

Abstract
While galvanic exchange is commonly applied to metallic nanoparticles, recently its applicability was expanded to metal-oxides. Here the galvanic exchange is studied in metal/metal-oxide core/shell nanocrystals. In particular Sn/SnO2 is treated by Ag+, Pt2+, Pt4+, and Pd2+. The conversion dynamics is monitored by in situ synchrotron X-ray diffraction. The Ag+ treatment converts the Sn cores to the intermetallic AgxSn (x ∼ 4) phase, by changing the core’s crystal structure. For the analogous treatment by Pt2+, Pt4+, and Pd2+, such a galvanic exchange is not observed. This different behavior is caused by the semipermeability of the naturally formed SnO2 shell, which allows diffusion of Ag+ but protects the nanocrystal cores from oxidation by Pt and Pd ions.
Introduction
Galvanic replacement1−14 and cation exchange reactions15−23 similar to it represent simple and versatile tools to achieve nanoarchitectures and compositions of colloidal nanocrystals not readily accomplished by other methods.8,24−27 Galvanic replacement has been applied to metal nanoparticles1−9,25−27 and recently also to metal-oxide nanocrystals.10 The former attracted a lot of attention due to their high potential for biomedical4 as well as for surface enhanced Raman scattering applications,5 whereas the latter have been demonstrated to exhibit good performance as anode materials for lithium ion batteries.10 Since only certain intermetallic compounds can be produced by direct synthesis, often exploiting nonequilibrium processes,28−31 the post growth treatment is an elegant alternative to produce intermetallic compound nanostructures, whose morphology can be tuned.9,25−27
Here the galvanic replacement method is expanded to metal/metal-oxide core/shell nanocrystals. Metal/metal-oxide nanocrystals based on Sn, Al, Fe, Ni, or Cu and covered by the corresponding oxide shells are synthesized for various applications, including plasmonics, photocatalysis, and electrochemical production of hydrogen, as well as magnetic targeting, magnetic resonance imaging, and near-infrared photothermal therapy.32−38 In this study we focus our interest on Sn-based nanocrystals and their alloys, which are promising materials in lithium ion batteries.39−44 In particular, spherical Sn/SnO2 nanocrystals are exposed either to (i) Ag+, (ii) Pt2+, (iii) Pt4+, or (iv) Pd2+ ions to study the formation of intermetallic alloys, and the influence of the oxide shell on the galvanic replacement reaction. Rather surprising results are obtained: For i, only the core material is heavily affected by the galvanic exchange, whereas the shell is almost fully retained. In cases ii–iv, no indications for any galvanic exchange reactions are observed. Condition i is further a special case because of the following: (a) The nanocrystal shape is preserved, which is commonly only the case in cation exchange reactions due to anion framework conservation,17 whereas galvanic exchange usually results in hollow morphologies.2−10 (b) The galvanic exchange is performed in organic solvents, whereas in literature it is done predominantly in aqueous solutions. (c) The galvanic exchange results in a change of the core’s crystal structure and forms an intermetallic AgxSn phase with x ∼ 4. The latter enables the in situ monitoring of the exchange reaction by synchrotron X-ray diffraction (XRD), revealing the exchange dynamics taking place on the time scale of a couple of minutes. The absence of galvanic exchange in the cases ii–iv is explained by the key role of the oxide shell, which we identify to prohibit the Pt and Pd ions from passing. Qualitatively the same result is obtained when performing equivalent reactions with Sn shots in water. The natural oxide formed on the surface of the Sn shots prevents the exchange reaction in all of these conditions, except the Ag+ case. Removing the oxide shell in an acidic solution, however, enables the galvanic exchange in all cases.
Sn nanocrystals have been converted to intermetallic M–Sn nanocrystals before, by solution-based “conversion chemistry”, due to their relevance as catalysts or their antiferromagnetic properties.24 In these cases, for instance, Fe, Ni, and Co were used as M. Galvanic exchange, however, was ruled out to be the reason for the observed conversion of the Sn to intermetallic nanocrystals. Instead it was attributed rather to a diffusion-based process, which was obtained by treating Sn template nanocrystals with appropriate metal salts under highly reducing conditions.24 In contrast to this previous conversion chemistry with Sn nanocrystals, in this work no reducing agents are added, and much lower reaction temperatures are applied. Furthermore, instead of transition metals, noble metals are applied for the conversion, having higher reduction potentials of the redox couples. This makes galvanic exchange reactions more favorable, and as a result, the chemical conversion of the Sn nanocrystals is obtained on time scales which are much shorter than those applied in the previous conversion processes obtained by reduction of metal salts and subsequent diffusion.
Experimental Section
Nanocrystal Synthesis
For the synthesis of Sn nanocrystals, we have adopted our previous synthesis of highly monodisperse and size controlled InSn nanocrystals,45 simply by omitting the precursor for In and by optimizing the growth temperature. In a typical experiment, 20 g of oleylamine was loaded into a three neck flask and heated under vacuum for drying and purification (20 min@125 °C). Subsequently, the reaction flask was vented with argon, and the temperature was increased to 170 °C. Meanwhile, two solutions were loaded into syringes inside a glovebox. The first solution contained anhydrous octadecene (3 mL), 0.1 mL of bis[bis(trimethylsilyl)amino]tin(II), also called Sn-hexamethyldisilazide (Sn-HMDS), and 0.434 g of Li-HMDS, whereas the second solution was a mixture of 0.5 mL of 1.0 M Li[Et3BH] in tetrahydrofuran together with 0.6 mL of oleylamine. Solution number 1 was rapidly injected to the hot oleylamine in the reactor-flask. After the temperature was stabilized at 165 °C, also the second solution was injected into the reaction mixture. As is described in ref (40), the important role of the Li-HMDS in this synthesis is to deprotonate the oleylamine. The resulting Li-oleylamide can form together with the Sn-HMDS Sn-oleylamide complexes, or Sn-HMDS can react directly with oleylamine generating Sn-oleylamide, which is the actual precursor for the final reduction step. The nucleation and growth of the Sn nanocrystals is therefore analogous to the mechanism described in detail in ref (46). The reaction steps are illustrated in Figure S1 in the Supporting Information. During the second injection, the color of the solution rapidly changed from bright yellow-orange to dark-brown, indicating the formation of tin nanocrystals. After 10 s of reaction at 165 °C, the growth was interrupted by cooling of the flask by a cold water bath. To the crude solution of nanocrystals, 10 mL of toluene and 40 mL ethanol were added, and the mixture was centrifuged at 6000 rpm for 5 min. After the supernatant was removed, all precipitates were dispersed in 7 mL of toluene and 0.5 mL of oleic acid were added. Adding oleic acid is crucial to maintain complete coverage by the ligand molecules and therefore maintain the colloidal stability. A 25 mL portion of ethanol was added again to the solution, and a further centrifugation and redispersion in toluene was performed. The washing cycles were repeated three times. Finally, the nanocrystals were dispersed in toluene without additional oleic acid, to obtain a clear, black colloidal solution. During the washing procedure the nanocrystals are in contact with air, and a natural oxide is formed resulting in Sn/SnO2 core–shell nanocrystals.47
Galvanic Exchange
Toward Silver Nanocrystals
A 0.12 g portion of silver trifluoroacetate (TFA) was added in a three neck flask to 20 mL of oleylamine. The reaction mixture was evacuated and under vigorous stirring heated for 20 min to 90 °C. After venting the flask by argon, 20 mg of tin nanocrystals in 2 mL of chloroform was injected. The solution was kept for certain treatment times t t (specified in the text and when referring to specific samples) at the reaction temperature and afterward cooled with a cold water bath. Immediately after cooling, the crude solution was diluted 4 times by ethanol and centrifuged for 5 min at 6000 rpm. To avoid any remaining precursors, which could influence the elemental analysis, 5–7 washing cycles were applied. Figure 1a sketches the exchange reaction indicating the exchange of Sn atoms by Ag.
Figure 1.
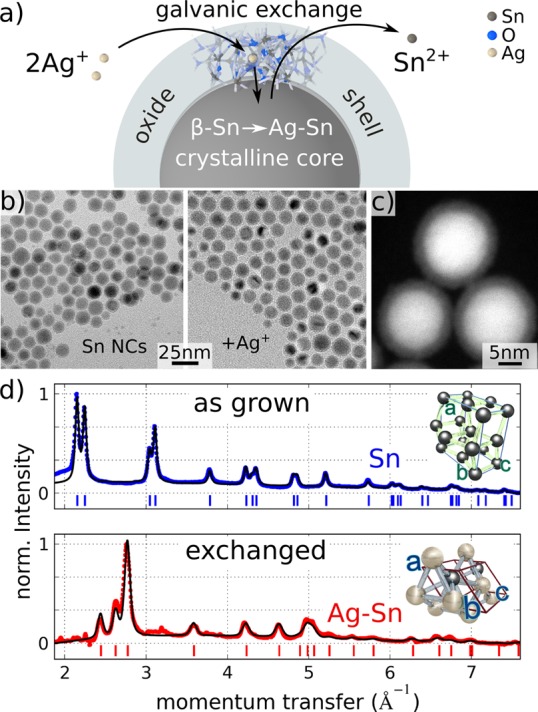
(a) Schematic of the galvanic exchange reaction through the oxide shell of the Sn/SnO2 core–shell nanocrystals. (b) Transmission electron microscopy images of the spherical particles after the synthesis and after the exchange reaction (15 min at 100 °C). (c) High angle annular dark field STEM image of the core/shell nanocrystals. (d) X-ray diffraction patterns from Sn nanocrystals after synthesis and Ag–Sn nanocrystals after galvanic exchange reaction with silver for 33 min at 90 °C. Shown are the experimental data (blue and red line) together with Rietveld refinements (black line). The patterns show the change of the crystal structure from the β-Sn (I41/amd(141), a = 5.837 Å and c = 3.187 Å) to the AgxSn (P63/mmc(194), a = 2.973 Å and c = 4.785 Å) structure upon the reaction. Bragg peak positions of the respective structures are shown as vertical lines.
Treatments by Platinum and Palladium
The galvanic exchange reactions for each type of Pt or Pd salt were attempted in analogy to that of Ag, by replacing the 0.12 g of silver trifluoroacetate with 0.132 g of platinum acetylacetonate, 0.12 g of PtCl2, 0.135 g of PtCl4, or 0.072 g of PdCl2.
In Situ Exchange During X-ray Diffraction
For the in situ experiments the reaction solutions were filled into Kapton capillaries with 1 mm diameter. First, Sn nanocrystals solved in hexadecylamine (HDA), kept at a temperature slightly above its melting point of ∼45–50 °C, were introduced into the capillaries to half-fill them. In a second step, after the Sn nanocrystal solution was frozen, the capillaries were topped up by silver trifluoroacetate which was also dissolved in HDA. The nanocrystal–HDA solution was prepared by adding 1 g of HDA to a 1.5 mL chloroform solution of tin nanocrystals (concentration of 30 mg/mL). The chloroform was then evaporated at 45 °C under vigorous stirring. The silver precursor mixture was prepared by dissolving 0.5 g of silver trifluoroacetate in 2 g of HDA at 50 °C.
Galvanic Exchange in Water
Additional galvanic exchange reactions with bulk Sn shots were attempted in aqueous environments. We used neutral and acidic water solutions for the salt types also used during the treatment of the nanocrystals. For this purpose, neutral water solutions were prepared by dissolution in 1 mL of deionized water: 0.0532 g PtCl2 and 0.035 g NaCl; or 0.0674 g PtCl4 and 0.035 g NaCl; or 0.0354 g PdCl2 and 0.035 g NaCl; or 0.0442 g Ag-TFA. The acidic solutions were prepared in analogy to the neutral ones by further adding 0.035 mL of 68% nitric acid, which results in 3% acid concentration. The millimeter sized pure Sn shots (99.9%) were loaded into each solution for 5 min in the case of Ag-TFA and PtCl4 solutions, and for 20 min for PtCl2 and PdCl2 solutions.
X-ray Diffraction
Synchrotron X-ray powder diffraction measurements were performed at the powder diffraction beamline P02.1 at PETRA III/DESY Hamburg with 60 keV X-ray energy.48 For the in situ experiments, a thermocouple mounted close to the sample was used to monitor its temperature while it was heated by a gas stream. Figure S2 shows a photograph of the diffraction setup. Data collection was done in transmission geometry with a PerkinElmer XRD1621 (400 × 400 mm2) two-dimensional detector at a distance of 1000 mm. For the data analysis, 1D diffraction patterns were obtained from those detector images (see Figure S3) by angular integration performed by xrayutilities.49 The obtained intensities are plotted versus the momentum transfer Q defined as (4π/λ) sin(2θ/2) with the angle 2θ measured between the scattered and primary beam. The galvanic exchange reaction was monitored by continuously taking 2D powder diffraction patterns, which in 1 s second exposition allows the collection of all necessary data. Rietveld refinement of the powder patterns was done with the analysis software MAUD.50
Energy-Dispersive X-ray Spectroscopy and Transmission Electron Microscopy Investigations
Chemical analysis and transmission electron microscopy (TEM) images were performed in a FEI TITAN3 80–300 electron microscope at 300 kV equipped with a CS-image-corrector. The energy-dispersive X-ray spectroscopy (EDXS) line-profiles were recorded drift corrected in scanning mode (STEM) with the FEI TIA software using a 30 mm2 EDAX detector. The recorded spectra were noise filtered by principal component analysis.51,52 The profiles were analyzed by the TIA software using the standardless k-factor method to quantify the O, Ag, Sn, and also the C content. Background correction was carried out semimanually, and automatic peak-fitting of the TIA software was used. The O-profile was adjusted by subtracting the constant O-content, which was determined from the C/O-ratio measured in the vicinity of the nanocrystal. Average shell contributions were subtracted weighted for the inner parts of the nanocrystal to separate the shell and the core contributions as described in the Supporting Information. The high resolution images were recorded CS-corrected in the TEM mode.
Results and Discussion
The synthesized Sn/SnO2 core/shell nanocrystals were inspected by TEM which provides a mean size of the spherically shaped nanocrystals of 13 nm, and provide evidence for their core/shell structure (Figure 1b,c). By synchrotron X-ray diffraction (XRD), in contrast, only peaks corresponding to the Sn cores are recorded, while no signals from any oxide shells are observed (Figure 1d). The XRD pattern corresponds to that of metallic β-Sn with lattice parameters, in agreement with the values in bulk.53,54 The Rietveld refinements also provide the average crystallite size of the metallic Sn-core of 12 nm, which is in agreement with the TEM data.
The driving force for a galvanic replacement reaction comes from the difference in reduction potentials of the two involved metals.9 For the exchange the reducing potential of the metal to be deposited has to be higher than the one which is corroded. Therefore, under standard conditions Sn (reduction potential −0.13 V in respect to a standard hydrogen electrode) should for example be replaced by Ag (reduction potential 0.8 V) and even more efficiently by Pt (reduction potential 1.18 V). The as-synthesized Sn nanocrystals are, however, not soluble in aqueous solutions due to their hydrophobic ligand shell, so that the galvanic exchange has to be performed under nonstandard conditions, for which predictions based on reduction potentials under standard conditions are not applicable. In our first series of experiments, the galvanic exchange was performed by injecting a chloroform solution of Sn nanocrystals into a heated solution (Ag-TFA) solved in oleylamine. Aliquots were taken for various treatment times t t , and the galvanic exchange was stopped by cooling the solution to room temperature. The galvanic exchange process hardly changed the size and shape of the nanocrystals, even for t t > 1 h (Figure 1b), but completely changed their crystal structure and chemical compositions. The XRD pattern taken after the treatment (Figure 1d) is obviously totally different than before the treatment. There are not any Bragg peaks remaining from the initial Sn diffraction pattern, and also no peaks indicating the presence of pure Ag are detected. The diffraction peaks could be indexed by a hexagonal structure of a silver rich intermetallic AgxSn, with lattice parameters a = 2.973 Å, c = 4.785 Å. If this new phase is either the ζ-phase (Ag4Sn crystal structure with space group P63/mmc(194)54,55) or the ε-phase (Ag3Sn with orthorhombic unit cell56), which is commonly used as a lead free solder,57 cannot be exactly determined within the accuracy of the XRD data. While the ε-phase has a distinct composition with Sn content of 24–25 at. %, the ζ-phase exists within the range of compositions between 12% and 22%, and both phases coexist in a certain range of compositions. Due to the similarity of these two crystal structures and the observed line broadening resulting from the finite size of the nanocrystals, the measurements could be equally well-indexed by the hexagonal ζ-phase and the orthorhombic unit cell of the ε-phase. However, the Sn to Ag conversion, performed for nanorods by interdiffusion, evidenced also the appearance of the Ag4Sn/ζ phase.58 The galvanic exchange reaction for the full nanocrystals can therefore be expressed as Sn3y + xyAg+ → (AgxSn)y + 2ySn2+, where y is a variable expressing the total amount of the Sn atoms in the nanocrystals. Using this equation and the 37% smaller van der Waals volume of Ag as compared to Sn, one can estimate the expected size change of the nanocrystals. For x in the range between 3 and 4 one expects that the nanocrystal size stays almost unchanged, in agreement with the observations in Figure 1b and the corresponding size histograms in Figure S4. The observed intermetallic AgxSn phase is stable against oxidation and does not undergo any changes at longer treatment times tt or at higher temperature (up to 150 °C, Figure S5). Note that the long time stability of the nanocrystal solutions is significantly improved after the galvanic exchange process. While untreated nanocrystal colloidal solutions stored under ambient conditions changed their color to transparent white and precipitated after six months, treated nanocrystals solutions show no degradation under the same storage conditions.
Recently, it was concluded from in situ monitoring the cation exchange reaction between CdSe and Cu2Se that the Cd to Cu exchange process is not diffusion limited, as might be intuitively expected, but it is rather a co-operative process.59,60 Any initial Cu+ doping of a CdSe nanocrystal enhances the likelihood for further Cu+ doping. This cooperativity caused the CdSe exciton luminescence to quench on a 100 ms time range, after a certain waiting time of several seconds, which was required to initiate the cation exchange. Since the galvanic exchange in the present metallic nanocrystals does not affect any easily accessible optical properties, here in situ synchrotron X-ray diffraction experiments were performed to study the dynamics of the exchange reaction. For this purpose the galvanic replacement was performed within small Kapton capillaries by placing the X-ray beam close to the interface between two interdiffusing liquids. One contains the nanocrystals, and the other one contains the Ag-TFA, the starting material for the galvanic exchange. Hexadecylamine is used as solvent for both, which is solid at room temperature (melting point 44 °C), so that the galvanic exchange reaction was started simply by heating, performed by a nitrogen stream with controlled temperature. For a sample temperature of 60 °C, the evolution of the nanocrystal Bragg peaks is shown in Figure 2a. As long as the nanocrystals and the Ag-TFA are immobilized in the frozen hexadecylamine, the Bragg peaks correspond to pure β-Sn. As soon as the two initially separate starting solutions melt and interdiffuse, the intensity of the peaks corresponding to the Sn phase decrease, and at the same time the reflections of the Ag–Sn intermetallic phase arise. Since the area under the Bragg peaks corresponds to the scattering volume and the peak width is related to the size of crystalline domains (see also the discussion in the Supporting Information), from the diffraction data the growth dynamics of the intermetallic Ag–Sn phase can be followed (Figure 2b).
Figure 2.
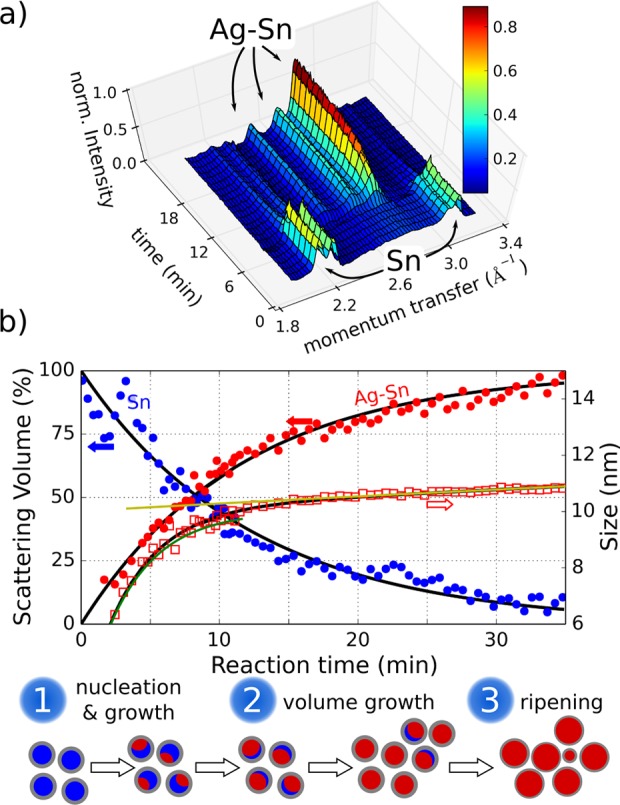
(a) X-ray diffraction patterns of Sn and Ag–Sn nanocrystals recorded in situ during the galvanic exchange reaction. Shown is a subset of Bragg peaks, which visualize the transition from the Sn phase to the Ag–Sn compound phase. (b) Scattering volume and nanocrystal size of the Sn and Ag–Sn phase during the galvanic exchange reaction extracted from the X-ray diffraction patterns. The time evolution of the scattering volume can be fit by exponential decay, and increase functions are plotted as solid black lines. The time evolution of the crystallite size of the Ag–Sn phase is described by an exponential growth together with ripening. These two contributions are shown as separate green and yellow lines, respectively. The reaction proceeds in different phases which are illustrated.
In bulk material the transformation of one phase into another one at constant temperature and as a function of time is described by the JMAK (Johnson–Mehl–Avrami–Kolmogorov) equation.61 It proposes an exponential decrease of the amount V of the initial phase, in our case the Sn, and a corresponding increase of the new phase by time t, following V(t) = 1–exp(−t/τ)n. Indeed, the time evolution of the scattering volumes of both the Sn and the AgxSn phases provides consistently the same time constant τ of ∼10 min for a reaction temperature of 60 °C. An Avrami exponent close to n = 1 is found to best describe our data, and we therefore fixed the value during the fitting. While the evolution of the scattering volume is well-described by this single time constant over the whole time span of the experiment, the evolution of particle size is more complex. Only within the first few minutes does the reaction fit well to the JMAK-type of equation (green line in Figure 2b), however, with a distinct time constant τ ∼ 3 min representing the nucleation and initial growth of the Ag–Sn phase. In contrast, at longer treatment times a different expression provides favorable agreement with the experimental data, namely, (Kt + S03)1/3, derived within the Lifshitz–Slyozov–Wagner theory62−64 to describe Ostwald ripening (yellow line in Figure 2b). More details about the fitting procedure can be found in the Supporting Information.
Thus, in total, the phase transformation during galvanic exchange from Sn to AgxSn can be roughly divided into three stages, within which either nucleation, growth, or ripening (Figure 2b) is the dominant process. The time scale on which the whole transformation takes place is also important, and is significantly longer than that observed for the cooperative cation exchange in Cd-chalcogenide nanocrystals.59 A reason for this might be that in the present case also the diffusion of precursors within the liquid medium and of the Ag+ ions through the protecting oxide shell of the Sn nanocrystals significantly contributes to the dynamics of the observed galvanic exchange process.
Besides the appearance of new crystallographic phases during cation exchange, it is also important to prove the chemical composition within the core/shell nanocrystals. For that purpose, EDXS in a scanning TEM was performed. The EDXS sum-spectrum shown in Figure 3a was calculated from the line-profile along the dashed line in Figure 3b. The sum-spectrum clearly provides evidence of the presence of both Ag and Sn in the nanocrystal after galvanic exchange, as well as traces of O, Cu, and Si. The Cu is ascribed to the sample support whereas the Si is an indication for the presence of residuals of HMDS. The high angle annular dark-field (HAADF) STEM image of the investigated nanocrystal (Figure 3b) unambiguously shows the core/shell structure with a core diameter of 19 nm and a shell thickness of 3 nm. The chemical composition profile of Figure 3c was calculated by using the Ag-L, Sn-L, and O-K peaks of the EDXS line-profile indicated in Figure 3b. Within the shell concentration gradients of all three elements are found. While at the surface the shell is oxygen rich and free of Ag, the Ag concentration increases to more than 20% at the core/shell interface. The Sn concentration increases within the shell from 22%, measured at the shell surface up to 32% at the interface to the core. To determine the core’s composition (from −9 to +9 nm, indicated by dashed lines in Figure 3c), the EDXS line scan was corrected by subtracting the shell signal proportionately to the shell/core electron path fraction. As a result the core composition is found to be homogeneous and to consist of 68% Ag and 32% Sn, with an estimated uncertainty of 5%. In a second, somewhat smaller, analyzed nanocrystal from the same batch, a different core composition of 81% Ag and 19% Sn was detected (Figure S7). Thus, as from the XRD data, the Ag to Sn ratio could correspond to either Ag4Sn/ζ or Ag3Sn/ε intermetallic phases. The most important insight from this analysis is, however, that the Ag concentration obviously increases within the SnOx shell. From the nanocrystal surface toward the core, the Ag signal rises from 0% to more than 60%, even though during the galvanic exchange treatment the Ag+ ions have to penetrate through the SnO2 shell. This observation indicates the decisive role of the oxide shell in the galvanic exchange process.
Figure 3.
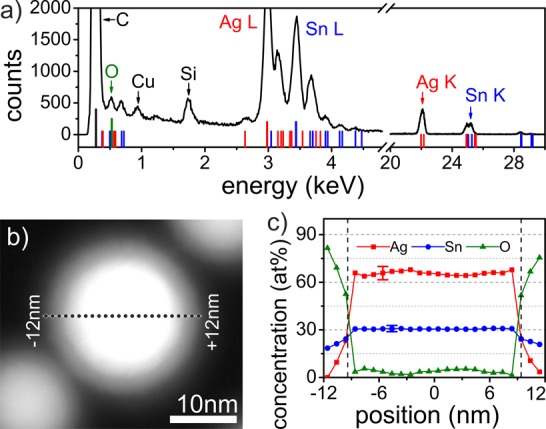
(a) Summed EDXS spectra of a Ag–Sn core/shell nanocrystal after the exchange reaction. Indicated red, blue, and green lines are the characteristic X-ray lines of Ag, Sn, and O. (b) HAADF STEM image of the investigated nanocrystals. A dashed line shows the position of the taken EDXS line scan. (c) Atomic concentrations extracted from the EDXS line scan of the nanocrystal shown in panel b. In the core region (between the dashed lines) the contribution from the shell was subtracted as described in the text. Two representative error bars for the Ag and Sn concentrations define the 2σ-confidence levels of the analyzed peaks.
While the XRD data and the elemental analysis confirm undoubtedly the galvanic exchange from Sn toward an Ag–Sn intermetallic phase, this exchange is also in reasonable agreement with predictions based on Pearson’s hard and soft acids and bases (HSAB) theory. This theory is frequently applied to systems for which parameters like chemical activity or reduction potential are not determined, and it provides empirical principles65−67(Figure S8 and Table S1) to propose basic interactions between solvent and solute or between ions and ligands. Since Sn2+ and the used solvent oleylamine in the term of HSAB represent a hard acid–base couple, these compounds tend to bind strongly, and to form ionic complexes, resulting in good solvation of Sn2+ by oleylamine. In contrast, Ag+ is a soft acid, which has weak interactions with the hard base solvent and therefore tends to phase separate, for example by precipitation on the surface of the nanocrystals.65−67 The same arguments can be applied to the soft acid Pt2+ and Pd2+, which according to HSAB argumentation would even more likely perform the galvanic exchange in oleylamine to Sn (Figure S8). The galvanic exchange treatment attempted with Pt-acetylacetonate instead of the Ag precursor, however, did not result in any change of the nanocrystal composition at all. This is proven by the EDXS data in Figure S9 taken before and after the Pt2+ treatment which are almost identical and do not show any Pt related signals. Also, making use of more reactive PtCl2, PtCl4, and PdCl2 did not result in any exchange reactions as seen in Figure S6 where the X-ray diffraction signal after the treatment still shows a diffraction signal of only β-Sn. This behavior apparently is due to a different effect not explained by the HSAB theory, resulting from the nanocrystal’s oxide shell.
It should be noted that oxidation of Sn by Ag+ ions, as it takes place during the galvanic exchange reaction (Sn0 + 2Ag+ → Sn2+ + 2Ag0), will not affect the SnO2 shell significantly, because there the Sn is already in the fully oxidized 4+ state. Ag+ ions only diffuse through the shell toward the metallic Sn core, where the galvanic exchange will take place. A closer analysis of the shell structure was performed by high resolution TEM imaging, by using an aberration-corrected transmission electron microscope. The images in Figure 4 reveal the monocrystalline nature of the nanocrystal cores, which are covered by either an amorphous or a polycrystalline shell. In both types of shells, the diffusivity is much higher as compared to that in monocrystalline materials, because it is either mediated by dangling bonds,68 or facilitated by pipe diffusion.69 The Ag+, Pt2+, and Pd2+ ions are too large to allow sufficient diffusion via interstitials, even in the lower density of the amorphous shell. The most probable material transport through the shell is therefore given by diffusive jumps occurring via an exchange of positions with an adjacent vacancy.69 This diffusion of cationic Frenkel pairs70 can account for both the transport of Sn2+ from the nanocrystal core toward its surface as well as for the diffusion of Ag+ from the surface toward the core. In both cases, the diffusion is also related to the formation and diffusion of dangling bonds, since the Sn4+ cations in the SnO2 crystal structure have higher coordination than Sn2+ and Ag+ in SnO and Ag2O, respectively. Such a diffusion pathway is at the given process temperature, however, inhibited for Pt and Pd ions, due to their inert properties in respect to oxygen binding. Thus, the amorphous SnO2 shell is semipermeable, and protects the nanocrystal cores from galvanic exchange, depending on the oxidative properties of the exchanging cations.
Figure 4.

High resolution transmission electron microscope images of a Sn/SnO2 core/shell nanocrystal with (a) amorphous and (b) polycrystalline shell. In both cases the core is single crystalline.
To confirm the key role of the oxide shell we have performed reference experiments with millimeter sized Sn shots in water. Also on these Sn shots a naturally formed oxide exists, which according to our arguments above should prevent the galvanic exchange in the Pt and Pd case. Figure 5 shows the corresponding photographs of the Sn shots after the respective treatment. While the silvery-white color of Sn is preserved after treatments with PtCl2, PtCl4, and PdCl2 in neutral water, the color is visually changed after treatment with Ag-TFA. The picture is radically changing when instead of the water 3% nitric acid is used which removes the oxide shell and treatment with all used salts results in a strong color change. We chose nitric acid instead of hydrochloric to avoid formation of hydrogen gas during the reaction with metallic tin, which otherwise would reduce noble metal salts to metals. The interpretation based on the visual appearance is supported by EDXS analysis shown in Figures S10–12, which for treatment with PtCl2, PtCl4, and PdCl2 a change of the chemical composition is detected only after treatment in acidic solution. On the other hand the equivalent EDXS analysis after treatment with Ag-TFA (Figure S13) detects incorporation of Ag also for the treatment in neutral water, supporting the interpretation of our nanocrystal experiments.
Figure 5.

Photographs of Sn shots after treatment with PtCl2, PtCl4, PdCl2, and Ag-TFA salts. The two lines of images show the result of the treatment in neutral water and in 3% nitric acid, respectively.
The galvanic exchange is driven by differences in reduction potentials, which depend on the precursors types, their concentrations, the environment, and chemical activities, and is empirically described by the HSAB theory. The fact that the conversion from Sn toward Ag in the nanocrystals stopped as soon as the intermetallic Ag4Sn phase is reached indicates that the difference of reduction potential between Sn and Ag is not beneficial in this configuration. This might be caused by changes of the Sn and Ag reduction potentials due to their altered environment within the Ag4Sn phase, where the Sn ions are highly, 12-times, coordinated (Figure S14). Furthermore, it is worth considering that galvanic exchange often results in the formation of hollow structures, since the location of reduction of the exchanging ions is usually not the same as the location of the oxidation of the sacrificing ions. The nanocrystals’ growth and dissolution on different locations change their morphology.8 In the present case, however, the sites of reduction and oxidation on the nanocrystal core material are restricted by cation diffusion channels through the oxide shell. This makes a one to one replacement of ions at the same site much more probable, which does then not result in any morphology transformation.
Conclusion
The galvanic exchange method was applied to metallic nanocrystals covered by a protective metal-oxide shell, which affects the exchange in several aspects. In particular, Sn/SnO2 nanocrystals were treated by Ag+, Pt2+, Pt4+, and Pd2+ ions in organic colloidal solutions, providing completely different results. In the first case, the core composition is changed during galvanic exchange from pure Sn toward an intermetallic Ag–Sn phase, whereas the shell composition is hardly changed. The galvanic exchange, converting the core composition on time scales of several minutes, requires vacancy assisted diffusion of Ag+ through the oxide shell. The latter is apparently inhibited for Pt and Pd ions, and thus, the core composition remains unaffected by the treatment. Similar experiments performed for bulk Sn shots confirm our results on the nanocrystals and also identify the key role of the oxide shell. This shows that the semipermeability of the metal-oxide shell in respect to the diffusion of metals ions has to be taken into account when galvanic exchange treatments are performed. This is not only relevant for Sn nanocrystals, but also for a series of base metals, forming natural oxide shells (such as Al, Fe, Ni, or Cu) and can open the way to novel alloyed core/shell combinations fabricated via galvanic exchange, with optimized properties for a wide range of applications spanning from nano-optics and magnetics to hydrogen evolution.
Acknowledgments
We acknowledge the help of the beamline staff at beamline BM20 at the ESRF (Carsten Bähtz and Olga Roshchupkina) and staff at beamline P02.1 at DESY, for assistance with the synchrotron experiments. DESY is a member of the Helmholtz Association (HGF). Financial support was given by the Austrian Science Fund FWF (IRON SFB25 and J3317-N27) and Czech Science Foundation (14-08124S). D.K. acknowledges support by the Austrian Academy of Sciences (DOC-fellowship). H.G. acknowledges the financial support by the Austrian Federal Ministry of Economy, Family and Youth and the National Foundation for Research, Technology and Development. A part of the research was also performed at the Energie Campus Nürnberg and supported by funding through the “Aufbruch Bayern” initiative of the state of Bavaria.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcc.6b06405.
Experimental procedures and characterization details as well as additional figures (PDF)
Author Contributions
∇ D.K. and M.S. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Sun Y.; Xia Y. Shape-controlled Synthesis of Gold and Silver Nanoparticles. Science 2002, 298, 2176–2179. 10.1126/science.1077229. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Mayers B. T.; Xia Y. Template-Engaged Replacement Reaction: A One-Step Approach to the Large-Scale Synthesis of Metal Nanostructures with Hollow Interiors. Nano Lett. 2002, 2, 481–485. 10.1021/nl025531v. [DOI] [Google Scholar]
- Sun Y.; Xia Y. Alloying and Dealloying Processes Involved in the Preparation of Metal Nanoshells through a Galvanic Replacement Reaction. Nano Lett. 2003, 3, 1569–1572. 10.1021/nl034765r. [DOI] [Google Scholar]
- Chen J.; Saeki F.; Wiley B. J.; Cang H.; Cobb M. J.; Li Z.-Y.; Au L.; Zhang H.; Kimmey M. B.; Li X.; et al. Gold Nanocages: Bioconjugation and their Potential Use as Optical Imaging Contrast Agents. Nano Lett. 2005, 5, 473–477. 10.1021/nl047950t. [DOI] [PubMed] [Google Scholar]
- Chen J.; Wiley B.; McLellan J.; Xiong Y.; Li Z.-Y.; Xia Y. Optical Properties of Pd-Ag and Pt-Ag Nanoboxes Synthesized via Galvanic Replacement Reactions. Nano Lett. 2005, 5, 2058–2062. 10.1021/nl051652u. [DOI] [PubMed] [Google Scholar]
- Lou X. W. D.; Archer L. A.; Yang Z. Hollow Micro-/Nanostructures: Synthesis and Applications. Adv. Mater. 2008, 20, 3987–4019. 10.1002/adma.200800854. [DOI] [Google Scholar]
- Au L.; Lu X.; Xia Y. A Comparative Study of Galvanic Replacement Reactions Involving Ag Nanocubes and AuCl(2) or AuCl(4). Adv. Mater. 2008, 20, 2517–2522. 10.1002/adma.200800255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González E.; Arbiol J.; Puntes V. F. Carving at the Nanoscale: Sequential Galvanic Exchange and Kirkendall Growth at Room Temperature. Science 2011, 334, 1377–1380. 10.1126/science.1212822. [DOI] [PubMed] [Google Scholar]
- Xia X.; Wang Y.; Ruditskiy A.; Xia Y. 25th Anniversary Article: Galvanic Replacement: a Simple and Versatile Route to Hollow Nanostructures with Tunable and Well-Controlled Properties. Adv. Mater. 2013, 25, 6313–6333. 10.1002/adma.201302820. [DOI] [PubMed] [Google Scholar]
- Oh M. H.; Yu T.; Yu S.-H.; Lim B.; Ko K.-T.; Willinger M.-G.; Seo D.-H.; Kim B. H.; Cho M. G.; Park J.-H.; et al. Galvanic Replacement Reactions in Metal Oxide Nanocrystals. Science 2013, 340, 964–968. 10.1126/science.1234751. [DOI] [PubMed] [Google Scholar]
- Li Q.; Xu P.; Zhang B.; Wu G.; Zhao H.; Fu E.; Wang H.-L. Self-Supported Pt Nanoclusters via Galvanic Replacement from Cu2O Nanocubes as Efficient Electrocatalysts. Nanoscale 2013, 5, 7397–7402. 10.1039/c3nr02243a. [DOI] [PubMed] [Google Scholar]
- Yalcin A. O.; Fan Z.; Goris B.; Li W.-F.; Koster R. S.; Fang C.-M.; van Blaaderen A.; Casavola M.; Tichelaar F. D.; Bals S.; et al. Atomic Resolution Monitoring of Cation Exchange in CdSe-PbSe Heteronanocrystals during Epitaxial Solid-Solid-Vapor Growth. Nano Lett. 2014, 14, 3661–3667. 10.1021/nl501441w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goris B.; Polavarapu L.; Bals S.; Van Tendeloo G.; Liz-Marzán L. M. Monitoring Galvanic Replacement through Three-Dimensional Morphological and Chemical Mapping. Nano Lett. 2014, 14, 3220–3226. 10.1021/nl500593j. [DOI] [PubMed] [Google Scholar]
- Nafria R.; Genç A.; Ibáñez M.; Arbiol J.; Ramírez de la Piscina P.; Homs N.; Cabot A. Co-Cu Nanoparticles: Synthesis by Galvanic Replacement and Phase Rearrangement during Catalytic Activation. Langmuir 2016, 32, 2267–2276. 10.1021/acs.langmuir.5b04622. [DOI] [PubMed] [Google Scholar]
- Son D. H.; Hughes S. M.; Yin Y.; Alivisatos A. P. Cation Exchange Reactions in Ionic Nanocrystals. Science 2004, 306, 1009–1012. 10.1126/science.1103755. [DOI] [PubMed] [Google Scholar]
- Luther J. M.; Zheng H.; Sadtler B.; Alivisatos A. P. Synthesis of PbS Nanorods and Other Ionic Nanocrystals of Complex Morphology by Sequential Cation Exchange Reactions. J. Am. Chem. Soc. 2009, 131, 16851–16857. 10.1021/ja906503w. [DOI] [PubMed] [Google Scholar]
- Jain P. K.; Amirav L.; Aloni S.; Alivisatos A. P. Nanoheterostructure Cation Exchange: Anionic Framework Conservation. J. Am. Chem. Soc. 2010, 132, 9997–9999. 10.1021/ja104126u. [DOI] [PubMed] [Google Scholar]
- Li H.; Brescia R.; Krahne R.; Bertoni G.; Alcocer M. J. P.; D’Andrea C.; Scotognella F.; Tassone F.; Zanella M.; De Giorgi M.; et al. Blue-UV-Emitting ZnSe(Dot)/ZnS(Rod) Core/Shell Nanocrystals Prepared from CdSe/CdS Nanocrystals by Sequential Cation Exchange. ACS Nano 2012, 6, 1637–1647. 10.1021/nn204601n. [DOI] [PubMed] [Google Scholar]
- Rivest J. B.; Jain P. K. Cation Exchange on the Nanoscale: an Emerging Technique for New Material Synthesis, Device Fabrication, and Chemical Sensing. Chem. Soc. Rev. 2013, 42, 89–96. 10.1039/C2CS35241A. [DOI] [PubMed] [Google Scholar]
- Beberwyck B. J.; Surendranath Y.; Alivisatos A. P. Cation Exchange: A Versatile Tool for Nanomaterials Synthesis. J. Phys. Chem. C 2013, 117, 19759–19770. 10.1021/jp405989z. [DOI] [Google Scholar]
- Sytnyk M.; Kirchschlager R.; Bodnarchuk M. I.; Primetzhofer D.; Kriegner D.; Enser H.; Stangl J.; Bauer P.; Voith M.; Hassel A. W.; et al. Tuning the Magnetic Properties of Metal Oxide Nanocrystal Heterostructures by Cation Exchange. Nano Lett. 2013, 13, 586–593. 10.1021/nl304115r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akkerman Q. A.; Genovese A.; George C.; Prato M.; Moreels I.; Casu A.; Marras S.; Curcio A.; Scarpellini A.; Pellegrino T.; et al. From Binary Cu2S to Ternary Cu-In-S and Quaternary Cu-In-Zn-S Nanocrystals with Tunable Composition via Partial Cation Exchange. ACS Nano 2015, 9, 521–531. 10.1021/nn505786d. [DOI] [PubMed] [Google Scholar]
- van der Stam W.; Bladt E.; Rabouw F. T.; Bals S.; Donega C. d. M. Near-Infrared Emitting CuInSe*/CuInS* Dot Core/Rod Shell Heteronanorods by Sequential Cation Exchange. ACS Nano 2015, 9, 11430–11438. 10.1021/acsnano.5b05496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou N. H.; Schaak R. E. Shape-Controlled Conversion of Beta-Sn Nanocrystals into Intermetallic M-Sn (M = Fe, Co, Ni, Pd) Nanocrystals. J. Am. Chem. Soc. 2007, 129, 7339–7345. 10.1021/ja069032y. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Zhang Q.; Xie J.; Lee J. Y. Engineering the Architectural Diversity of Heterogeneous Metallic Nanocrystals. Nat. Commun. 2013, 4, 1454. 10.1038/ncomms2474. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Zhang Q.; Yao Q.; Xie J.; Lee J. Y. Guiding Principles in the Galvanic Replacement Reaction of an Underpotentially Deposited Metal Layer for Site-Selective Deposition and Shape and Size Control of Satellite Nanocrystals. Chem. Mater. 2013, 25, 4746–4756. 10.1021/cm402734r. [DOI] [Google Scholar]
- Yu Y.; Zhang Q.; Yao Q.; Xie J.; Lee J. Y. Architectural Design of Heterogeneous Metallic Nanocrystals–Principles and Processes. Acc. Chem. Res. 2014, 47, 3530–3540. 10.1021/ar5002704. [DOI] [PubMed] [Google Scholar]
- Sra A. K.; Ewers T. D.; Schaak R. E. Direct Solution Synthesis of Intermetallic AuCu and AuCu3 Nanocrystals and Nanowire Networks. Chem. Mater. 2005, 17, 758–766. 10.1021/cm0484450. [DOI] [Google Scholar]
- Cable R. E.; Schaak R. E. Low-Temperature Solution Synthesis of Nanocrystalline Binary Intermetallic Compounds Using the Polyol Process. Chem. Mater. 2005, 17, 6835–6841. 10.1021/cm0520113. [DOI] [Google Scholar]
- Vasquez Y.; Luo Z.; Schaak R. E. Low-Temperature Solution Synthesis of the Non-Equilibrium Ordered Intermetallic Compounds Au3Fe, Au3Co, and Au3Ni as Nanocrystals. J. Am. Chem. Soc. 2008, 130, 11866–11867. 10.1021/ja804858u. [DOI] [PubMed] [Google Scholar]
- Bondi J. F.; Misra R.; Ke X.; Sines I. T.; Schiffer P.; Schaak R. E. Optimized Synthesis and Magnetic Properties of Intermetallic Au3Fe1–x, Au3Co1–x, and Au3Ni1–x Nanoparticles. Chem. Mater. 2010, 22, 3988–3994. 10.1021/cm100705c. [DOI] [Google Scholar]
- Shavel A.; Rodríguez-González B.; Spasova M.; Farle M.; Liz-Marzán L. Synthesis and Characterization of Iron/Iron Oxide Core/Shell Nanocubes. Adv. Funct. Mater. 2007, 17, 3870–3876. 10.1002/adfm.200700494. [DOI] [Google Scholar]
- Pedersen D. B.; Wang S.; Liang S. H. Charge-Transfer-Driven Diffusion Processes in Cu@Cu-Oxide Core–Shell Nanoparticles: Oxidation of 3.0 ± 0.3 nm Diameter Copper Nanoparticles. J. Phys. Chem. C 2008, 112, 8819–8826. 10.1021/jp710619r. [DOI] [Google Scholar]
- Xu Y.-F.; Gao M.-R.; Zheng Y.-R.; Jiang J.; Yu S.-H. Nickel/Nickel(II) Oxide Nanoparticles Anchored onto Cobalt(IV) Diselenide Nanobelts for the Electrochemical Production of Hydrogen. Angew. Chem., Int. Ed. 2013, 52, 8546–8550. 10.1002/anie.201303495. [DOI] [PubMed] [Google Scholar]
- Zhou Z.; Sun Y.; Shen J.; Wei J.; Yu C.; Kong B.; Liu W.; Yang H.; Yang S.; Wang W. Iron/Iron Oxide Core/Shell Nanoparticles for Magnetic Targeting MRI and Near-Infrared Photothermal Therapy. Biomaterials 2014, 35, 7470–7478. 10.1016/j.biomaterials.2014.04.063. [DOI] [PubMed] [Google Scholar]
- McClain M. J.; Schlather A. E.; Ringe E.; King N. S.; Liu L.; Manjavacas A.; Knight M. W.; Kumar I.; Whitmire K. H.; Everitt H. O.; et al. Aluminum Nanocrystals. Nano Lett. 2015, 15, 2751–2755. 10.1021/acs.nanolett.5b00614. [DOI] [PubMed] [Google Scholar]
- Sobhani A.; Manjavacas A.; Cao Y.; McClain M. J.; García de Abajo F. J.; Nordlander P.; Halas N. J. Pronounced Linewidth Narrowing of an Aluminum Nanoparticle Plasmon Resonance by Interaction with an Aluminum Metallic Film. Nano Lett. 2015, 15, 6946–6951. 10.1021/acs.nanolett.5b02883. [DOI] [PubMed] [Google Scholar]
- Zhou L.; Zhang C.; McClain M. J.; Manjavacas A.; Krauter C. M.; Tian S.; Berg F.; Everitt H. O.; Carter E. A.; Nordlander P.; et al. Aluminum Nanocrystals as a Plasmonic Photocatalyst for Hydrogen Dissociation. Nano Lett. 2016, 16, 1478–1484. 10.1021/acs.nanolett.5b05149. [DOI] [PubMed] [Google Scholar]
- Xu L.; Kim C.; Shukla A. K.; Dong A.; Mattox T. M.; Milliron D. J.; Cabana J. Monodisperse Sn Nanocrystals as a Platform for the Study of Mechanical Damage during Electrochemical Reactions with Li. Nano Lett. 2013, 13, 1800–1805. 10.1021/nl400418c. [DOI] [PubMed] [Google Scholar]
- Kravchyk K.; Protesescu L.; Bodnarchuk M. I.; Krumeich F.; Yarema M.; Walter M.; Guntlin C.; Kovalenko M. V. Monodisperse and Inorganically Capped Sn and Sn/SnO2 Nanocrystals for High-Performance Li-ion Battery Anodes. J. Am. Chem. Soc. 2013, 135, 4199–4202. 10.1021/ja312604r. [DOI] [PubMed] [Google Scholar]
- Wang H.; Rogach A. L. Hierarchical SnO2 Nanostructures: Recent Advances in Design, Synthesis, and Applications. Chem. Mater. 2014, 26, 123–133. 10.1021/cm4018248. [DOI] [Google Scholar]
- Zhu Z.; Wang S.; Du J.; Jin Q.; Zhang T.; Cheng F.; Chen J. Ultrasmall Sn Nanoparticles Embedded in Nitrogen-Doped Porous Carbon as High-Performance Anode for Lithium-Ion Batteries. Nano Lett. 2014, 14, 153–157. 10.1021/nl403631h. [DOI] [PubMed] [Google Scholar]
- Bodnarchuk M. I.; Kravchyk K. V.; Krumeich F.; Wang S.; Kovalenko M. V. Colloidal Tin-Germanium Nanorods and their Li-ion Storage Properties. ACS Nano 2014, 8, 2360–2368. 10.1021/nn4058227. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Jiang L.; Wang C. Preparation of a Porous Sn@C Nanocomposite as a High-Performance Anode Material for Lithium-Ion Batteries. Nanoscale 2015, 7, 11940–11944. 10.1039/C5NR03093E. [DOI] [PubMed] [Google Scholar]
- Yarema M.; Pichler S.; Kriegner D.; Stangl J.; Yarema O.; Kirchschlager R.; Tollabimazraehno S.; Humer M.; Häringer D.; Kohl M.; et al. From Highly Monodisperse Indium and Indium Tin Colloidal Nanocrystals to Self-Assembled Indium Tin Oxide Nanoelectrodes. ACS Nano 2012, 6, 4113–4121. 10.1021/nn3005558. [DOI] [PubMed] [Google Scholar]
- Yarema M.; Caputo R.; Kovalenko M. V. Precision Synthesis of Colloidal Inorganic Nanocrystals using Metal and Metalloid Amides. Nanoscale 2013, 5, 8398–8410. 10.1039/c3nr02076b. [DOI] [PubMed] [Google Scholar]
- Protesescu L.; Rossini A. J.; Kriegner D.; Valla M.; de Kergommeaux A.; Walter M.; Kravchyk K. V.; Nachtegaal M.; Stangl J.; Malaman B.; et al. Unraveling the Core–Shell Structure of Ligand-Capped Sn/SnOxNanoparticles by Surface-Enhanced Nuclear Magnetic Resonance, Mössbauer, and X-ray Absorption Spectroscopies. ACS Nano 2014, 8, 2639–2648. 10.1021/nn406344n. [DOI] [PubMed] [Google Scholar]
- Dippel A.-C.; Liermann H.-P.; Delitz J. T.; Walter P.; Schulte-Schrepping H.; Seeck O. H.; Franz H. Beamline P02.1 at PETRA III for High-Resolution and High-Energy Powder Diffraction. J. Synchrotron Radiat. 2015, 22, 675–687. 10.1107/S1600577515002222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegner D.; Wintersberger E.; Stangl J. Xrayutilities: A Versatile Tool for Reciprocal Space Conversion of Scattering Data Recorded with Linear and Area Detectors. J. Appl. Crystallogr. 2013, 46, 1162–1170. 10.1107/S0021889813017214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutterotti L.; Matthies S.; Wenk H.-R. MAUD: A Friendly Java Program for Material Analysis using Diffraction. IUCr: Newsletter of the CPD 1999, 21, 14–15. [Google Scholar]
- Kotula P. G.; Keenan M. R.; Michael J. R. Automated Analysis of SEM X-Ray Spectral Images: A Powerful New Microanalysis Tool. Microsc. Microanal. 2003, 9, 1–17. 10.1017/S1431927603030058. [DOI] [PubMed] [Google Scholar]
- Keenan M. R.; Kotula P. G. Accounting for Poisson Noise in the Multivariate Analysis of ToF-SIMS Spectrum Images. Surf. Interface Anal. 2004, 36, 203–212. 10.1002/sia.1657. [DOI] [Google Scholar]
- Wyckhoff R. W. G.Crystal Structures; Interscience Publishers: New York, 1964. [Google Scholar]
- Madelung O.Ag-Sn (Silver-Tin). Landolt-Börnstein—Group IV Physical Chemistry; SpringerMaterials—The Landolt-Börnstein Database; Predel B., Ed.; http://www.springermaterials.com; 1991. [Google Scholar]
- King H. W.; Massalski T. B. Lattice Spacing Relationships and the Electronic Structure of H.C.P. ζ Phases Based on Silver. Philos. Mag. 1961, 6, 669–682. 10.1080/14786436108244417. [DOI] [Google Scholar]
- Fairhurst C. W.; Cohen J. B. The Crystal Structures of Two Compounds Found in Dental Amalgam: Ag2Hg3 and Ag3Sn. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1972, 28, 371–378. 10.1107/S0567740872002432. [DOI] [Google Scholar]
- Frear D. R.; Jang J. W.; Lin J. K.; Zhang C. Pb-Free Solders for Flip-Chip Interconnects. JOM 2001, 53, 28–33. 10.1007/s11837-001-0099-3. [DOI] [Google Scholar]
- Chou N. H.; Schaak R. E. A Library of Single-Crystal Metal–Tin Nanorods: Using Diffusion as a Tool for Controlling the Morphology of Intermetallic Nanocrystals. Chem. Mater. 2008, 20, 2081–2085. 10.1021/cm703640u. [DOI] [Google Scholar]
- White S. L.; Smith J. G.; Behl M.; Jain P. K. Co-Operativity in a Nanocrystalline Solid-State Transition. Nat. Commun. 2013, 4, 2933. 10.1038/ncomms3933. [DOI] [PubMed] [Google Scholar]
- Routzahn A. L.; Jain P. K. Single-Nanocrystal Reaction Trajectories Reveal Sharp Cooperative Transitions. Nano Lett. 2014, 14, 987–992. 10.1021/nl4044289. [DOI] [PubMed] [Google Scholar]
- Avrami M. Kinetics of Phase Change. I General Theory. J. Chem. Phys. 1939, 7, 1103–1112. 10.1063/1.1750380. [DOI] [Google Scholar]
- Lifshitz I. M.; Slyozov V. V. The Kinetics of Precipitation from Supersaturated Solid Solutions. J. Phys. Chem. Solids 1961, 19, 35–50. 10.1016/0022-3697(61)90054-3. [DOI] [Google Scholar]
- Wagner C. Theorie der Alterung von Niederschlägen durch Umlösen (Ostwald-Reifung). Z. Elektrochem. 1961, 65, 581–591. [Google Scholar]
- Kahlweit M. Ostwald Ripening of Precipitates. Adv. Colloid Interface Sci. 1975, 5, 1–35. 10.1016/0001-8686(75)85001-9. [DOI] [Google Scholar]
- Ahrland S.; Chatt J.; Davies N. R. The Relative Affinities of Ligand Atoms for Acceptor Molecules and Ions. Q. Rev., Chem. Soc. 1958, 12, 265–276. 10.1039/qr9581200265. [DOI] [Google Scholar]
- Pearson R. G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. 10.1021/ja00905a001. [DOI] [Google Scholar]
- Misono M.; Ochiai E.; Saito Y.; Yoneda Y. A New Dual Parameter Scale for the Strength of Lewis Acids and Bases with the Evaluation of their Softness. J. Inorg. Nucl. Chem. 1967, 29, 2685–2691. 10.1016/0022-1902(67)80006-X. [DOI] [Google Scholar]
- Mirabella S.; de Salvador D.; Bruno E.; Napolitani E.; Pecora E. F.; Boninelli S.; Priolo F. Mechanism of Boron Diffusion in Amorphous Silicon. Phys. Rev. Lett. 2008, 100, 155901. 10.1103/PhysRevLett.100.155901. [DOI] [PubMed] [Google Scholar]
- Borg R. J.; Dienes G. J.. An Introduction to Solid State Diffusion; Academic Press, Harcourt Brace Jovanovich Publishers: Boston, 1988. [Google Scholar]
- Groeneveld E.; Witteman L.; Lefferts M.; Ke X.; Bals S.; Van Tendeloo G.; Donega C. d. M. Tailoring ZnSe-CdSe Colloidal Quantum Dots via Cation Exchange: from Core/Shell to Alloy Nanocrystals. ACS Nano 2013, 7, 7913–7930. 10.1021/nn402931y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.