

Significance
Amyotrophic lateral sclerosis (ALS) can be caused by mutations in superoxide dismutase (SOD1), which lead to the accumulation of misfolded SOD1 proteins and to the death of motor neurons. Here we show that endogenous macrophage migration inhibitory factor (MIF) acts as a chaperone for misfolded SOD1 in vivo, because completely eliminating MIF in a mutant SOD1 mouse model of familial ALS enhanced the accumulation of misfolded SOD1, accelerated disease onset and late disease progression, and shortened the lifespan of mice expressing mutant SOD1. This study thus sheds light on the important implications of modulating MIF levels and provides insight into the potential therapeutic role of MIF in suppressing the selective accumulation of misfolded SOD1 in ALS.
Keywords: ALS, mutant SOD1 mouse, mutant SOD1, misfolded SOD1, MIF
Abstract
Mutations in superoxide dismutase (SOD1) cause amyotrophic lateral sclerosis (ALS), a fatal neurodegenerative disease characterized by the loss of upper and lower motor neurons in the brain and spinal cord. It has been suggested that the toxicity of mutant SOD1 results from its misfolding and accumulation on the cytoplasmic faces of intracellular organelles, including the mitochondria and endoplasmic reticulum (ER) of ALS-affected tissues. Recently, macrophage migration inhibitory factor (MIF) was shown to directly inhibit the accumulation of misfolded SOD1 and its binding to intracellular membranes, but the role of endogenous MIF in modulating SOD1 misfolding in vivo remains unknown. To elucidate this role, we bred MIF-deficient mice with SOD1G85R mice, which express a dismutase-inactive mutant of SOD1 and are considered a model of familial ALS. We found that the accumulation of misfolded SOD1, its association with mitochondrial and ER membranes, and the levels of sedimentable insoluble SOD1 aggregates were significantly higher in the spinal cords of SOD1G85R-MIF−/− mice than in their SOD1G85R-MIF+/+ littermates. Moreover, increasing MIF expression in neuronal cultures inhibited the accumulation of misfolded SOD1 and rescued from mutant SOD1-induced cell death. In contrast, the complete elimination of endogenous MIF accelerated disease onset and late disease progression and shortened the lifespan of the SOD1G85R mutant mice. These findings indicate that MIF plays a significant role in the folding and misfolding of SOD1 in vivo, and they have implications for the potential therapeutic role of up-regulating MIF within the nervous system to modulate the selective accumulation of misfolded SOD1.
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by a selective loss of upper and lower motor neurons in the brain and spinal cord. Most cases of ALS are sporadic and lack any apparent genetic linkage, but 10% of cases are dominantly inherited, known as familial ALS (fALS) (1). Of these familial cases, ∼20% have been attributed to mutations in a gene encoding for the ubiquitous cytoplasmic copper/zinc superoxide dismutase (SOD1) (2), and overexpression of the human mutant SOD1 in mouse models of fALS invariably results in motor neuron loss, muscle wasting, and hindlimb paralysis (3).
Although the mechanism underlying SOD1-mediated toxicity is still unknown, many of the pathways that were hypothesized to underlie motor neuron degeneration in ALS involve damage incurred by the accumulation of misfolded SOD1 (4), as determined by using antibodies that recognize epitopes unavailable in the natively folded protein, and that bind preferentially or exclusively to misfolded conformers (5–7).
Whereas the wild-type (WT) SOD1 is a ubiquitous cytoplasmic protein, a common feature of the SOD1 mutants is that they are localized to the mitochondria (8–13) and/or endoplasmic reticulum (ER) (14–17), specifically in nervous system tissues. For instance, an association between mutant SOD1 and the ER has been implicated in the induction of ER stress (14–17), and misfolded mutant SOD1 has been found in fractions enriched for mitochondria derived from ALS-affected tissues, but not from unaffected ones (8, 10, 12, 13, 18, 19). In addition, misfolded mutant SOD1 in its nonaggregated, soluble form has been found deposited on the cytoplasmic face of the outer membrane of spinal cord mitochondria (10, 12), and this deposition was accompanied by altered mitochondrial shape and distribution (19). These phenomena may be caused, at least in part, by binding of misfolded SOD1 directly to the mitochondrial voltage-dependent anion channel 1 (VDAC1), because such binding inhibits the ability of VDAC1 to transfer adenine nucleotides across the outer mitochondrial membrane (9). Another possible cause is an interaction between misfolded SOD1 and other components in the outer membrane of the mitochondria, including Bcl-2 (20) and the protein import machinery (21).
The molecular determinants that underlie the selective accumulation and binding of misfolded mutant SOD1 to the spinal cord mitochondria and ER remain unknown; however, we recently found that macrophage migration inhibitory factor (MIF) acts as a cytosolic chaperone that inhibits mutant SOD1 misfolding onto the mitochondria and ER, with extremely low MIF levels within the cytosol of motor neurons (22).
MIF knockout (KO) mice, in which exons 2 and 3 of MIF are disrupted (23), did not develop obvious phenotypes when backcrossed onto a C57BL6 background (24). In the present study, we used these MIF KO mice and the transgenic mutant SOD1G85R mice (25) to study how endogenous MIF affects the course of disease and the accumulation and localization of misfolded SOD1. We report here that overexpression of MIF in neuronal cultures suppresses the accumulation of misfolded SOD1 and rescues from mutant SOD1-induced cell death. In contrast, completely eliminating MIF significantly enhances the accumulation of misfolded SOD1 and its association with mitochondrial and ER membranes and ultimately accelerates disease onset and decreases survival in SOD1G85R mice.
Results
Increased MIF Expression Suppresses the Accumulation of Misfolded SOD1 and Enhances the Survival of Neurons Expressing Mutant SOD1G93A.
To test in vitro whether increased synthesis of MIF can prevent the accumulation of misfolded SOD1 and protect against its toxicity in neurons, human SH-SY5Y neuroblastoma cells were transfected to express the human WT (SOD1WT) or mutant (SOD1G93A) SOD1 transgenes, with or without cotransfection with a plasmid encoding for the human MIF. The accumulation of misfolded SOD1 was detected by immunoprecipitation (IP) with B8H10, a monoclonal antibody that recognizes epitopes within exon 3 that are exposed only on misfolding or denaturation of SOD1 (6, 26) and thus allows the detection of misfolded SOD1 forms by IP or immunofluorescence. Cell survival was quantified with the XTT (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide) assay or, in a different set of experiments (wherein GFP-tagged versions of SOD1WT or SOD1G93A were used), by counting cells with the Operetta High-Content Imaging System (PerkinElmer).
Whereas expressing SOD1G93A without MIF resulted in the intracellular accumulation of misfolded SOD1, coexpressing it with MIF reduced the accumulation of misfolded SOD1 without affecting the overall level of SOD1 (Fig. 1A). Concomitantly, expressing mutant SOD1G93A without MIF reduced cell survival by ∼30% compared with the expression of SOD1WT, which did not affect cell survival, whereas coexpressing it with MIF rescued the cells from this toxic effect (Fig. 1B). Similar results were obtained when MIF was expressed in motor neuron-like NSC-34 cells in the presence of GFP-tagged versions of SOD1WT or mutant SOD1G93A (Fig. 1 C and D).
Fig. 1.

Increased MIF expression enhances the survival of neurons expressing a mutant SOD1 by inhibiting the accumulation of misfolded SOD1. SH-SY5Y neuroblastoma cells were transfected to express the human SOD1WT, the human mutant SOD1G93A, or neither (control), in each case either with (+) or without (–) cotransfection with MIF. (A) Misfolded SOD1 was detected by immunoblotting of immunoprecipitates produced with the B8H10 antibody, which recognizes only the misfolded forms of SOD1. (B) A cell viability analysis, performed with the CellTiter 96 AQueous One-Solution cell proliferation assay with ELISA at 490 nm. Quantitative analysis from triplicates of different biological repeats (n = 3) was performed using Student’s t test. ***P < 0.001. (C) Representative images of NSC-34 motor neuron-like cells transfected with GFP-tagged SOD1WT or SOD1G93A, with or without cotransfection with MIF. At 48 h after transfection, the cells were nuclear-stained with DAPI and then segmented with the Operetta High-Content Imaging System, using the Find Nuclei method. (Scale bars: 200 µm.) (D) The number of cells was quantified using the Operetta system. Quantitative analysis from triplicates of different biological repeats (n = 3) was performed using Student’s t test. ***P < 0.001.
Endogenous MIF Suppresses the Association of Misfolded SOD1 to Spinal Cord Mitochondria and ER Membranes and Reduces Its Intracellular Aggregation.
To determine in vivo whether the accumulation of misfolded SOD1 alters the course and pathogenesis of fALS, we bred the dismutase-inactive SOD1G85R transgenic mice with MIF−/− (KO) mice, which completely lack MIF expression (24) (Fig. S1). The SOD1G85R mouse line used in this study (25) develops a slowly progressive adult-onset fatal paralysis, which results from the expression of the mutant SOD1G85R. Importantly, levels of SOD1G85R accumulation in these mice are similar to those of endogenous mouse SOD1, thus closely mimicking the levels of mutant SOD1 accumulation in human fALS patients.
Fig. S1.
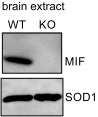
Characterization of MIF KO mice. Immunoblotting of brain extracts recovered by MIF+/+ and MIF−/− mice and probed with anti-MIF or anti-SOD1 antibodies.
We also determined the intracellular localization of MIF in the spinal cords of these mice. Endogenous MIF clearly colocalized with mutant SOD1 in the cytosol of some, but not all, spinal cord cell types (Fig. S2). For example, MIF accumulation was very low within spinal neuronal cells (Fig. S3), confirming our previous observations using rat spinal cord tissues (22).
Fig. S2.

Endogenous MIF colocalizes with SOD1 in the spinal cord of mutant SOD1G85R mice. Representative micrographs of lumbar spinal cord sections from SOD1G85R mice at the presymptomatic disease stage, processed for immunofluorescence using antibodies to MIF (A and D), antibodies to SOD1 (B and E), and the merge of both, including DAPI staining in blue (C and F). (Scale bar: 25 μm.)
Fig. S3.

Endogenous MIF accumulates at very low levels in the spinal neurons of mutant SOD1G85R mice. Shown are representative micrographs of lumbar spinal cord sections from SOD1G85R mice at the presymptomatic stage of the disease, processed for immunofluorescence using antibodies to MIF (A and D), antibodies to NeuN (for the identification of neurons) (B and E), and the merge of both, including DAPI staining (C and F). (Scale bars: A–C, 100 μm; D–F, 20 μm.)
To test whether endogenous MIF inhibits the association of mutant SOD1 with intracellular membranes, we collected spinal cord or liver tissue specimens from SOD1G85R/MIF−/− and SOD1G85R/MIF+/+ mice at different stages of the disease and isolated the mitochondrial and ER membranes (see schematic in Fig. 2A). At disease onset, a significant amount of SOD1G85R accumulated in mitochondrial (Fig. 2 B and C) and ER (Fig. 2 E and F) membranes isolated from the spinal cords of SOD1G85R/MIF−/− mice, but accumulation was much lower in their SOD1G85R/MIF+/+ littermates. In contrast, in the symptomatic disease stage, SOD1G85R levels were not increased in ER membranes and were increased only slightly (in mitochondrial membranes) in SOD1G85R/MIF−/− mice, whereas a dramatic increase was observed in their SOD1G85R/MIF+/+ littermates. At both disease stages, the deletion of endogenous MIF did not affect the amounts of SOD1G85R in liver mitochondrial membranes (Fig. 2D) and very slightly increased SOD1G85R levels in liver ER membranes (Fig. 2G).
Fig. 2.

Endogenous MIF inhibits the association of misfolded SOD1 with spinal cord intracellular membranes. (A) Schematic drawing of the protocol used to test whether the deletion of endogenous MIF in SOD1G85R mice enhances the association of the mutant SOD1G85R with mitochondria and ER membranes. (B, D, E, and G) Immunoblotting of spinal cord mitochondria (B), spinal cord ER (E), liver mitochondria (D), and liver ER (G) recovered from SOD1G85R/MIF+/+ (WT) or SOD1G85R/MIF−/− (KO) mice, as detailed in A, and assayed for the presence of mutant SOD1 in these fractions at disease onset or in its symptomatic stages. Immunoblots were probed with an anti-hSOD1 antibody. Immunoblotting for VDAC1 was used to verify the amount of mitochondria added/recovered. Ponceau staining was used to verify the amount of ER membranes recovered. (C and F) Quantitative analysis of mutant SOD1 associated with spinal cord mitochondria and ER performed from triplicates analyzed from two or three different mice (for each genotype at each disease stage) using ImageJ software and Student’s t test. *P < 0.05.
To determine whether endogenous MIF plays a role in the aggregation of mutant SOD1 in vivo, we removed spinal cords from SOD1G85R/MIF−/− mice and their SOD1G85R/MIF+/+ littermates at different disease stages, and then homogenized and separated them in detergent-soluble and -insoluble fractions (Fig. S4A). At both disease onset and the symptomatic stage, the accumulation of SOD1G85R aggregates in the spinal cords was much higher in the SOD1G85R/MIF−/− mice compared with their SOD1G85R/MIF+/+ littermates (Fig. S4B).
Fig. S4.

Deletion of endogenous MIF enhances the mutant SOD1-insoluble fraction in the spinal cords of mutant SOD1G85R mice at disease onset. (A) Schematic drawing of a protocol for testing whether the deletion of endogenous MIF in SOD1G85R mice enhances the accumulation of mutant SOD1 aggregates. (B) Spinal cord extracts were recovered from SOD1G85R/MIF+/+ (WT) and SOD1G85R/MIF−/− (KO) mice at disease onset or in its symptomatic stage. The amount of SOD1 aggregation was determined by immunoblotting of the soluble and insoluble fractions isolated as described in A. Immunoblots were probed with an anti-SOD1 antibody.
Endogenous MIF Inhibits the Accumulation of Misfolded SOD1 in the Spinal Cords of SOD1G85R Mice.
To examine whether MIF deletion enhances the accumulation of misfolded SOD1 in different tissues of SOD1G85R mice, we used the B8H10 antibody to identify misfolded SOD1. An IP study (Fig. 3A) revealed that compared with their SOD1G85R/MIF+/+ littermates, SOD1G85R/MIF−/− mice showed increased misfolded SOD1 accumulation in the spinal cord at all disease stages (Fig. 3B), in the brain at the symptomatic and end stages (Fig. 3C), and even (albeit less evidently) in the liver (Fig. 3D). An immunofluorescence study revealed misfolded SOD1 accumulation in motor neurons and in other spinal cord cells already at the presymptomatic stage in SOD1G85R/MIF−/− mice (Fig. 4 A–C and G), but not in their SOD1G85R/MIF+/+ littermates Fig. 4 D–G).
Fig. 3.

Endogenous MIF suppresses the accumulation of misfolded SOD1 in the SOD1G85R mouse model of ALS. (A) Protocol used to determine whether the elimination of endogenous MIF enhances the accumulation of misfolded SOD1 detectable with the B8H10 antibody. (B–D) Accumulation of misfolded SOD1 was determined by immunoblotting of immunoprecipitates with the B8H10 antibody after incubation of spinal cord (B), brain (C), or liver (D) extracts (70 µg) recovered from hSOD1G85R/MIF+/+ (WT) and SOD1G85R/MIF−/− (KO) mice at the presymptomatic, symptomatic, or end-stage disease phase. Immunoblotting was also used to determine the presence and absence of MIF and the SOD1 levels remaining in the unbound fraction of each IP assay. Quantitative analysis of misfolded SOD1 was performed from duplicates or triplicates from different mice (between two and four mice for each genotype at each disease stage) using ImageJ software and Student’s t test. **P < 0.01.
Fig. 4.

Eliminating endogenous MIF significantly enhances the accumulation of misfolded SOD1 at the presymptomatic stage. (A–F) Representative micrographs of lumbar spinal cord sections from SOD1G85R/MIF−/− (A–C) and SOD1G85R/MIF+/+ (D–F) mice at the presymptomatic disease stage, processed for immunofluorescence using the B8H10 antibody, which detects misfolded SOD1 (A and D), and the NeuN antibody, which detects neuronal cells (B and E). (Scale bar: 25 μm.) (G) Quantification of the relative fluorescence intensity of the B8H10 staining of lumbar spinal cord from SOD1G85R/MIF−/− and SOD1G85R/MIF+/+ mice at a presymptomatic disease stage. Approximately 30–35 different areas from different mice of each genotype were analyzed. The bar graph represents mean SEM. ***P < 0.001 (Student’s t test).
MIF Deletion Accelerates Disease Onset and Progression in Mutant SOD1G85R Mice.
After establishing that (i) MIF acts as a chaperone for misfolded SOD1 (22) and protects from mutant SOD1-induced cell death (Fig. 1), (ii) endogenous MIF inhibits the association of misfolded SOD1 with intracellular membranes (Fig. 2), and (iii) endogenous MIF suppresses the accumulation of misfolded SOD1 (Figs. 3 and 4), we examined how the deletion of endogenous MIF affects the course of disease by following disease onset and progression in SOD1G85R/MIF−/− mice (n = 21) and SOD1G85R/MIF+/+ mice (n = 19) (Fig. 5). The SOD1G85R/MIF−/− mice, compared with their SOD1G85R/MIF+/+ littermates, showed a 22-d acceleration in disease onset (285 ± 7 d vs. 307 ± 7 d, respectively; Fig. 5 A and D), a 21-d acceleration in the progression to an early point (i.e., 10% weight loss) of the disease (316 ± 8 d vs. 337 ± 7 d, respectively; Fig. 5B), an 11-d acceleration in the progression from the early point of the disease to its end stage (14 ± 3 d vs. 25 ± 2 d, respectively; Fig. 5F), and a 32-d acceleration in the age of disease end stage (330 ± 9 d vs. 362 ± 7 d, respectively; Fig. 5C). The progression from disease onset to an early disease point was not different between the two groups of mice (Fig. 5E).
Fig. 5.

Eliminating endogenous MIF accelerates disease onset and late disease progression and shortens the survival of SOD1G85R mice. (A–C) Mean age (± SD) of disease onset, defined as the time when mice reached peak body weight (P < 0.05) (A); early disease, defined as the time when mice lost 10% of their maximal weight (P < 0.05) (B); and disease end stage, defined as the time when the mouse could not right itself within 20 s when placed on its side (P < 0.01) (C) of SOD1G85R/MIF+/+ mice (red) and their SOD1G85R/MIF−/− littermates (blue). (D–F) Mean (± SD) age of disease onset (P < 0.05) (D), duration of early disease (from onset to 10% weight loss; P = 0.406) (E), and duration of late disease (from 10% weight loss to end stage; P < 0.01) (F). At each time point, the P value was determined by Student’s t test. Error bars denote SD.
Discussion
One of the most important unsolved questions in ALS pathogenesis is what determines the selective, age-dependent degeneration of motor neurons. In cases related to mutant SOD1, such a degeneration is accompanied by the misfolding of mutant SOD1 and its association with intracellular membranes. We recently determined that the association of mutant SOD1 with the mitochondria and ER can be suppressed by cytosolic MIF, which inhibits the accumulation of misfolded SOD1 (22). In addition, we have shown that MIF levels are low within the cell bodies of motor neurons, and that increasing MIF levels extends the survival of motor neurons in culture. The low levels of MIF in motor neurons correlate with the accumulation of misfolded SOD1 species and with their increased association with various intracellular organelles.
In the present study, we demonstrate that completely eliminating the expression of endogenous MIF in vivo accelerated disease onset and late disease progression and shortened the lifespan of the SOD1 mutant mice. Importantly, the acceleration of disease onset was accompanied by the accumulation of misfolded SOD1 as early as the presymptomatic stage. In addition, the association of the mutant SOD1 with mitochondrial and ER membranes in the spinal cords of MIF-deficient mice was strongly increased, and the levels of sedimentable insoluble SOD1 aggregates were higher. Late disease progression was also accelerated in these mice, suggesting the involvement of endogenous MIF in preventing the toxicity of misfolded SOD1 within nonneuronal cells as well. In that context, the accumulation of misfolded SOD1 in glial cells has been proposed previously (27), and its involvement in late disease progression is well established (4, 28).
MIF is a 12-kDa protein that has been implicated in both extracellular and intracellular functions and is synthesized as a cytoplasmic protein (22). The cytokine activity of MIF is achieved by posttranslational sequestration of the cytoplasmic MIF into vesicles, followed by its release, through an as-yet unidentified mechanism, in response to a variety of signals (29). Intracellularly, MIF was previously shown to act as a chaperone protein (30) and as a thiol-protein oxidoreductase (31).
Although MIF KO mice have been widely used in the context of various diseases, here we have studied the effects of MIF in a neurodegenerative disease model. Given the critical involvement of MIF in processes related to misfolding and neurodegeneration, as reported herein, it will be interesting to test whether MIF also can function as a protein modifier in other neurologic diseases in which misfolded proteins play a central role, such as Alzheimer’s, Parkinson’s, and Huntington’s diseases.
Of note, a previous study has shown that reducing the levels of aggregated misfolded SOD1 by deleting cyclophilin D does not ameliorate the pathogenesis of ALS in mutant SOD1 mouse models (32). Therefore, SOD1 toxicity in vivo appears to derive from the soluble form of the misfolded SOD1, rather than from its highly aggregated form. Indeed, we demonstrate here that reducing MIF levels accelerates disease onset and progression, and that this acceleration is accompanied by increased levels of the soluble misfolded SOD1, which accumulates and associates with mitochondrial and ER membranes. Altering the expression levels of other chaperones previously linked to SOD1, including hsp70, hsp90, hsp27, and aB-crystallin, failed to significantly affect the disease course in different mutant SOD1 mouse models (33–37); however, it was recently shown that overexpression of hsp110 in neurons extends the survival of SOD1G85R-YFP and SOD1G93A mice (38). Importantly, there are only very few studies in which the course of disease was altered in the SOD1 model that we used here, which expresses mutant SOD1 at low levels similar to those of the endogenous protein, and in which disease onset was observed at approximately 10 mo, with a very rapid disease progression (25). Here we propose that the reduced chaperone-like activity of MIF in motor neurons plays a pivotal role in the accumulation of misfolded SOD1 and its subsequent toxicity. In addition, with the recently proposed mechanism for cell-to-cell spread of misfolded SOD1 as a means of disease propagation (5, 39, 40), chaperone activity by extracellular MIF may act to limit such spreading.
Finally, accumulation of misfolded SOD1 has been reported by several groups also in sporadic ALS (27, 41–47), although other groups have reached the opposite conclusion (48–51). The identification of MIF as a cytosolic chaperone that stimulates the folding or refolding of misfolded SOD1 and inhibits the aggregation of mutant SOD1 in vivo suggests new avenues for therapy in ALS, mediated by increasing intracellular MIF levels in the nervous system.
Materials and Methods
Transgenic and KO Mice.
Transgenic mice expressing the human SOD1G85R were as described previously (25). MIF KO mice have been developed in which exons 2 and 3 of MIF are disrupted (23). These MIF KO mice, backcrossed onto a C57BL6 background (24), were used in this study. Importantly, all mouse lines were on a pure C57BL6 background to eliminate confounding genetic influences.
Survival Analysis.
MIF null mice (MIF−/−) were mated to heterozygous SOD1G85R ALS mice, and the resulting SOD1G85R/MIF+/− mice were mated to MIF+/− mice to obtain the experimental cohorts of SOD1G85R/MIF−/− mice (n = 19; 9 females and 10 males) that were compared with SOD1G85R/MIF+/+ littermates (n = 21; 10 females and 11 males). Mice were weighed weekly as an objective and unbiased measure of disease course. The time of disease onset was determined retrospectively as the time at which mice reached peak body weight, which is observed before any motor performance decline. The time of early disease was defined as the age at which the animals had lost 10% of their maximal weight. Disease end stage was defined by paralysis so severe that the animal could not right itself within 20 s when placed on its side, an endpoint frequently used for SOD1 mutant-expressing mice. Mice were genotyped by PCR of DNA extracted from a tail biopsy specimen. All mice were maintained using standard protocols in the animal facility of Ben-Gurion University of the Negev. All procedures involving animals were consistent with the requirements of the Animal Care and Use Committee of Ben-Gurion University of the Negev.
Statistics.
Values are reported throughout as mean ± SEM. Comparisons of two datasets were performed using the Student’s t test, after a normal distribution was confirmed by the Shapiro-Wilk normality test. Significance was set at a confidence level of 0.05. In all figures, *P < 0.05, **P < 0.01, and ***P < 0.001. All statistical analysis were performed with SigmaPlot 13.1 (Systat Software).
SI Materials and Methods
Immunoprecipitation.
Spinal cord, brain, or liver fractions (70 µg) or whole-cell extracts (100 µg) were solubilized in IP buffer (50 mM Tris⋅HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40, plus 1× protease inhibitors) and incubated overnight with B8H10 (MédiMabs) antibodies previously cross-linked to Dynabeads protein G (Invitrogen) with dimethyl pimelimidate (Pierce) according to the manufacturer’s instructions. The beads were magnetically isolated and washed three times with IP buffer. Samples were eluted by boiling in a 2× sample buffer.
SOD1 Aggregation Assay by Differential Extraction.
The protocol for SOD1 aggregation by differential extraction was similar to that described previously (52). Spinal cords were homogenized with a homogenizer in 1:10 (wt/vol) 1 TEN (10 mM Tris, 1 mM EDTA, and 100 mM NaCl), then mixed with an equal volume of 2× extraction buffer 1 (10 mM Tris, 1 mM EDTA, 100 mM NaCl, 1% Nonidet P-40, and 1× protease inhibitor mixture) and homogenized as described above. The resulting lysate was centrifuged for 5 min at 100,000 × g to separate the nonionic detergent-insoluble pellet (P1) from the supernatant (S1). S1 was then decanted and stored for analysis. P1 was resuspended in 200 µL of 1× extraction buffer 2 (10 mM Tris, 1 mM EDTA, 100 mM NaCl, 0.5% Nonidet P-40, and 1× protease inhibitor mixture) and then sonicated to resuspend. The extract was then centrifuged for 5 min at 100,000 × g in a Beckman Airfuge to separate the pellet (P2) from the supernatant. The P2 fraction was then resuspended in buffer 3 (10 mM Tris, 1 mM EDTA, 100 mM NaCl, 0.5% Nonidet P-40, 0.25% SDS, 0.5% deoxycholic acid, and 1× protease inhibitor mixture) by sonication and stored for analysis. Protein concentrations were measured in the S1 and P2 fractions by the BCA method (Pierce), according to the manufacturer’s protocol. The proteins of S1 and P2 fractions were run in a 13% and a 15% SDS/PAGE gel, respectively.
Mitochondria and ER Purification.
Mitochondria were purified as described previously (22). Spinal cords were homogenized on ice in 5 volumes of ice-cold homogenization buffer (HB), composed of 210 mM mannitol, 70 mM sucrose, 1 mM EDTA-(Tris), and 10 mM Tris⋅HCl pH 7.2. Homogenates were centrifuged at 1,000 × g for 10 min. The supernatants were then recovered, and the pellets were washed with half-volume HB and centrifuged at 1,000 × g for 5 min. The supernatants were pooled and centrifuged at 17,000 × g for 15 min to yield a crude mitochondrial pellet. The supernatant was used to make cytosolic and ER fractions by further centrifugation at 100,000 × g for 1 h. The mitochondria were gently resuspended in a 12–14% OptiPrep density gradient medium diluted in HB and centrifuged at 17,000 × g for 15 min. Myelin from the spinal cords was removed, and mitochondria were collected from the pellet, washed once with HB to remove the OptiPrep, and centrifuged at 17,000 × g for 15 min. Then the supernatant was discarded, and the pellet was resuspended in a small volume of HB. The proteins were quantified and run in a 13% SDS/PAGE gel.
Immunoblotting.
Proteins were separated on a 13% SDS/PAGE gel, transferred to nitrocellulose membranes, and probed with various antibodies, including goat anti-SOD1 (C-17; Santa Cruz Biotechnology), sheep anti-SOD1 (Calbiochem), monoclonal anti-VDAC/porin 31HL (Calbiochem), goat anti-MIF (N-18; Santa Cruz Biotechnology), rabbit anti-MIF (FL-115; Santa Cruz Biotechnology), rabbit anti-human SOD1 (ab52950; Abcam), and rabbit anti-VDAC (ab154856; Abcam). Horseradish peroxidase-conjugated anti-mouse, anti-rabbit, anti-sheep, or anti-goat IgG secondary antibodies (Jackson ImmunoResearch) were used and detected by ECL (GE Healthcare).
Immunofluorescence.
Mice were anesthetized by inhalation of 1.5–3% isoflurane, followed by a transcardial perfusion of 250 mL of 0.1 M PBS, which was then switched to 4% paraformaldehyde in 0.1 M PBS. The spinal cords were dissected out and postfixed in 4% formaldehyde at 4 °C overnight, cryoprotected in 20% sucrose (48 h at 4 °C), and then stored at 4 °C with 0.02% sodium azide. Free-floating sections (35 µm thick) were blocked with a blocking peroxidase buffer (0.1 M PBS, 20% methanol, 0.2% Triton-X100, and 1.5% H2O2) to reduce endogenous peroxidase activity. Slices were stained following standard protocols. Sections were blocked for 1 h in a blocking solution (1× PBS, 5% free fatty acid BSA, and 0.3% Triton-X100), immunostained for 48 h at 4 °C with antibodies made in 1× PBS, 2% free fatty acid BSA with 0.3% Triton-X100, including mouse anti-neuronal nuclei antigen (1:500, NeuN; Millipore), monoclonal anti-misfolded SOD1 (1:100, B8H10; MédiMabs), or goat anti-MIF (1:100, N18; Santa Cruz Biotechnology).
After the first antibody incubation, sections were washed three times in 0.1 M PBS, then incubated for 2 h at room temperature with a fluorescent-conjugated secondary goat anti-mouse (1:5,000, Alexa Fluor 405; Invitrogen), chicken anti-rabbit (1:5,000, Alexa Fluor 647; Invitrogen), or chicken anti-goat (1:5,000, Alexa Fluor 488; Invitrogen) antibodies, and with DAPI for nuclear staining. Images were acquired on a Nikon C2Plus laser unit docked to a Nikon Ti eclipse unit of a confocal microscope using 20× and 60× oil objectives. Scanning settings were reused across the samples.
Cell Culture and Plasmids.
To generate pCDNA-hMIF, the cDNA of the human MIF (obtained from Jurgen Bernhagen, University Hospital RWTH, Aachen, Germany) was amplified by PCR and inserted into pCDNA3.1(−) plasmid using the BamHI and XbaI sites. pEGFP-hSOD1wt, pEGFP-hSOD1G93A, and pEGFP-hSOD1G85R were kindly provided by Jean Pierre Julien (Laval University, Canada), and pCI-hSOD1WT, pCI-hSOD1G93A, and pCI-hSOD1G85R were generated by inserting human SOD1 constructs into the pCI-NEO vector (Promega), between the EcoRI and the NotI sites. NSC-34 and SH-SY5Y cells were grown at 37 °C and 5% CO2 in DMEM supplemented with 10% tetracycline-free FBS, 2 mM l-glutamine, and 100 U/mL penicillin/0.1 mg/mL streptomycin (all reagents from Biological Industries).
Transfection was performed using TurboFect reagents (Thermo Fisher Scientific) according to the manufacturer’s protocol. When cotransfections were performed, empty plasmids were always transfected as controls. At 48 h after transfection, the cells were stained with DAPI (according to the manufacturer’s protocol) and then analyzed with Operetta. Cell viability was analyzed using the CellTiter 96 AQueous One-Solution cell proliferation assay (Promega) and ELISA at 490 nm, according to the manufacturer’s protocol.
Acknowledgments
We thank all of the members of the A.I. laboratory for helpful comments and suggestions and Tom Shani and Alexandra Stavsky for technical help. This work was supported by Israeli Science Foundation Grant 124/14, Binational Science Foundation Grant 2013325, Seventh Framework Programme Marie Curie Actions Career Integration Grant 333794, German-Israeli Foundation Grant I-2320-1089.13, and National Institute for Psychobiology in Israel Grant b133-14/15.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1604600113/-/DCSupplemental.
References
- 1.Da Cruz S, Cleveland DW. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr Opin Neurobiol. 2011;21(6):904–919. doi: 10.1016/j.conb.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 3.Turner BJ, Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog Neurobiol. 2008;85(1):94–134. doi: 10.1016/j.pneurobio.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187(6):761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grad LI, et al. Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc Natl Acad Sci USA. 2011;108(39):16398–16403. doi: 10.1073/pnas.1102645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gros-Louis F, Soucy G, Larivière R, Julien JP. Intracerebroventricular infusion of monoclonal antibody or its derived Fab fragment against misfolded forms of SOD1 mutant delays mortality in a mouse model of ALS. J Neurochem. 2010;113(5):1188–1199. doi: 10.1111/j.1471-4159.2010.06683.x. [DOI] [PubMed] [Google Scholar]
- 7.Rakhit R, et al. An immunological epitope selective for pathological monomer-misfolded SOD1 in ALS. Nat Med. 2007;13(6):754–759. doi: 10.1038/nm1559. [DOI] [PubMed] [Google Scholar]
- 8.Deng HX, et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci USA. 2006;103(18):7142–7147. doi: 10.1073/pnas.0602046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Israelson A, et al. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron. 2010;67(4):575–587. doi: 10.1016/j.neuron.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J, et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43(1):5–17. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 11.Mattiazzi M, et al. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277(33):29626–29633. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- 12.Vande Velde C, Miller TM, Cashman NR, Cleveland DW. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc Natl Acad Sci USA. 2008;105(10):4022–4027. doi: 10.1073/pnas.0712209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vijayvergiya C, Beal MF, Buck J, Manfredi G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J Neurosci. 2005;25(10):2463–2470. doi: 10.1523/JNEUROSCI.4385-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mori A, et al. Derlin-1 overexpression ameliorates mutant SOD1-induced endoplasmic reticulum stress by reducing mutant SOD1 accumulation. Neurochem Int. 2011;58(3):344–353. doi: 10.1016/j.neuint.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 15.Homma K, et al. SOD1 as a molecular switch for initiating the homeostatic ER stress response under zinc deficiency. Mol Cell. 2013;52(1):75–86. doi: 10.1016/j.molcel.2013.08.038. [DOI] [PubMed] [Google Scholar]
- 16.Fujisawa T, et al. A novel monoclonal antibody reveals a conformational alteration shared by amyotrophic lateral sclerosis-linked SOD1 mutants. Ann Neurol. 2012;72(5):739–749. doi: 10.1002/ana.23668. [DOI] [PubMed] [Google Scholar]
- 17.Nishitoh H, et al. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008;22(11):1451–1464. doi: 10.1101/gad.1640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergemalm D, et al. Overloading of stable and exclusion of unstable human superoxide dismutase-1 variants in mitochondria of murine amyotrophic lateral sclerosis models. J Neurosci. 2006;26(16):4147–4154. doi: 10.1523/JNEUROSCI.5461-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vande Velde C, et al. Misfolded SOD1 associated with motor neuron mitochondria alters mitochondrial shape and distribution prior to clinical onset. PLoS One. 2011;6(7):e22031. doi: 10.1371/journal.pone.0022031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pedrini S, et al. ALS-linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl-2. Hum Mol Genet. 2010;19(15):2974–2986. doi: 10.1093/hmg/ddq202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Q, et al. ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc Natl Acad Sci USA. 2010;107(49):21146–21151. doi: 10.1073/pnas.1014862107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Israelson A, et al. Macrophage migration inhibitory factor as a chaperone inhibiting accumulation of misfolded SOD1. Neuron. 2015;86(1):218–232. doi: 10.1016/j.neuron.2015.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bozza M, et al. Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J Exp Med. 1999;189(2):341–346. doi: 10.1084/jem.189.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor JA, et al. Null mutation in macrophage migration inhibitory factor prevents muscle cell loss and fibrosis in partial bladder outlet obstruction. Am J Physiol Renal Physiol. 2006;291(6):F1343–F1353. doi: 10.1152/ajprenal.00144.2006. [DOI] [PubMed] [Google Scholar]
- 25.Bruijn LI, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18(2):327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 26.Pickles S, et al. Mitochondrial damage revealed by immunoselection for ALS-linked misfolded SOD1. Hum Mol Genet. 2013;22(19):3947–3959. doi: 10.1093/hmg/ddt249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forsberg K, Andersen PM, Marklund SL, Brännström T. Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2011;121(5):623–634. doi: 10.1007/s00401-011-0805-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boillée S, Vande Velde C, Cleveland DW. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52(1):39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 29.Merk M, et al. The Golgi-associated protein p115 mediates the secretion of macrophage migration inhibitory factor. J Immunol. 2009;182(11):6896–6906. doi: 10.4049/jimmunol.0803710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cherepkova OA, Lyutova EM, Eronina TB, Gurvits BY. Chaperone-like activity of macrophage migration inhibitory factor. Int J Biochem Cell Biol. 2006;38(1):43–55. doi: 10.1016/j.biocel.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Kleemann R, et al. Disulfide analysis reveals a role for macrophage migration inhibitory factor (MIF) as thiol-protein oxidoreductase. J Mol Biol. 1998;280(1):85–102. doi: 10.1006/jmbi.1998.1864. [DOI] [PubMed] [Google Scholar]
- 32.Parone PA, et al. Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J Neurosci. 2013;33(11):4657–4671. doi: 10.1523/JNEUROSCI.1119-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karch CM, Borchelt DR. An examination of alpha B-crystallin as a modifier of SOD1 aggregate pathology and toxicity in models of familial amyotrophic lateral sclerosis. J Neurochem. 2010;113(5):1092–1100. doi: 10.1111/j.1471-4159.2010.06572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krishnan J, et al. Over-expression of Hsp27 does not influence disease in the mutant SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J Neurochem. 2008;106(5):2170–2183. doi: 10.1111/j.1471-4159.2008.05545.x. [DOI] [PubMed] [Google Scholar]
- 35.Liu J, Shinobu LA, Ward CM, Young D, Cleveland DW. Elevation of the Hsp70 chaperone does not effect toxicity in mouse models of familial amyotrophic lateral sclerosis. J Neurochem. 2005;93(4):875–882. doi: 10.1111/j.1471-4159.2005.03054.x. [DOI] [PubMed] [Google Scholar]
- 36.Sharp PS, et al. Protective effects of heat shock protein 27 in a model of ALS occur in the early stages of disease progression. Neurobiol Dis. 2008;30(1):42–55. doi: 10.1016/j.nbd.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 37.Xu G, et al. Substantially elevating the levels of αB-crystallin in spinal motor neurons of mutant SOD1 mice does not significantly delay paralysis or attenuate mutant protein aggregation. J Neurochem. 2015;133(3):452–464. doi: 10.1111/jnc.13022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagy M, Fenton WA, Li D, Furtak K, Horwich AL. Extended survival of misfolded G85R SOD1-linked ALS mice by transgenic expression of chaperone Hsp110. Proc Natl Acad Sci USA. 2016 doi: 10.1073/pnas.1604885113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grad LI, Cashman NR. Prion-like activity of Cu/Zn superoxide dismutase: Implications for amyotrophic lateral sclerosis. Prion. 2014;8(1):33–41. doi: 10.4161/pri.27602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Münch C, O’Brien J, Bertolotti A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc Natl Acad Sci USA. 2011;108(9):3548–3553. doi: 10.1073/pnas.1017275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grad LI, et al. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc Natl Acad Sci USA. 2014;111(9):3620–3625. doi: 10.1073/pnas.1312245111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bosco DA, et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13(11):1396–1403. doi: 10.1038/nn.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Forsberg K, et al. Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS One. 2010;5(7):e11552. doi: 10.1371/journal.pone.0011552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guareschi S, et al. An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc Natl Acad Sci USA. 2012;109(13):5074–5079. doi: 10.1073/pnas.1115402109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kabashi E, Valdmanis PN, Dion P, Rouleau GA. Oxidized/misfolded superoxide dismutase-1: The cause of all amyotrophic lateral sclerosis? Ann Neurol. 2007;62(6):553–559. doi: 10.1002/ana.21319. [DOI] [PubMed] [Google Scholar]
- 46.Pokrishevsky E, et al. Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PLoS One. 2012;7(4):e35050. doi: 10.1371/journal.pone.0035050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zetterström P, Andersen PM, Brännström T, Marklund SL. Misfolded superoxide dismutase-1 in CSF from amyotrophic lateral sclerosis patients. J Neurochem. 2011;117(1):91–99. doi: 10.1111/j.1471-4159.2011.07177.x. [DOI] [PubMed] [Google Scholar]
- 48.Brotherton TE, et al. Localization of a toxic form of superoxide dismutase 1 protein to pathologically affected tissues in familial ALS. Proc Natl Acad Sci USA. 2012;109(14):5505–5510. doi: 10.1073/pnas.1115009109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ayers JI, et al. Conformational specificity of the C4F6 SOD1 antibody: Low frequency of reactivity in sporadic ALS cases. Acta Neuropathol Commun. 2014;2(1):55. doi: 10.1186/2051-5960-2-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kerman A, et al. Amyotrophic lateral sclerosis is a non-amyloid disease in which extensive misfolding of SOD1 is unique to the familial form. Acta Neuropathol. 2010;119(3):335–344. doi: 10.1007/s00401-010-0646-5. [DOI] [PubMed] [Google Scholar]
- 51.Liu HN, et al. Lack of evidence of monomer/misfolded superoxide dismutase-1 in sporadic amyotrophic lateral sclerosis. Ann Neurol. 2009;66(1):75–80. doi: 10.1002/ana.21704. [DOI] [PubMed] [Google Scholar]
- 52.Karch CMBD, Borchelt DR. A limited role for disulfide cross-linking in the aggregation of mutant SOD1 linked to familial amyotrophic lateral sclerosis. J Biol Chem. 2008;283(20):13528–13537. doi: 10.1074/jbc.M800564200. [DOI] [PMC free article] [PubMed] [Google Scholar]