

Abstract

Directly disrupting the Keap1-Nrf2 protein–protein interaction (PPI) has emerged as an attractive way to activate Nrf2, and Keap1-Nrf2 PPI inhibitors have been proposed as potential agents to relieve inflammatory and oxidative stress diseases. In this work, we investigated the diacetic moiety around the potent Keap1-Nrf2 PPI inhibitor DDO1018 (2), which was reported by our group previously. Exploration of bioisosteric replacements afforded the ditetrazole analog 7, which maintains the potent PPI inhibition activity (IC50 = 15.8 nM) in an in vitro fluorescence polarization assay. Physicochemical property determination demonstrated that ditetrazole replacement can improve the drug-like property, including elevation of pKa, log D, and transcellular permeability. Additionally, 7 is more efficacious than 2 on inducing the expression of Nrf2-dependent gene products in cells. This study provides an alternative way to replace the diacetic moiety and occupy the polar subpockets in Keap1, which can benefit the subsequent development of Keap1-Nrf2 PPI inhibitor.
Keywords: Keap1, Nrf2, protein−protein interaction
Oxidative stresses exacerbate many chronic diseases, including cancer, cardiovascular diseases, chronic inflammation, and neurodegenerative diseases.1 The constant stresses imposed by electrophiles and oxidants can increase the levels of oxidized proteins, phospholipids, and DNA, which finally contribute to the neoplastic and chronic degenerative diseases.2 The transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2), a member of the Cap’N’collar family of proteins, is the primary regulator of cell defensive mechanisms against oxidative stresses. Nrf2 binds to the antioxidant response element (ARE) and regulates the transcription of a battery of cell protective genes (about 200), including phase 1 and 2 enzymes, and glutathione-based and thioredoxin-based antioxidant systems.3
Kelch-like ECH-associated protein-1 (Keap1), a Bric-a-brac/Tram-track/Broad (BTB) protein, is responsible for the regulation of Nrf2 activity. Under normal conditions, Keap1 acts as a substrate adaptor component in the assembly of the Cullin3 (Cul3)-based ubiquitin E3 ligase complex. This E3 ligase complex shepherds Nrf2 toward the polyubiquitination and degradation by the 26S proteasome, which limits the Nrf2 activity at a basal level.4−6 Under stress conditions, the cysteine residues in the Keap1 protein are covalently modified by oxidative and/or electrophilic agents, which cause the conformational changes of the complex.7,8 These changes relieve Nrf2 from Keap1-meditated depression and activate the Nrf2-ARE pathway to induce the transcription of downstream genes, which finally activate the cell defensive system to protect cells from endogenous and exogenous stresses. Thus, compounds that up-regulate the expression of ARE gene products may have therapeutic potential in a range of disease types, including inflammatory conditions, chronic kidney diseases, and cancer chemoprevention.9−11
A wide variety of dietary and synthetic compounds have been identified as potent activators of ARE-regulated genes, e.g. sulforaphane, chalcone, and curcumin.12 These traditional Nrf2-ARE activators are all electrophiles that form a covalent bond to a cysteine residue in the target Keap1 protein.13 The electrophilic nature of these activators could increase the uncertainty in further clinical development. Thus, directly disrupting the Keap1-Nrf2 protein–protein interaction (PPI) has been regarded as an innovative strategy to activate Nrf2. Owing to the competitive and reversible nature, the Keap1-Nrf2 PPI inhibitors may show some advantages toward the electrophilic Nrf2 activators.14
Recently, a series of Keap1-Nrf2 PPI inhibitors, including peptides15−17 and small molecules,18−23 have been identified. Among these agents, the diacetate compound series showed the most predominant PPI inhibition activity. The discovery of DDO1002 (1), the first nanomolar Keap1-Nrf2 PPI inhibitor, confirmed that the necessity of fulfilling the five subpockets in Keap1 and making multiple polar interactions with key arginines in the Keap1 binding cavity should be stressed in the design of potent Keap1-Nrf2 PPI inhibitors.24 Further optimization of 1 resulted in DDO1018 (2) with higher solubility and better Nrf2 inducing activity. The in vivo experiments also confirmed the anti-inflammatory effects of 2.24,25 This progress provided useful tools to further investigate the pharmacological effects of the Keap1-Nrf2 PPI inhibitors. However, the diacetate moiety of these compounds may have some potential risks for further development. Thus, it is quite necessary to investigate whether the diacetate moiety can be replaced without sacrificing the activity.
Design and Synthesis of Compounds with Various Bioisosteric Substituents of Diacetic Groups
Our previous studies have identified that the P1 and P2 polar subpockets of the Keap1 cavity are the determinants for the potent Keap1-Nrf2 PPI inhibitors.26,27 Accordingly, we successfully developed the diacetate compound series, which utilized the two carboxylic acid groups to occupy the P1 and P2 subpockets (as shown in Figure 1).24,25 The diacetate moiety can form multiple polar interactions, including hydrogen bonds and salt bridges, with key polar residues, especially arginines in the P1 and P2 subpockets. However, the diacetic acid moiety is highly polar and easily ionized, which may possess potential hurdles in druggability issues.
Figure 1.
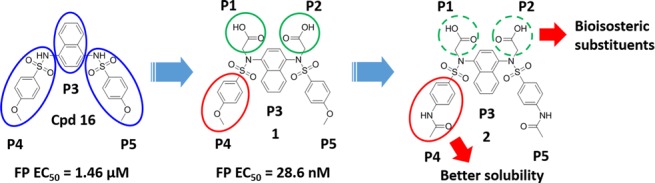
Discovery and optimization process of the diacetic compound series.
To investigate whether the diacetate moiety can be replaced without sacrificing the activity, a series of compounds with various bioisosteric substituents were designed and synthesized based upon our previously identified compound 2 (as shown in Table 1). These compounds were synthesized via the derivation of 3. The different side chains were introduced by the electrophilic substitution of the sulfonamides, which have been used to construct the diacetic compound series (Scheme 1).24,25
Table 1. IC50, Physicochemical Properties and ARE Induction Fold Results of the Compounds Containing Various Substituents.a.

Scheme 1.

Reagents and conditions: (a) NH2OH·HCl, 95% ethanol, MeOH, 60 °C, 2 h; (b) Pd/C, H2, THF, rt, 4 h; (c) 4-acetamido benzenesulfonyl chloride, toluene, pyridine, 100 °C, 2 h, 71%; (d) DMF, K2CO3, bromoacetonitrile, rt, 3 h, 53%; (e) DMF, K2CO3, 3-bromoprop-1-yne, rt, 3 h, 59%; (f) DMF, K2CO3, methyl bromoacetate, rt, 3 h, 54%; (g) DMF, K2CO3, 2-bromoacetamide, rt, 3 h, 64%; (h) DMF, NH4Cl, NaN3, N2, 100 °C, 6 h, 67%; (i) DMF, NH4Cl, NaN3, N2, 100 °C, 6 h, 63%; (j) NH2OH·HCl, NaOH (aq), MeOH, rt, 6 h, 64%; (k) LiAlH4, THF, NaOH (aq), rt, 3 h, 51%; (l) LiOH, CH3OH/H2O, rt, 6 h, 68%.
Ditetrazole Analog Can Maintain the Potent PPI Inhibition Activity
These compounds were evaluated for their Keap1-Nrf2 PPI inhibition activity in a previously described fluorescence polarization (FP) assay.24,28 As shown in Figure 2, compound 7 bearing the tetrazole groups maintained the PPI inhibition activity with an IC50 of 15.8 nM, which is comparable to that of 2 (IC50 = 13.4 nM). While the introduction of other bioisosteric substituents led to the dramatic decrease in activity, only 8 with 1,2,3-triazole groups displayed the nanomole activity (IC50 = 609 nM). The prominent performance of the tetrazole and 1,2,3-triazole groups indicated that an electron-rich aromatic ring may be an alternative pattern for occupying the P1 and P2 subpockets. The carboxamide group, which can form a similar hydrogen bond but has a significantly high pKa value, resulted in the inactive compound. This is consistent with the results provided by Jnoff et al.29 However, the carboxamide group can also be active in certain cases. The research of Jain et al.30 showed that when the substituent on the side-chain phenyl ring was the p-methoxy group, the diacetamide moiety can insert into the P1 and P2 subpockets and good inhibition activity can be retained. The hydroxamic acid analog also showed moderate activity. Considering the similar properties of the two functional groups in forming hydrogen bonds, the difference in acidity may be the main reason for the loss of activity. The methyl ether of 2 did not show activity, providing a possible choice for the prodrug design of the Keap1-Nrf2 PPI inhibitor.
Figure 2.

Dose–response inhibition curves of 2 and 7 determined by the FP-based binding assay.
Binding Mode Investigation Indicated Ditetrazole Moiety Has Novel “Sandwich”-like Binding Pattern
In order to compare the role of the diacetic moiety and tetrazole groups in Keap1 binding, we carried out molecular docking studies between the Keap1 Kelch domain and the two active compounds 2 and 7. The LigandFit tool, which has been proven suitable for the Keap1 target and has been applied in our previous work,20,24 was used to propose the binding pose. As shown in Figure 3A, the diacetic moiety of 2 can insert into the P1 and P2 subpockets and interact with key arginines by hydrogen bonds and salt bridges. Although the tetrazole groups of 7 can occupy the P1 and P2 subpockets, the binding pattern showed some differences. In the case of 7, most of the hydrogen bonds and salt bridge interactions were retained. Besides, the ditetrazole moiety can make extra π–charge interactions with the guanidyl group of arginines. As shown in Figure 3B, the ditetrazole moiety packed between Arg380 and Arg415 and formed multiple electrostatic interactions, including charge–charge and π–charge interactions, which seems like a “sandwich” binding mode, further enhancing the binding in the P1 and P2 subpockets. In order to match up this binding pose, the core naphthalene ring also had a little bit of deflection, but the hydrophobic interactions with Arg415 and Ala556 were retained. The movement of the naphthalene ring also induced the displacement of two phenyl rings. But the overall binding pattern was similar, and the key π–π interactions were kept. Overall, the docking results indicated that the tetrazole moiety not only interacted with key arginines by hydrogen bonds and salt bridges, but also formed favorable π–charge interactions. The “sandwich”-like binding pattern taken by the ditetrazole moiety provided an alternative for the design of potent Keap1-Nrf2 PPI inhibitors.
Figure 3.

Binding mode investigation of 2 and 7 using molecular docking. Parts A and B represent the binding modes of 2 and 7, respectively. The surface of Keap1 is colored as the property of hydrogen bonds. The ligand is represented as sticks. Green dashed lines represent hydrogen bonds, yellow solid lines represent electrostatic interactions, the pink dashed lines represent π–π interactions, and the violet dashed lines represent hydrophobic interactions.
Physicochemical Property Determination Confirms Ditetrazole Replacement Can Elevate pKa and log DpH=7.4
We further evaluated the impact of bioisosteric substituents on the physicochemical properties of 2 and 4–11. As shown in Table 1, the pKa and log DpH=7.4 of 2 were 4.79 and 1.02, respectively, which further confirmed 2 as an easily ionizable and highly polar compound. All of the compounds showed higher pKa and log DpH=7.4 values than those of 2, which reflected that the replacement of the diacetic moiety can improve the physicochemical properties. The pKa and log DpH=7.4 of the most potent analogue in the PPI inhibition experiment, compound 7, were 5.12 and 2.31, respectively. They were both higher than those of 2, but lower than the other compounds, especially for the pKa value. This indicated that the P1 and P2 subpockets favor the easily negatively charged groups, and the electrostatic interactions are important for Keap1–ligand binding. Although the pKa and log DpH=7.4 changed, 7 retained the advantages in solubility, similar to 2. Taken together, the higher pKa and log DpH=7.4 of 7 could enhance the passive cell membrane permeability, which may be beneficial for the cellular activity.
Ditetrazole Replacement Resulted in Better Cell Membrane Permeability
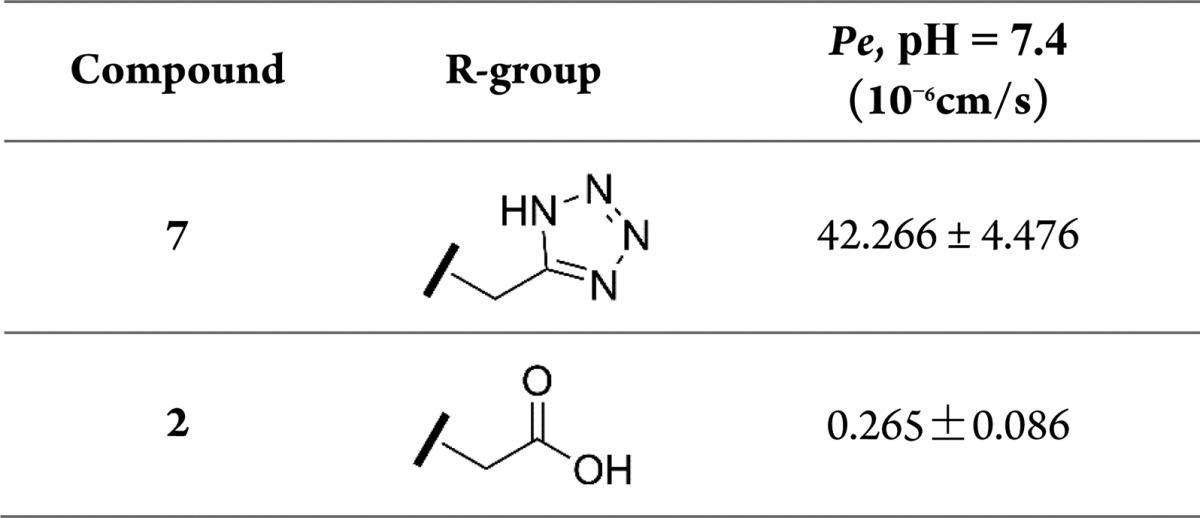
Considering the significant change of the physicochemical property induced by the polar recognition group, we then examined whether ditetrazole replacement could affect the cell membrane permeability. Permeability coefficients (Pe) were determined using a standard parallel artificial membrane permeability assay (PAMPA) on a PAMPA Explorer instrument (pION). PAMPA is used as an in vitro model of passive, transcellular permeability.
Surprisingly, 7 with the ditetrazole showed a Pe value of 42.3 × 10–6 cm/s, while 2 with the diacetic group only gave a Pe value of 0.3 × 10–6 cm/s (Table 2). The results clearly showed that the ditetrazole analog can permeate the cell membrane through the passive diffusion mechanism, while the diacetic analog can hardly accomplish the transmembrane transport by this mechanism. This confirmed that the ditetrazole replacement can improve the cell membrane permeability, which is beneficial for the cellular activity.
Table 2. Membrane Permeability of the Selected Compounds.
Ditetrazole Analog 7 Is More Potent in the Cellular ARE-Luciferase Reporter Assay
In view of the promising character of 7, we investigated the cellular activities of these compounds using the ARE-luciferase reporter assay in HepG2-ARE-C8 cells, which has been applied extensively to determine the Nrf2 inducing ability of small molecules. As shown in Table 1, 7, which is optimal in both PPI inhibition activity and physicochemical properties, showed significant improvement in the Nrf2-ARE inducing activity. The level of ARE to about 15-fold can be achieved by 7 at 5 μM, while 2 can only achieve about 11-fold at the same concentration. These results showed that 7 has obvious advantages compared with 2 in the cellular ARE-luciferase reporter assay.
qRT-PCR Analysis Confirms 7 Can More Significantly Induce the Expression of Nrf2-Driven Genes
We then investigated whether 7 is more potent in elevating the transcription of Nrf2-driven genes. Quantitative real-time PCR (qRT-PCR) analysis was performed to monitor the mRNA level of three selected target genes: GCLM (glutamate-cysteine ligase, modifier subunit), NQO1 (NAD(P)H: Quinone Oxidoreductase 1), and HO-1 (Heme oxygenase 1). Both of the two compounds concentration-dependently increased the transcription of Nrf2-regulated genes. In the case of 2 at the highest concentration (10 μM), the levels of HO-1 and GCLM were about 21- and 14-fold, respectively (Figure 4A and B). Remarkably, for the optimized compound 7, there was approximately 30- and 20- fold increase in the mRNA level of HO-1 and GCLM, respectively (Figure 4A and B). 7 also showed superiority in the elevation of NQO1 mRNA. Treatment with 7 can induce about 5-fold at the concentration of 5 μM, while similar elevation effects were achieved by 2 at 10 μM (Figure 4C). Taken together, these results indicated that 7 can obviously activate the transcriptional activity of Nrf2 at a lower concentration than 2.
Figure 4.

Expression of Nrf2-regulated genes after treatment with 2 and 7. HCT116 cells were treated with these two compounds respectively at the indicated concentrations for 12 h. The histogram showed qRT-PCR analysis of (A) HO-1, (B) GCLM, and (C) NQO1 expression in HCT116 cells after treatment with compound 2 and 7, respectively. The expression of Nrf2 targeted genes used β-actin for normalization (n = 3).
Compound 7 Is More Efficacious on Up-Regulating the Protein Level of Nrf2 Targeted Genes
Then we examined the concentration-dependent effects of 7 on the protein level of Nrf2 targeted genes including HO-1, NQO1, and γ-GCS. The effects of 2 were also evaluated as a comparison. As shown in Figure 5, the up-regulation of downstream protein NQO1 was not obvious at 8 h, which may result from the high basal level of NQO1 in HCT116 cells.31,32 Compared to NQO1, the elevation of HO-1 and γ-GCS was much more obvious. Compound 7 doubled the expression of HO-1 and γ-GCS at the concentration of 10 μM at 8 h, while 2 only increased the expression of these two genes at 1.5- and 1.6-fold, respectively, and, at 16 h, compound 7 showed significant superiority in the elevation of NQO1, HO-1, and γ-GCS (Figure 5 for the protein band and Figure S1 for the semiquantitative data). These data further substantiated the higher cellular Nrf2 inducing activity of 7 bearing tetrazole groups.
Figure 5.

Dose-dependent increase of Nrf2-regulated genes NQO1, HO-1 and γ-GCS in HCT116 cells after treatment with 2 and 7, respectively.
In summary, with the aim to improve the physicochemical properties and cell membrane permeability, we used the bioisosteric groups to replace the diacetate groups of 2, which is a potent Keap1-Nrf2 PPI inhibitor with less-than-ideal drug-like properties. Among the compounds with various bioisosteric substituents, 7, with tetrazole groups, retains the Keap1-Nrf2 PPI inhibition potency with an IC50 of 15.8 nM in the FP assay, but has improved logP and pKa profiles compared to 2. Moreover, the transcellular permeability assay showed that 7 can easily cross the membrane by the passive diffusion. Compared to compound 2, compound 7 showed better cellular ARE activity and was more efficacious on up-regulating both the mRNA and the protein level of Nrf2 targeted genes. Collectively, these data substantiate that the ditetrazole moiety not only fits the Keap1 P1 and P2 polar subpockets, but also has better drug-like properties, which would benefit the subsequent development of Keap1-Nrf2 PPI inhibitors.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00407.
Experimental materials and methods, and the densitometric analysis of the relative ratios of each protein in Western blot assay (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
This work is supported by the projects 81230078 (key program) and 81173087 of National Natural Science Foundation of China; 2014ZX09507002-005-015 and 2013ZX09402102-001-005 of the National Major Science and Technology Project of China (Innovation and Development of New Drugs); Specialized Research Fund for the Doctoral Program of Higher Education (SRFDP, 20130096110002); the Fundamental Research Funds for the Central Universities 2016ZPY016; and the Project Program of State Key Laboratory of Natural Medicines, China Pharmaceutical University (No.SKLNMZZCX201611).
The authors declare no competing financial interest.
Supplementary Material
References
- Kensler T. W.; Wakabayashi N.; Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Baird L.; Dinkova-Kostova A. T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–72. 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]
- Hayes J. D.; Dinkova-Kostova A. T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- Zhang D. D.; Lo S. C.; Cross J. V.; Templeton D. J.; Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–53. 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A.; Kang M. I.; Watai Y.; Tong K. I.; Shibata T.; Uchida K.; Yamamoto M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–9. 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong K. I.; Katoh Y.; Kusunoki H.; Itoh K.; Tanaka T.; Yamamoto M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol. Cell. Biol. 2006, 26, 2887–900. 10.1128/MCB.26.8.2887-2900.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird L.; Lleres D.; Swift S.; Dinkova-Kostova A. T. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 15259–64. 10.1073/pnas.1305687110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird L.; Swift S.; Lleres D.; Dinkova-Kostova A. T. Monitoring Keap1-Nrf2 interactions in single live cells. Biotechnol. Adv. 2014, 32, 1133–1144. 10.1016/j.biotechadv.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T.; Motohashi H.; Yamamoto M. Toward clinical application of the Keap1–Nrf2 pathway. Trends Pharmacol. Sci. 2013, 34, 340–346. 10.1016/j.tips.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Lu M.-C.; Ji J.-A.; Jiang Z.-Y.; You Q.-D.. The Keap1-Nrf2-ARE Pathway As a Potential Preventive and Therapeutic Target: An Update. Med. Res. Rev., published online May 18, 2016; DOI: 10.1002/med.21396. [DOI] [PubMed] [Google Scholar]
- Lu M.-C.; Ji J.-A.; Jiang Y.-L.; Chen Z.-Y.; Yuan Z.-W.; You Q.-D.; Jiang Z.-Y. An inhibitor of the Keap1-Nrf2 protein-protein interaction protects NCM460 colonic cells and alleviates experimental colitis. Sci. Rep. 2016, 6, 26585. 10.1038/srep26585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magesh S.; Chen Y.; Hu L. Small Molecule Modulators of Keap1-Nrf2-ARE Pathway as Potential Preventive and Therapeutic Agents. Med. Res. Rev. 2012, 32, 687–726. 10.1002/med.21257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A. J.; Kerns J. K.; Callahan J. F.; Moody C. J. Keap calm, and carry on covalently. J. Med. Chem. 2013, 56, 7463–76. 10.1021/jm400224q. [DOI] [PubMed] [Google Scholar]
- Richardson B. G.; Jain A. D.; Speltz T. E.; Moore T. W. Non-electrophilic modulators of the canonical Keap1/Nrf2 pathway. Bioorg. Med. Chem. Lett. 2015, 25, 2261–8. 10.1016/j.bmcl.2015.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock R.; Schaap M.; Pfister H.; Wells G. Peptide inhibitors of the Keap1-Nrf2 protein-protein interaction with improved binding and cellular activity. Org. Biomol. Chem. 2013, 11, 3553–7. 10.1039/c3ob40249e. [DOI] [PubMed] [Google Scholar]
- Hancock R.; Bertrand H. C.; Tsujita T.; Naz S.; El-Bakry A.; Laoruchupong J.; Hayes J. D.; Wells G. Peptide inhibitors of the Keap1-Nrf2 protein-protein interaction. Free Radical Biol. Med. 2012, 52, 444–51. 10.1016/j.freeradbiomed.2011.10.486. [DOI] [PubMed] [Google Scholar]
- Zhuang C.; Miao Z.; Sheng C.; Zhang W. Updated Research and Applications of Small Molecule Inhibitors of Keap1-Nrf2 Protein-Protein Interaction: a Review. Curr. Med. Chem. 2014, 21, 1861–70. 10.2174/0929867321666140217104648. [DOI] [PubMed] [Google Scholar]
- Hu L.; Magesh S.; Chen L.; Wang L.; Lewis T. A.; Chen Y.; Khodier C.; Inoyama D.; Beamer L. J.; Emge T. J.; Shen J.; Kerrigan J. E.; Kong A. N.; Dandapani S.; Palmer M.; Schreiber S. L.; Munoz B. Discovery of a small-molecule inhibitor and cellular probe of Keap1-Nrf2 protein-protein interaction. Bioorg. Med. Chem. Lett. 2013, 23, 3039–43. 10.1016/j.bmcl.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte D.; Zeng W.; Hus J.-C.; McKenzie A.; Hession C.; Jin P.; Bergeron C.; Lugovskoy A.; Enyedy I.; Cuervo H.; Wang D.; Atmanene C.; Roecklin D.; Vecchi M.; Vivat V.; Kraemer J.; Winkler D.; Hong V.; Chao J.; Lukashev M.; Silvian L. Small molecules inhibit the interaction of Nrf2 and the Keap1 Kelch domain through a non-covalent mechanism. Bioorg. Med. Chem. 2013, 21, 4011–4019. 10.1016/j.bmc.2013.04.019. [DOI] [PubMed] [Google Scholar]
- Sun H.-P.; Jiang Z.-Y.; Zhang M.-Y.; Lu M.-C.; Yang T.-T.; Pan Y.; Huang H.-Z.; Zhang X.-J.; You Q.-d. Novel protein-protein interaction inhibitor of Nrf2-Keap1 discovered by structure-based virtual screening. MedChemComm 2014, 5, 93–98. 10.1039/C3MD00240C. [DOI] [Google Scholar]
- Zhuang C.; Narayanapillai S.; Zhang W.; Sham Y. Y.; Xing C. Rapid identification of Keap1-Nrf2 small-molecule inhibitors through structure-based virtual screening and hit-based substructure search. J. Med. Chem. 2014, 57, 1121–6. 10.1021/jm4017174. [DOI] [PubMed] [Google Scholar]
- Davies T. G.; Wixted W. E.; Coyle J. E.; Griffiths-Jones C.; Hearn K.; McMenamin R.; Norton D.; Rich S. J.; Richardson C.; Saxty G.; Willems H. M. G.; Woolford A. J. A.; Cottom J. E.; Kou J.-P.; Yonchuk J. G.; Feldser H. G.; Sanchez Y.; Foley J. P.; Bolognese B. J.; Logan G.; Podolin P. L.; Yan H.; Callahan J. F.; Heightman T. D.; Kerns J. K. Monoacidic Inhibitors of the Kelch-like ECH-Associated Protein 1: Nuclear Factor Erythroid 2-Related Factor 2 (KEAP1:NRF2) Protein–Protein Interaction with High Cell Potency Identified by Fragment-Based Discovery. J. Med. Chem. 2016, 59, 3991–4006. 10.1021/acs.jmedchem.6b00228. [DOI] [PubMed] [Google Scholar]
- Lu M.-C.; Yuan Z.-W.; Jiang Y.-L.; Chen Z.-Y.; You Q.-D.; Jiang Z.-Y. A systematic molecular dynamics approach to the study of peptide Keap1-Nrf2 protein-protein interaction inhibitors and its application to p62 peptides. Mol. BioSyst. 2016, 12, 1378–1387. 10.1039/C6MB00030D. [DOI] [PubMed] [Google Scholar]
- Jiang Z.-Y.; Lu M.-C.; Xu L. L.; Yang T.-T.; Xi M.-Y.; Xu X.-L.; Guo X.-K.; Zhang X.-J.; You Q.-D.; Sun H.-P. Discovery of Potent Keap1–Nrf2 Protein–Protein Interaction Inhibitor Based on Molecular Binding Determinants Analysis. J. Med. Chem. 2014, 57, 2736–2745. 10.1021/jm5000529. [DOI] [PubMed] [Google Scholar]
- Jiang Z. Y.; Xu L. L.; Lu M. C.; Chen Z. Y.; Yuan Z. W.; Xu X. L.; Guo X. K.; Zhang X. J.; Sun H. P.; You Q. D. Structure-Activity and Structure-Property Relationship and Exploratory in Vivo Evaluation of the Nanomolar Keap1-Nrf2 Protein-Protein Interaction Inhibitor. J. Med. Chem. 2015, 58, 6410–6421. 10.1021/acs.jmedchem.5b00185. [DOI] [PubMed] [Google Scholar]
- Jiang Z.-Y.; Xu L.-L.; Lu M.-C.; Pan Y.; Huang H.-Z.; Zhang X.-J.; Sun H.-P.; You Q.-D. Investigation of the intermolecular recognition mechanism between the E3 ubiquitin ligase Keap1 and substrate based on multiple substrates analysis. J. Comput.-Aided Mol. Des. 2014, 28, 1233–1245. 10.1007/s10822-014-9799-y. [DOI] [PubMed] [Google Scholar]
- Lu M.; Chen Z.; Wang Y.; Jiang Y.; Yuan Z.; You Q.; Jiang Z. Binding Thermodynamics and Kinetics Guided Optimization of Potent Keap1-Nrf2 Peptide Inhibitors. RSC Adv. 2015, 5, 85983–85987. 10.1039/C5RA16262A. [DOI] [Google Scholar]
- Inoyama D.; Chen Y.; Huang X.; Beamer L. J.; Kong A. N.; Hu L. Optimization of fluorescently labeled Nrf2 peptide probes and the development of a fluorescence polarization assay for the discovery of inhibitors of Keap1-Nrf2 interaction. J. Biomol. Screening 2012, 17, 435–47. 10.1177/1087057111430124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jnoff E.; Albrecht C.; Barker J. J.; Barker O.; Beaumont E.; Bromidge S.; Brookfield F.; Brooks M.; Bubert C.; Ceska T.; Corden V.; Dawson G.; Duclos S.; Fryatt T.; Genicot C.; Jigorel E.; Kwong J.; Maghames R.; Mushi I.; Pike R.; Sands Z. A.; Smith M. A.; Stimson C. C.; Courade J. P. Binding mode and structure-activity relationships around direct inhibitors of the Nrf2-Keap1 complex. ChemMedChem 2014, 9, 699–705. 10.1002/cmdc.201300525. [DOI] [PubMed] [Google Scholar]
- Jain A. D.; Potteti H.; Richardson B. G.; Kingsley L.; Luciano J. P.; Ryuzoji A. F.; Lee H.; Krunic A.; Mesecar A. D.; Reddy S. P.; Moore T. W. Probing the structural requirements of non-electrophilic naphthalene-based Nrf2 activators. Eur. J. Med. Chem. 2015, 103, 252–268. 10.1016/j.ejmech.2015.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi M.-y.; Sun Z.-y.; Sun H.-p.; Jia J.-m.; Jiang Z.-y.; Tao L.; Ye M.; Yang X.; Wang Y.-j.; Xue X.; Huang J.-j.; Gao Y.; Guo X.-k.; Zhang S.-l.; Yang Y.-r.; Guo Q.-l.; Hu R.; You Q.-d. Synthesis and bioevaluation of a series of α-pyrone derivatives as potent activators of Nrf2/ARE pathway (part I). Eur. J. Med. Chem. 2013, 66, 364–371. 10.1016/j.ejmech.2013.06.007. [DOI] [PubMed] [Google Scholar]
- Xi M. Y.; Jia J. M.; Sun H. P.; Sun Z. Y.; Jiang J. W.; Wang Y. J.; Zhang M. Y.; Zhu J. F.; Xu L. L.; Jiang Z. Y.; Xue X.; Ye M.; Yang X.; Gao Y.; Tao L.; Guo X. K.; Xu X. L.; Guo Q. L.; Zhang X. J.; Hu R.; You Q. D. 3-Aroylmethylene-2,3,6,7-tetrahydro-1H-pyrazino[2,1-a]isoquinolin-4(11bH)-ones as Potent Nrf2/ARE Inducers in Human Cancer Cells and AOM-DSS Treated Mice. J. Med. Chem. 2013, 56, 7925–7938. 10.1021/jm400944k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.