonize CsrA binding to mRNA in a competitive manner (11–13). These sRNAs act as “protein sponges,” which contain multiple binding sites that permit sequestration, storage, and release of CsrA (13–15). Although the underlying mechanisms of the sRNA-dependent regulation of CsrA are well understood, modulation of CsrA activity by the regulatory protein FliW lags behind.
FliW regulates CsrA to homeostatically restrict translation of the flagellar filament protein flagellin (encoded by the hag gene) (reviewed in refs. 16 and 17). In Bacillus subtilis, CsrA inhibits translation of flagellin by binding to two sites that are present within the 5′UTR of the hag mRNA (10, 18). FliW inhibits binding of CsrA to the 5′UTR of the hag mRNA, allowing the translation of flagellin (19, 20). FliW, which can also bind to flagellin, is sequestered as cytoplasmic levels of flagellin rise, allowing CsrA again to inhibit translation of the hag mRNA. This cycle is thought to enable homeostasis of cytoplasmic flagellin concentrations over a low and narrow threshold (19). A recent study in Campylobacter jejuni has shown that CsrA regulation by FliW might be much more conserved among flagellated eubacteria than anticipated so far (21). Additional experiments demonstrated that the hag mRNA is not the only target of CsrA, but rather the most abundant in a pool of several motility-related mRNAs that fall under CsrA regulation (21).
To delineate the molecular mechanism by which FliW antagonizes CsrA binding to the 5′UTR of the hag mRNA, we determined the crystal structure of FliW alone and in complex with CsrA. Our structural analysis shows that FliW interacts with a C-terminal extension of CsrA (CsrA-C). In contrast to sRNAs that act as competitive inhibitors of mRNA-binding to CsrA, FliW acts in a noncompetitive manner by allosterically modulating CsrA.
Results
Crystal Structure of FliW Reveals a Minimalistic β-Barrel Fold.
We wanted to determine the structure of FliW because it shares no similarity with any other proteins on the primary sequence level. Because thermophilic proteins are often better suited for crystallography, we used FliW from the moderate thermophile Geobacillus thermodenitrificans (Gt) (Fig. S1). The GtFliW was produced in Escherichia coli BL21(DE3) and purified by Ni-ion affinity, followed by size-exclusion chromatography (SEC). SEC of FliW revealed an apparent molecular mass of 17 kDa corresponding to a FliW monomer (Fig. 1A).
Fig. S1.

Amino acid sequence alignment of FliW primary sequences from B. halodurans, B. subtilis, and Geobacillus thermodenitrificans.
Fig. 1.

Crystal structure of FliW. (A) SEC chromatograms of GtFliW (green), GtCsrA (gray), and the GtCsrA–FliW complex (black) (Right). The Inset shows a Coomassie-stained SDS/PAGE of the GtCsrA–FliW peak fraction. (B) The crystal structure of GtFliW is shown in rainbow colors from the N to the C terminus indicated by “N” and “C,” respectively. (C) The electrostatic surface representation of FliW shows that one side of the molecule is highly negatively charged, as indicated by red colors.
Two crystal forms of GtFliW were observed that diffracted to 1.45- and 1.8-Å resolution (Table 1). During the course of our study, the structure of a contractile protein from Bacillus halodurans (Bh) became available through a structural-genomics consortium (PDB ID code 2AJ7). This protein shared high sequence similarity with FliW and could be successfully used as a search model for molecular replacement. We conclude that PDB ID code 2AJ7 is a FliW homolog. Moreover, both crystal structures of GtFliW and that of BhFliW are almost identical (Fig. S2A, also compare to PDB ID code 2AJ7). In agreement with the SEC (Fig. 1A), both crystal forms of GtFliW suggest that FliW is a monomer (Fig. S2B).
Table 1.
Data collection and refinement statistics for GtFliW and GtCsrA–FliW
| Data collection and refinement | GtFliW | GtFliW | GtCsrA–FliW |
| Data collection | |||
| Space group | P21 | P22121 | C2 |
| Cell dimensions | |||
| a, b, c (Å) | 47.54 | 31.24 | 108.88 |
| 42.52 | 77.99 | 61.68 | |
| 71.92 | 87.05 | 42.90 | |
| α, β, γ (°) | 90.00 | 90.00 | 90.00 |
| 102.66 | 90.00 | 98.08 | |
| 90.00 | 90.00 | 90.00 | |
| Energy (keV) | |||
| Resolution (Å) | 46.38–1.45 | 43.52–1.80 | 42.48–2.30 |
| (1.53–1.45) | (1.95–1.80) | (2.42–2.30) | |
| Rmerge | 0.056 (0.555) | 0.052 (0.51) | 0.056 (0.518) |
| I/σI | 9.5 (1.9) | 18.7 (3.5) | 12.0 (1.8) |
| Completeness (%) | 99.4 (96.6) | 100.0 (99.8) | 99.5 (99.6) |
| Redundancy | 3.3 (3.1) | 7.2 (7.5) | 3.1 (2.9) |
| Refinement | |||
| Resolution (Å) | 46.38–1.45 | 43.52–1.80 | 42.48–2.30 |
| No. reflections | 49,625 | 20,382 | 11,552 |
| Rwork/ Rfree | 20.4 | 22.4 | 22.4 |
| 23.6 | 24.0 | 28.8 | |
| No. atoms | 2,658 | 1,226 | 1,767 |
| Protein | 2,355 | 1,131 | 1,694 |
| Ligand | 0 | 0 | 0 |
| Water | 303 | 95 | 73 |
| Rmsd | |||
| Bond lengths (Å) | 0.01 | 0.01 | 0.01 |
| Bond angles (°) | 1.1 | 1.4 | 1.81 |
| Ramachandran (%) | |||
| Preferred | 97.49 | 98.00 | 95.00 |
| Allowed | 1.43 | 2.0 | 4.52 |
| Outliers | 1.08 | 0.00 | 0.48 |
Values in parentheses are for highest-resolution shell.
Fig. S2.

Structures of GtFliW were obtained from two crystal forms. (A) Superimposition of the two crystal structures in two orientations. The P22121-form is shown in green and the P21 structure is colored in blue. The overall rmsd of the Cα-atoms is 0.766, showing no significant differences between the two structures. (B) Crystallographic symmetry of the two GtFliW structures. The main difference between the two crystal lattices is the amount of molecules within the asymmetric unit. The P22121 form contains only one molecule, whereas the P21 contains two molecules in the asymmetric unit.
Search for FliW-related structures by the DALI server (22) did not result in obvious domain similarities. FliW exhibits a minimalistic β-barrel–like fold consisting of six β-strands (Fig. 1B). The strands β3 to β5 form one-half, whereas strands β6, β8, and β9 close the β-barrel structure. A short α-helical segment of 1.5 turns and several loops, as well as three short β-strands (β1, β2, β7) extend from the central β-barrel to the outside of the molecule. Surface analysis of FliW revealed several extended grooves that could represent interaction sites (Fig. 1C). Furthermore, one side of FliW exhibits a high density of negative charges that might play a role in the electrostatic repulsion of negatively charged molecules (e.g., RNA) (Fig. 1C). Taking these data together, our structural analysis of FliW reveals a minimal β-barrel–like fold that might possess the ability to interact with other partners.
Crystal Structure of the CsrA/FliW Heterotetramer.
To understand the mechanisms of CsrA regulation by FliW, we determined the crystal structure of the CsrA–FliW complex. The GtCsrA–FliW complex was produced by coexpression of a hexahistidine-tagged FliW and a native CsrA in E. coli BL21(DE3). The GtCsrA–FliW complex was purified by Ni-ion affinity chromatography, followed by SEC. On SEC, the CsrA–FliW complex showed the apparent molecular weight of a heterotetramer (Fig. 1A). Crystals of GtCsrA–FliW grew overnight and its structure was determined at 2.3-Å resolution by molecular replacement, with the structures of GtFliW (present study) and Pseudomonas fluorescens CsrA (8) as search models (Table 1).
As concluded from SEC, the crystal structure of GtCsrA/FliW shows a heterotetramer consisting of two CsrA and two FliW molecules (Fig. 2A). CsrA forms a symmetric homodimer along the crystallographic twofold axis. The interface of the CsrA homodimer is established by five β-strands, with β1 being part of the opposing monomer. This finding is in perfect agreement with many other structures of CsrA homodimers obtained by X-ray crystallography and NMR (6–8, 15). The CsrA–FliW interface is primarily established through the C-terminal extension of CsrA (CsrA-C) (Fig. 2A). Each of the CsrA half sites interacts with one FliW molecule through approximately ∼1,405 Å2 of buried surface area (Fig. 2B). The binding site of CsrA-C localizes opposite to the negative charged area of FliW (Fig. 2B). CsrA-C consists of two α-helices, α1 (residues: 44–59) and α2 (residues 60–74), that are connected by a short loop. CsrA-C binds into an extended groove at the surface of FliW via an extensive network of polar and hydrophobic interactions (Fig. 2B and Fig. S3). Successive deletion of helices α1 and α2 from CsrA abolishes its interaction with FliW in vitro (Fig. 2C), and variations of FliW residues critical for CsrA binding abolish the in vitro formation of the CsrA/FliW complex (Fig. 2 D and E). Taken together, our structural and biochemical analysis shows that FliW interacts with CsrA-C. Thus, the CsrA homodimer is able to recruit two FliW molecules, yielding a wing-shaped heterotetramer.
Fig. 2.

Crystal structure of CsrA–FliW. (A) Cartoon representation of the GtCsrA–FliW crystal structure shows that the CsrA homodimer (gray) binds two FliW molecules (green). “N” and “C” indicate N and C termini, respectively. The dashed line indicates the twofold symmetry axis. (B) Electrostatic surface representation of FliW and cartoon representation of a CsrA monomer rainbow-colored from the N to the C terminus. Helices α1 and α2 of CsrA-C bind into an extended groove of FliW (Left) located opposite to the negatively charged area of FliW (Right) (compare also to Fig. 1C). (C) Coomassie-stained SDS/PAGE of an in vitro pulldown assay using GtFliW as bait and GtCsrA, GtCsrAΔα2, and GtCsrAΔα1/α2 as prey. (D) Detailed view of the interaction of FliW with α1 (Upper) and α2 (Lower) of CsrA-C. Residues in FliW that were tested in the in vitro pull-down assay are shown in red (compare with E). (E) Coomassie-stained SDS/PAGE of an in vitro pulldown assay using wild-type GtFliW and its variants as bait and GtCsrA as prey.
Fig. S3.

Detailed interaction network of CsrA and FliW. (A) The helix α1 of CsrA is mainly coordinated by polar and electrostatic interactions. Asn55 of FliW and Gln123 of FliW establish a hydrogen bond; Lys134 of FliW coordinates Glu54. (B) Changing the orientation of helix α1 of CsrA by 90° allows a closer look on the residues of FliW that coordinate CsrA. Ile51 is part of a hydrophobic patch. (C) Interaction surface of helix α2 with FliW. Lys62 establishes a hydrogen bonding to the backbone of Tyr7 FliW; Asn70 is coordinated by the backbone of Asp41 and Leu66 of CsrA is part of a hydrophobic patch at FliW.
FliW Distorts CsrA Binding to the 5′UTR.
To understand the molecular mechanism by which FliW antagonizes CsrA binding to the Shine–Dalgarno-containing 5′UTR of the hag mRNA, we compared the structure of GtCsrA–FliW with that of mRNA-bound CsrA. We chose the structure of CsrA bound to a Shine–Dalgarno present within the 5′UTRs of hcnA mRNA [PDB ID code 2JPP (8)]. This structural comparison is justified on the grounds that the Shine–Dalgarno sequences of the hcnA and hag mRNAs share a high degree of sequence conservation within their “CANGGANG” CsrA-binding motifs (Fig. 3A). Both structures were superimposed on the CsrA homodimers that share high structural similarity with an rmsd of 0.746 over all Cα atoms (Fig. 3B, Right).
Fig. 3.

FliW and RNA interaction with CsrA. (A) Sequence alignment of the 5′UTR regions of the hag and hcnA mRNAs from B. subtilis (Bs) and P. fluorescens (Ps), respectively. A black bar indicates the Shine–Dalgarno (SD) sequence. The red triangle indicates the central base G11. (B) Side-by-side views of the structures of CsrA (red) bound to hcnA (orange) (Left), CsrA (gray) bound to FliW (green) (Center) and the superimposition of both structures (Right). The Upper and Lower panels show two orientations of the same that are rotated by 90 ° relative to each other. (C) Detailed view on the overlapping binding sites of FliW (green) and hcnA (orange) at CsrA (gray). (D) Detailed view of how FliW (green) hinders interaction of CsrA (gray) with hcnA (orange) in a steric (Left) and electrostatic manner (Right).
Our structural comparison shows that the Shine–Dalgarno and FliW share overlapping interfaces at CsrA (Fig. 3B). Moreover, Shine–Dalgarno and FliW engross similar spatial entities and charge distributions relative to CsrA. The overlapping binding sites at CsrA involve residues 125–129 of FliW and bases C9 and G10 of the Shine–Dalgarno (Fig. 3C). Additionally, the negatively charged side of FliW (i.e., amino acids E70, D72, D74, D75, and E79) resides in close proximity at CsrA, where the phosphate backbone of the Shine–Dalgarno would otherwise localize (Fig. 3D). This negative patch of FliW will lead to electrostatic repulsions with the phosphate backbone of the Shine–Dalgarno and therefore hinder RNA binding.
Discussion
Different Mechanisms of CsrA Regulation by FliW and sRNA.
In B. subtilis, CsrA inhibits translation of flagellin by binding to two adjacent sites present within the 5′UTR of hag (18). Interaction of FliW with CsrA enables translation of flagellin, which itself can also sequester FliW (19, 20). This pattern of interactions permits FliW to antagonize initiation of flagellin translation (via CsrA) in a flagellin-dependent manner. FliW thus appears to participate in the homeostatic maintenance of flagellin concentrations.
Here we have investigated the structural basis for how FliW antagonizes CsrA, enabling ribosomes to access the Shine–Dalgarno present within the 5′UTR of the hag mRNA. Our crystal structure shows that FliW binds to CsrA primarily through its C-terminal extension (CsrA-C), but also through contacts with the CsrA RNA-binding domain. FliW shares overlapping interfaces with the Shine–Dalgarno at CsrA, which makes the simultaneous binding of FliW and Shine–Dalgarno extremely unlikely (Figs. 3 and 4 A and B). Moreover, one-half of FliW is highly negatively charged. This side of the protein localizes in close proximity to where the RNA is found in Shine–Dalgarno/CsrA structures. This negatively charged surface of FliW provides strong electrostatic repulsions inhibiting RNA binding. Therefore, FliW allosterically antagonizes CsrA in a noncompetitive manner by excluding the 5′UTR from CsrA (Figs. 3 and 4A). This mechanism is in strong contrast to that of sRNAs also regulating CsrA. Recent structural analysis has elegantly demonstrated that the sRNA RsmZ sequesters CsrA by offering multiple CsrA binding sites that are similar to Shine–Dalgarno sequences (13, 14) (Fig. 4B). As such, sRNAs regulate CsrA by acting as competitive inhibitors of the original Shine–Dalgarno sequences. Our analysis shows that FliW acts, in contrast to sRNAs, by a noncompetitive mechanism through a highly specific binding site offered by CsrA-C.
Fig. 4.

Regulation of CsrA by sRNA and the protein FliW. (A) The different modes of CsrA (red) regulation by FliW (green, Right) and (B) sRNA (black, Right) are depicted. Both FliW and sRNA antagonize the CsrA-dependent repression of translation initiation (Left). (C) Schematic summary of the phylogenetic analysis of CsrA and FliW. Flagellated species containing FliW and CsrA are shown in green; a full circle marks the general presence of the genes in the respective clades. The flagellin protein serves as a marker for flagella-mediated motility. Some members of the Proteobacteria and Planctomycetes contain FliW, whereas some lack FliW (yellow), which is also indicated by the split circle (Right). sRNAs have only been experimentally confirmed within the class of γ-proteobacteria. The Aquificae only contain flagellin, but lack FliW and CsrA. Species at the bottom (red, half circle) lack all of the proteins with the exception of Chlamydiae and Cyanobacteria, in which some species possess a CsrA (Table S1).
Therefore, FliW is highly specific to CsrA, but also to the process of flagellar assembly via its interaction with flagellin. Although a precise molecular understanding of the FliW–flagellin interaction is lacking, a previous study indicated a role of the C terminus of flagellin for its interaction with FliW (23). FliW therefore links the presence of mature, cytoplasmic flagellin molecules with the production of new flagellin via CsrA. Such a mechanism might be useful in the light that flagellin is one of the most abundant proteins in flagellated bacteria (one flagellar filament consists of over 20,000 flagellin copies), and therefore, should be homeostatically controlled (Fig. 4A).
FliW Is an Ancient Regulator That Is Evolutionarily Coupled to CsrA.
Our study shows that the interaction of FliW and CsrA relies on CsrA-C. We therefore wanted to know whether the presence of CsrA-C relates to FliW among flagellated bacteria. To address this question, we compared the conservation of the CsrA–FliW module by performing a systematic search using the National Center for Biotechnology Information (NCBI) BLAST tool and subsequent phylogenetic inference (Fig. 4C and Table S1). Our analysis confirms that FliW and CsrA form an evolutionary conserved unit among flagellated bacteria, as has been suggested previously (19, 24, 25). Manual inspection of CsrA proteins from organisms that also contain FliW shows that these possess a C-terminal elongation (Fig. 4C, Fig. S4, and Table S1). Vice versa, CsrA proteins from flagellated species missing FliW also lack the C-terminal elongation, whereas the RNA-binding domain is highly conserved (Fig. S4). We therefore suspect that CsrA-C and FliW are subject to coevolution. Absence of CsrA-C and FliW in only some species suggests their secondary loss during the course of evolution. Taking these data together, we find that both proteins apparently form an ancient regulatory module of flagellated bacteria.
Table S1.
Summary of our phylogenetic inference
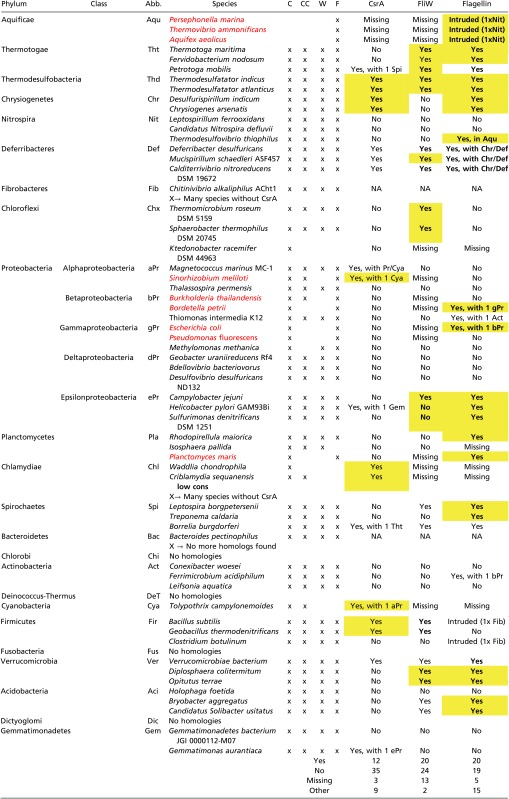 |
Yellow highlighting denotes Neighbor-Joining analysis with bootstrap support of 90% or larger; boldface denotes a Maximum-Likelihood inference with bootstrap support of 90% or larger. All species marked in red are exceptions to their clades, as they lack the fliw gene although it is present in other bacteria of the same clade. Abb, Abbreviation; C, CsrA; CC, CsrA-C; F, flagellin; NA, not applicable; W, FliW.
Fig. S4.

Sequence alignment of CsrA/RsmA homologs from representatives of different phyla sorted by length. The abbreviations are: BACSU_Fir: Bacillus subtilis (Firmicutes); BORPE_bPr: Bordetella petrii (β-proteobacteria); CAMJE_gPr: Campylobacter jejuni (ε-proteobacteria); ESCCO_gPr: Escherichia coli (γ-proteobacteria); GEMAU_Gem: Gemmatimonas aurantiaca (Gemmatimonadetes); HELPY_ePr: Helicobacter pylori (ε-proteobacteria); LEIAQ_Act: Leifsonia aquatic (Actinobacteria); NITDE_Nit: Candidatus Nitrospira defluvii (Nitrospira); PLAMA_Pla: Planctomyces maris (Planctomycetes); THAPE_aPr: Thalassospira permensis (γ-proteobacteria); VERBA_Ver: Verrucomicrobiae bacterium (Verrucomicrobia).
Regulation of CsrA by FliW or sRNAs.
Our study also raises the question how CsrA proteins lacking a FliW binding site are regulated. Phylogenetic analysis shows that CsrA proteins without an extended C terminus are primarily found within the γ-proteobacteria (that also lack FliW) (Fig. 4C and Table S1). In the γ-proteobacteria, CsrA activity is regulated through the CsrB/C and RsmX/Y/Z family members (4, 26–28). In contrast to FliW, these antagonizing sRNAs provide multiple CsrA binding sites and prevent CsrA binding to their mRNA target sites in a competitive manner (Fig. 4B). Physiologically, these sRNAs are related to a plethora of antagonizing effects associated with motility, metabolism, and biofilm formation (reviewed in refs. 3, 5, 29, and 30). Notably, in E. coli and Yersinia pseudotuberculosis, CsrA has been shown to interact directly with the 5′UTR of the flhDC mRNA and to stabilize the transcript (31–33). FlhDC represent the master regulatory proteins of flagella biosynthesis in these organisms. In Salmonella typhimurium, deletion of the CsrA interacting sRNAs CsrB/C show increased transcription of the mRNA encoding the major flagellin protein FliA (34). This finding is in contrast to B. subtilis and C. jejuni, where the deletion of FliW decreases the production of flagellin (19, 21). Thus, it could be concluded that a major target of CsrA regulation by FliW or sRNAs is the flagella assembly pathway with respect to the homeostatic control of flagellin. Phylogenetic analysis also suggests that regulation of CsrA by sRNAs is exclusive to the γ-proteobacteria (5, 35), whereas the CsrA–FliW regulon is widely distributed among flagellated bacteria (Fig. 4C). Why the γ-proteobacteria use sRNAs instead of FliW in regulating CsrA is unclear, but might be related to the adaptation of virulence mechanisms (3).
Another question is whether organisms exist using both FliW and sRNAs to regulate CsrA. Answering this question is hampered by the low sequence conservation of sRNAs that has already been documented even for closely related members of the γ-proteobacteria (5). Nevertheless, a bioinformatic analysis suggested that sRNAs in FliW-containing organisms, such as B. subtilis and Helicobacter pylori, might be present (36), although no experimental validation exists to our knowledge. Therefore, future studies need to address whether organisms exist that use both FliW and sRNAs to coregulate CsrA.
Taking these data together, we have shown that FliW and sRNAs control the activity of CsrA via two fundamentally different mechanisms. Although the sRNA-dependent mechanism seems to be a derived feature of γ-proteobacteria, the FliW-dependent regulation of CsrA might be dominant for all of the other clades of CsrA-containing, flagellated eubacteria.
Materials and Methods
Phylogenetic Analyses.
Homologs of B. subtilis CsrA, flagellin, and FliW bait sequences were collected using NCBI BLAST (blast.ncbi.nlm.nih.gov/Blast.cgi). We randomly selected three representative species per phylum (for accession numbers, see Table S1). Homologous hits were acquired by twilight zone-filtering according to ref. 37 and the requirement of a query coverage of 50%. Sequences were aligned using MAFFT (38) in the G-INS-i mode. Alignments were manually curated using Jalview (39), with the aim to remove positions of questionable quality. The most suitable evolutionary models were selected using ProtTest 3.4 (40) and turned out to be LG+I+G, Blosum62+I+G+F, and LG+I+G in the case for CsrA, flagellin, and FliW, respectively. Bayesian inference trees were inferred using MrBayes (41) with two hot and two cold chains and eight γ rates for 4 million generations or until the SD of split frequencies was <0.01 (Figs. S5 A and B and S6A). This cut-off criterion was reached for CsrA at 2,760,799 generations and for flagellin at 3,723,000 generations; for FliW the deviation after 4 million generations was 0.028. Visual inspection of generation vs. log probability showed no obvious trends; 150 trees each were discarded as burnin. Neighbor-Joining trees were inferred using Quicktree_sd with 1,000 bootstrap replicates (Figs. S6B and S7 A and B). Maximum-likelihood (ML) inference was conducted using RAxML (42) starting from a random tree, generating 20 distinct trees to select the one with the best-likelihood. Subsequently, 100 bootstrap inferences were carried out and the support values drawn on the best tree (Figs. S8 A and B and S9). Trees were visualized using FigTree (tree.bio.ed.ac.uk/software/figtree/).
Fig. S5.

(A) Midpoint-rooted Bayesian inference tree of CsrA, the numbers at the nodes represent posterior probabilities. (B) Midpoint-rooted Bayesian inference tree of FliW, the numbers at the nodes represent posterior probabilities.
Fig. S6.

(A) Midpoint-rooted Bayesian inference tree of flagellin, the numbers at the nodes represent posterior probabilities. (B) Midpoint-rooted neighbor-joining tree of CsrA, the numbers at the nodes represent bootstrap values.
Fig. S7.

(A) Midpoint-rooted neighbor-joining tree of FliW, the numbers at the nodes represent bootstrap values. (B) Midpoint-rooted neighbor-joining tree of flagellin, the numbers at the nodes represent bootstrap values.
Fig. S8.

(A) Midpoint-rooted ML tree of CsrA, the numbers at the nodes represent bootstrap values. (B) Midpoint-rooted ML tree of FliW, the numbers at the nodes represent bootstrap values.
Fig. S9.

Midpoint-rooted ML tree of flagellin; the numbers at the nodes represent bootstrap values.
Protein Production and Purification.
The genes encoding for CsrA and FliW proteins from G. thermodenitrificans NG-80 were amplified from genomic DNA by PCR and cloned into pET16b and pET24d vectors (Novagen) via the NcoI and BamHI restriction sites. FliW contained an N-terminal hexa-histidine tag. Proteins were produced in BL21 (DE3) (Novagen). After cell lysis by a Microfluidizer (M110-L, Microfluidics), cell debris was removed by high-speed centrifugation and proteins were purified by Ni-ion affinity- and size-exclusion chromatography, as described recently (43). The SEC buffer consisted of 20 mM Hepes-Na (pH 7.5), 200 mM NaCl, 20 mM KCl, and 20 mM MgCl2.
Crystallization and Structure Determination.
Crystallization was performed by the sitting-drop method at 20 °C in 600-nL drops consisting of equal parts of protein and precipitation solutions. GtFliW crystallized at 20-mg/mL concentration within 1–5 d in 0.1 M MES pH 6.0, 40% (vol/vol) PEG 400, 5% (wt/vol) PEG 3000 and 0.2 M sodium acetate, 0.1 M sodium cacolydate pH 6.5, and 30% (wt/vol) PEG 8000. GtCsrA–FliW crystallized at 15 mg/mL in a condition containing 0.2 M potassium fluoride, 20% (wt/vol) PEG 3350. Prior data collection, crystals were flash-frozen in liquid nitrogen using a cryo-solution that consisted of mother-liquor supplemented with 20% (vol/vol) glycerol. Data were collected under cryogenic conditions at the European Synchrotron Radiation Facility (Grenoble, France) at beamlines ID23-2 and ID30A-1.
Data were processed with XDS (44) and ccp4-implemented SCALA (45). Structures were determined by molecular replacement with PHASER (46), manually built in COOT (47), and refined with PHENIX (48). Search models were the structures of a putative contractile protein (FliW/bh3618; PDB ID code 2AJ7), GtFliW (present study), and RsmA (PDB ID code 2JPP). Figures were prepared with Pymol (www.pymol.org).
In Vitro Pull-Down Assays.
For in vitro pull-down assays CsrA, FliW, and their variants were coproduced in E. coli BL21(DE3). The bait protein contained a hexa-histidine tag at its N terminus, whereas the prey protein did not. Cells were resuspended in a lysis buffer containing 20 mM Hepes-Na, 200 mM NaCl, 20 mM MgCl2, 20 mM KCl, and 40 mM imidazole, pH 8.0. After cell lysis and removal of cell debris by high-speed centrifugation, the lysate was incubated with Ni-agarose beads for 5 min. After excessive washing of the beads with lysis buffer, bound protein was eluted in a buffer containing 20 mM Hepes-Na, 200 mM NaCl, 20 mM MgCl2, 20 mM KCl, and 500 mM imidazole, pH 8.0. Eluates were analyzed by Coomassie-stained SDS/PAGE.
Summary of Our Phylogenetic Inference
We considered the hag gene as a putative ancestral feature of the Eubacteria, and the phyla as defined by the Tree of Life project (tolweb.org/tree) as monophyletic. Hence, we expect the gene to be present in the three species selected (via BLAST search; see Materials and Methods) per phylum, and to form a tripartite monophyletic cluster comprising the sequences of that phylum. Exceptions to this rule would be considered as evidence for secondary loss of the gene in the case of lack of homologs, and as evidence for potential horizontal gene transfer (HGT) in the case of mixed clusters.
Our phylogenetic inference (Figs. S5–S9), summarized in Table S1, shows that of 27 phyla (including the 5 Proteobacteria classes in the count), 4—namely the Chlorobi, the Deinococcus-Thermus group, the Fusobacteria, and the Dictyoglomi—apparently secondarily lost the hag gene (as well as the csrA and fliW gene). All other 23 phyla contain the gene, supporting its status as an ancestral trait of the Eubacteria. Seven phyla exhibit confidently supported monophyletic clades [marked by “yes” in Table S1, showing the expected lineage-specific clustering with a Bayesian inference probability of 0.9 or higher; in addition, many of the results are also supported in a neighbor-joining analysis with bootstrap support of 90% or larger (marked by yellow highlighting in Table S1), respectively, in a ML inference with bootstrap support of 90% or larger (marked in bold face in Table S1)]. In four phyla, all sequences are clustered with less support than 0.9 (marked by “no” in Table S1). It should be noted, however, that a less-stringent support requirement would result in more monophyletic relationships being resolved for all three genes. Seven phyla contain hag clades that might be interpreted as evidence for HGT (the monophyletic Aquificae clade contains a Nitrospira sequence with high support; a Deferribacteres sequence cluster outside of the other two sequences and Chrysiogenetes; a β- and a γ-proteobacterium form a supported clade; a β-proteobacterium cluster with an Acidobacteria sequence; and the Firmicutes clade contains a Fiibrobacteres sequence). In summary, the hag gene has been retained in the majority of phyla and there is some evidence for potential HGT.
Of the 59 species that harbor at least one of the three genes (hag, csrA, fliW), only five lack the hag gene. Its monophyly is unanimously resolved in eight phyla, lacks support in four phyla, and shows HGT evidence in seven.
The csrA gene is lacking in only three species, namely all sampled Aquificae. Its monyphyly is unanimously resolved in three phyla, lacks support in nine phyla, and shows HGT evidence in six.
The fliW gene is lacking in 13 species (among them the 3 Aquificae that also lack the CsrA gene). Its monyphyly is unanimously resolved in 5 phyla, lacks support in 14 phyla, and shows no HGT evidence at all.
Our data support csrA and fliW to be an ancestral trait, because a comparatively low number of secondary losses (approximately 16) offer a more parsimonious explanation for the extant situation than a many-gains scenario (approximately 47).
Shared traits of all three genes include their lack in four phyla (mentioned above), as well as their unanimously supported monophyly in the Thermodesulfobacteria and their unsupported monophyly in the δ-proteobacteria. Shared pairwise traits of csrA and hag are their unanimously supported monophyly in the Chrysiogenetes and their unsupported monophyly in the Chloroflexi, whereas fliW and hag share unanimous support in the Thermotogae and Verrucomicrobia, and lack of such in the α-proteobacteria and Gemmatimonadetes as well; both genes have been lost from Chlamydiae and the Cyanobacterium. csrA and fliW have both been lost in the Aquificae, are supported in the Deferribacteres, and not supported in the Nitrospira, Planctomycetes, Actinobacteria, as well as β- and γ-proteobacteria. In summary, fliW shares more pairwise commonalities with hag than csrA does, and csrA and fliW share even more than fliW and hag. Interestingly, no HGT evidence is shared between any of the genes, providing evidence for the spurious and secondary nature of such events.
Acknowledgments
We thank the European Synchrotron Radiation Facility for support; Daniel B Kearns (Indiana University) for discussions; and Carey Nadell (Marburg, Germany) for critical reading of the manuscript. The LOEWE program of the state of Hesse funded this work (G.B.). We are grateful to SFB987 for support.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org [PDB ID codes 5DMD, 5JAK, and 5DMB for GtFliW in P2(1), GtFliW in P22(1)2(1), and GtCsrA/FliW, respectively].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1602425113/-/DCSupplemental.
References
- 1.Steitz JA, Jakes K. How ribosomes select initiator regions in mRNA: Base pair formation between the 3′ terminus of 16S rRNA and the mRNA during initiation of protein synthesis in Escherichia coli. Proc Natl Acad Sci USA. 1975;72(12):4734–4738. doi: 10.1073/pnas.72.12.4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shine J, Dalgarno L. Determinant of cistron specificity in bacterial ribosomes. Nature. 1975;254(5495):34–38. doi: 10.1038/254034a0. [DOI] [PubMed] [Google Scholar]
- 3.Vakulskas CA, Potts AH, Babitzke P, Ahmer BMM, Romeo T. Regulation of bacterial virulence by Csr (Rsm) systems. Microbiol Mol Biol Rev. 2015;79(2):193–224. doi: 10.1128/MMBR.00052-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Romeo T. Global regulation by the small RNA-binding protein CsrA and the non-coding RNA molecule CsrB. Mol Microbiol. 1998;29(6):1321–1330. doi: 10.1046/j.1365-2958.1998.01021.x. [DOI] [PubMed] [Google Scholar]
- 5.Heroven AK, Böhme K, Dersch P. The Csr/Rsm system of Yersinia and related pathogens: A post-transcriptional strategy for managing virulence. RNA Biol. 2012;9(4):379–391. doi: 10.4161/rna.19333. [DOI] [PubMed] [Google Scholar]
- 6.Gutiérrez P, et al. Solution structure of the carbon storage regulator protein CsrA from Escherichia coli. J Bacteriol. 2005;187(10):3496–3501. doi: 10.1128/JB.187.10.3496-3501.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heeb S, et al. Functional analysis of the post-transcriptional regulator RsmA reveals a novel RNA-binding site. J Mol Biol. 2006;355(5):1026–1036. doi: 10.1016/j.jmb.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 8.Schubert M, et al. Molecular basis of messenger RNA recognition by the specific bacterial repressing clamp RsmA/CsrA. Nat Struct Mol Biol. 2007;14(9):807–813. doi: 10.1038/nsmb1285. [DOI] [PubMed] [Google Scholar]
- 9.Rife C, et al. Crystal structure of the global regulatory protein CsrA from Pseudomonas putida at 2.05 A resolution reveals a new fold. Proteins. 2005;61(2):449–453. doi: 10.1002/prot.20502. [DOI] [PubMed] [Google Scholar]
- 10.Dubey AK, Baker CS, Romeo T, Babitzke P. RNA sequence and secondary structure participate in high-affinity CsrA-RNA interaction. RNA. 2005;11(10):1579–1587. doi: 10.1261/rna.2990205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weilbacher T, et al. A novel sRNA component of the carbon storage regulatory system of Escherichia coli. Mol Microbiol. 2003;48(3):657–670. doi: 10.1046/j.1365-2958.2003.03459.x. [DOI] [PubMed] [Google Scholar]
- 12.Sonnleitner E, Haas D. Small RNAs as regulators of primary and secondary metabolism in Pseudomonas species. Appl Microbiol Biotechnol. 2011;91(1):63–79. doi: 10.1007/s00253-011-3332-1. [DOI] [PubMed] [Google Scholar]
- 13.Duss O, et al. Structural basis of the non-coding RNA RsmZ acting as a protein sponge. Nature. 2014;509(7502):588–592. doi: 10.1038/nature13271. [DOI] [PubMed] [Google Scholar]
- 14.Duss O, Michel E, Diarra dit Konté N, Schubert M, Allain FH. Molecular basis for the wide range of affinity found in Csr/Rsm protein-RNA recognition. Nucleic Acids Res. 2014;42(8):5332–5346. doi: 10.1093/nar/gku141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morris ER, et al. Structural rearrangement in an RsmA/CsrA ortholog of Pseudomonas aeruginosa creates a dimeric RNA-binding protein, RsmN. Structure. 2013;21(9):1659–1671. doi: 10.1016/j.str.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mukherjee S, Kearns DB. The structure and regulation of flagella in Bacillus subtilis. Annu Rev Genet. 2014;48:319–340. doi: 10.1146/annurev-genet-120213-092406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altegoer F, Bange G. Undiscovered regions on the molecular landscape of flagellar assembly. Curr Opin Microbiol. 2015;28:98–105. doi: 10.1016/j.mib.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 18.Yakhnin H, et al. CsrA of Bacillus subtilis regulates translation initiation of the gene encoding the flagellin protein (hag) by blocking ribosome binding. Mol Microbiol. 2007;64(6):1605–1620. doi: 10.1111/j.1365-2958.2007.05765.x. [DOI] [PubMed] [Google Scholar]
- 19.Mukherjee S, et al. CsrA-FliW interaction governs flagellin homeostasis and a checkpoint on flagellar morphogenesis in Bacillus subtilis. Mol Microbiol. 2011;82(2):447–461. doi: 10.1111/j.1365-2958.2011.07822.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mukherjee S, Babitzke P, Kearns DB. FliW and FliS function independently to control cytoplasmic flagellin levels in Bacillus subtilis. J Bacteriol. 2013;195(2):297–306. doi: 10.1128/JB.01654-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dugar G, et al. The CsrA-FliW network controls polar localization of the dual-function flagellin mRNA in Campylobacter jejuni. Nat Commun. 2016;7:11667. doi: 10.1038/ncomms11667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holm L, Sander C. Dali: A network tool for protein structure comparison. Trends Biochem Sci. 1995;20(11):478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- 23.Titz B, Rajagopala SV, Ester C, Häuser R, Uetz P. Novel conserved assembly factor of the bacterial flagellum. J Bacteriol. 2006;188(21):7700–7706. doi: 10.1128/JB.00820-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Titz B, et al. The binary protein interactome of Treponema pallidum—The syphilis spirochete. PLoS One. 2008;3(5):e2292. doi: 10.1371/journal.pone.0002292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karna SLR, Prabhu RG, Lin Y-H, Miller CL, Seshu J. Contributions of environmental signals and conserved residues to the functions of carbon storage regulator A of Borrelia burgdorferi. Infect Immun. 2013;81(8):2972–2985. doi: 10.1128/IAI.00494-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alon U, et al. Response regulator output in bacterial chemotaxis. EMBO J. 1998;17(15):4238–4248. doi: 10.1093/emboj/17.15.4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y, Cui Y, Mukherjee A, Chatterjee AK. Characterization of a novel RNA regulator of Erwinia carotovora ssp. carotovora that controls production of extracellular enzymes and secondary metabolites. Mol Microbiol. 1998;29(1):219–234. doi: 10.1046/j.1365-2958.1998.00924.x. [DOI] [PubMed] [Google Scholar]
- 28.Valverde C, Heeb S, Keel C, Haas D. RsmY, a small regulatory RNA, is required in concert with RsmZ for GacA-dependent expression of biocontrol traits in Pseudomonas fluorescens CHA0. Mol Microbiol. 2003;50(4):1361–1379. doi: 10.1046/j.1365-2958.2003.03774.x. [DOI] [PubMed] [Google Scholar]
- 29.Lucchetti-Miganeh C, Burrowes E, Baysse C, Ermel G. The post-transcriptional regulator CsrA plays a central role in the adaptation of bacterial pathogens to different stages of infection in animal hosts. Microbiology. 2008;154(Pt 1):16–29. doi: 10.1099/mic.0.2007/012286-0. [DOI] [PubMed] [Google Scholar]
- 30.Romeo T, Vakulskas CA, Babitzke P. Post-transcriptional regulation on a global scale: Form and function of Csr/Rsm systems. Environ Microbiol. 2013;15(2):313–324. doi: 10.1111/j.1462-2920.2012.02794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heroven AK, Böhme K, Rohde M, Dersch P. A Csr-type regulatory system, including small non-coding RNAs, regulates the global virulence regulator RovA of Yersinia pseudotuberculosis through RovM. Mol Microbiol. 2008;68(5):1179–1195. doi: 10.1111/j.1365-2958.2008.06218.x. [DOI] [PubMed] [Google Scholar]
- 32.Wei BL, et al. Positive regulation of motility and flhDC expression by the RNA-binding protein CsrA of Escherichia coli. Mol Microbiol. 2001;40(1):245–256. doi: 10.1046/j.1365-2958.2001.02380.x. [DOI] [PubMed] [Google Scholar]
- 33.Yakhnin AV, et al. CsrA activates flhDC expression by protecting flhDC mRNA from RNase E-mediated cleavage. Mol Microbiol. 2013;87(4):851–866. doi: 10.1111/mmi.12136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jonas K, et al. Complex regulatory network encompassing the Csr, c-di-GMP and motility systems of Salmonella Typhimurium. Environ Microbiol. 2010;12(2):524–540. doi: 10.1111/j.1462-2920.2009.02097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lapouge K, Schubert M, Allain FHT, Haas D. Gac/Rsm signal transduction pathway of γ-proteobacteria: From RNA recognition to regulation of social behaviour. Mol Microbiol. 2008;67(2):241–253. doi: 10.1111/j.1365-2958.2007.06042.x. [DOI] [PubMed] [Google Scholar]
- 36.Kulkarni PR, Cui X, Williams JW, Stevens AM, Kulkarni RV. Prediction of CsrA-regulating small RNAs in bacteria and their experimental verification in Vibrio fischeri. Nucleic Acids Res. 2006;34(11):3361–3369. doi: 10.1093/nar/gkl439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rost B. Twilight zone of protein sequence alignments. Protein Eng. 1999;12(2):85–94. doi: 10.1093/protein/12.2.85. [DOI] [PubMed] [Google Scholar]
- 38.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25(9):1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27(8):1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ronquist F, et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stamatakis A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schuhmacher JS, et al. MinD-like ATPase FlhG effects location and number of bacterial flagella during C-ring assembly. Proc Natl Acad Sci USA. 2015;112(10):3092–3097. doi: 10.1073/pnas.1419388112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 48.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]