

ABSTRACT
Toxic shock syndrome toxin 1 (TSST-1) is a Staphylococcus aureus superantigen that has been implicated in both menstrual and nonmenstrual toxic shock syndrome (TSS). Despite the important role of TSST-1 in severe human disease, a comprehensive understanding of staphylococcal regulatory factors that control TSST-1 expression remains incomplete. The S. aureus exotoxin expression (Sae) operon contains a well-characterized two-component system that regulates a number of important exotoxins in S. aureus, although regulation of TSST-1 by the Sae system has not been investigated. We generated a defined deletion mutant of the Sae histidine kinase sensor (saeS) in the prototypic menstrual TSS strain S. aureus MN8. Mutation of saeS resulted in a complete loss of TSST-1 expression. Using both luciferase reporter experiments and quantitative real-time PCR, we demonstrate that the Sae system is an important transcriptional activator of TSST-1 expression. Recombinant SaeR was able to bind directly to the tst promoter to a region containing two SaeR consensus binding sites. Although the stand-alone SarA transcriptional regulator has been shown to be both a positive and a negative regulator of TSST-1, deletion of sarA in S. aureus MN8 resulted in a dramatic overexpression of TSST-1. As expected, mutation of agr also reduced TSST-1 expression, but this phenotype appeared to be independent of Sae. A double mutation of saeS and sarA resulted in the loss of TSST-1 expression. This work indicates that the Sae system is a dominant and direct transcriptional activator that is required for expression of TSST-1.
IMPORTANCE The TSST-1 superantigen is an exotoxin, produced by some strains of S. aureus, that has a clear role in both menstrual and nonmenstrual TSS. Although the well-characterized agr quorum sensing system is a known positive regulator of TSST-1, the molecular mechanisms that directly control TSST-1 expression are only partially understood. Our studies demonstrate that the Sae two-component regulatory system is a positive transcriptional regulator that binds directly to the TSST-1 promoter, and furthermore, our data suggest that Sae is required for expression of TSST-1. This work highlights how major regulatory circuits can converge to fine-tune exotoxin expression and suggests that the Sae regulatory system may be an important target for antivirulence strategies.
INTRODUCTION
Staphylococcus aureus is both a common commensal of humans and a prominent bacterial pathogen that is responsible for an assortment of illnesses ranging from self-limiting superficial infections to life-threatening invasive diseases (1). While most infections caused by S. aureus involve the coordinated expression of numerous virulence factors, a few select diseases, including staphylococcal scalded skin syndrome, food poisoning, and toxic shock syndrome (TSS), require the expression of specific staphylococcal exotoxins (2).
Staphylococcal TSS is a rare but devastating disease that is caused by S. aureus strains that secrete high levels of superantigens. Superantigen exotoxins function by binding directly to lateral surfaces of both T-cell receptor β-chains and major histocompatibility complex (MHC) class II molecules, which forces the massive activation of numerous T cells and can induce a T-cell-dependent cytokine storm (3). TSS symptoms are characterized by the acute onset of fever, rash formation, hypotension, multisystem involvement, and desquamation in patients that recover (4), although strict use of this definition likely underestimates the actual number of TSS cases (5). There are two major forms of TSS, including menstrual TSS (mTSS) and nonmenstrual TSS (3). Although the latter can occur from virtually any S. aureus infection, mTSS typically occurs within ∼2 days prior to or ∼2 days following menstruation and is often associated with the use of tampons (6, 7). The staphylococcal superantigen toxic shock syndrome toxin 1 (TSST-1) is believed to be responsible for nearly all cases of menstrual TSS (3, 8), as well as many cases of nonmenstrual TSS (5, 9).
Despite the well-recognized role of TSST-1 in both forms of TSS, a comprehensive understanding of the genetic elements required for the regulation and expression of TSST-1 remains incomplete. Although the prototypical positive regulator of TSST-1 is the well-characterized accessory gene regulator (agr) quorum sensing system (10), a number of additional regulatory factors also impact TSST-1 expression. For example, mutation of the staphylococcal respiratory response (srrAB) two-component system resulted in a decrease in TSST-1 production, particularly under conditions of low oxygen (11). However, overexpression of srrAB in trans massively upregulated RNAIII, the major effector molecule of the agr system, with a near complete repression of TSST-1, demonstrating that regulation of TSST-1 by agr can be uncoupled (11). This is also consistent with a study that could not correlate agr polymorphisms, including agrC-inactivating mutations, with varied production of TSST-1 (12). Also, the staphylococcal accessory regulator (SarA) has previously been demonstrated to bind directly to the tst promoter region (13), and depending upon the genetic background, has been reported to function as either a positive or negative regulator of TSST-1 expression (13–15). Furthermore, carbon catabolite protein A (CcpA) acts as a repressor of TSST-1 (16), which explains the initial observation that TSST-1 expression is repressed by glucose (17). Although mutation of the repressor of toxins (rot) transcriptional regulator was reported to have little effect on TSST-1 expression, overexpression of Rot in trans strongly repressed TSST-1 (15). In addition to these experiments, prior work from our laboratory has demonstrated that interspecies communication between Lactobacillus reuteri and S. aureus can result in a dramatic repression of TSST-1 expression (18). Although this phenotype correlated with repression of agr, and deletion of agr in S. aureus MN8 reduced TSST-1 expression, TSST-1 could be further repressed in the agr-null background by the L. reuteri signaling molecules. This additional repression of TSST-1 correlated with decreased transcription of the S. aureus exotoxin expression (Sae) system and increased transcription of stand-alone transcriptional regulator SarA (18). The aim of the present study was to investigate the role of both Sae and SarA in production of TSST-1 in S. aureus MN8, a prototypic menstrual TSS S. aureus strain.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains used in this study are listed in Table 1. All cloning was carried out utilizing Escherichia coli XL-1 Blue (Stratagene) grown on Luria-Bertani (LB) broth (Difco) or brain heart infusion (BHI) (Becton Dickinson) plates supplemented with 1.5% agar. For E. coli, antibiotics (Sigma-Aldrich) were added to growth media at the following concentrations: 200 μg/ml ampicillin (Amp), 150 μg/ml erythromycin (Erm), and 10 μg/ml chloramphenicol (Cm). S. aureus MN8 was isolated prior to 1980 and is a prototypic mTSS strain (17). S. aureus strains were routinely grown in tryptic soy broth (TSB) (Difco) or BHI broth or on solid agar with antibiotics as appropriate. For S. aureus, antibiotics were added to growth media at the following concentrations: 5 μg/ml Erm and 10 μg/ml Cm.
TABLE 1.
Bacterial strains used in this study
| Strain | Description | Source or reference |
|---|---|---|
| E. coli | ||
| XL-1 Blue | Cloning strain | Stratagene |
| SA30B | DNA methylation strain | 20 |
| BL21(DE3) | Protein expression strain | New England Biolabs |
| S. aureus | ||
| RN4220 | DNA methylation strain | 19 |
| MN8 | Prototypic menstrual TSS strain, tst+ | 17 |
| MN8 Δagr | S. aureus MN8 with agr operon replaced with 3-kb tetR marker | 18 |
| MN8 ΔsaeS | S. aureus MN8 containing a deletion within saeS | This study |
| MN8 ΔsaeS (+sae) | S. aureus MN8 ΔsaeS containing pALC2073::sae | This study |
| MN8 ΔsarA | S. aureus MN8 containing a deletion within sarA | This study |
| MN8 ΔsarA (+sarA) | S. aureus MN8 ΔsarA containing pALC2073::sarA | This study |
| MN8 ΔsaeS ΔsarA | S. aureus MN8 containing deletions in both saeS and sarA | This study |
Molecular cloning.
The plasmids and primers used in this work are listed in Tables 2 and 3, respectively. Plasmid DNA was isolated from E. coli using the Qiagen Miniprep kit. Routine PCR amplifications were carried out using an MJ Research PTC-200 instrument with primers obtained from Sigma. Digestions were carried out utilizing restriction enzymes provided by New England BioLabs or Roche. Ligations were performed utilizing T4 DNA ligase (New England BioLabs). E. coli cells were made competent using the RbCl2 method (3). Transformation of plasmids into S. aureus was carried out as described previously (18), and S. aureus RN4220 (19) and E. coli SA30B (20) were used as cloning intermediates to methylate DNA prior to electroporation in S. aureus MN8.
TABLE 2.
Plasmids used in this study
| Plasmid | Descriptiona | Source or reference |
|---|---|---|
| pMAD | Temp-sensitive integration vector with blue-white selection; Emr | 21 |
| pMAD::ΔsaeS | saeS deletion plasmid; Emr | This study |
| pKOR1 | Temp-sensitive integration vector with inducible counterselection; Emr | 22 |
| pKOR1::ΔsarA | sarA deletion plasmid; Emr | This study |
| pALC2073 | S. aureus complementation plasmid; Cmr | 23 |
| pALC2073::sae | Complementation plasmid containing saeQRS operon; Cmr | This study |
| pALC2073::sarA | Complementation plasmid containing sarA; Cmr | This study |
| pAmilux | Luminescence reporter plasmid; Cmr | 24 |
| pAmilux::Ptst | TSST-1 promoter reporter plasmid; Cmr | 18 |
| pET28::saeR | Protein expression clone containing full-length SaeR; Kanr | 25 |
| pMCSG19::saeSc | Protein expression clone encoding residues 93–351 of SaeS; Ampr | 25 |
Emr, erythromycin resistant; Cmr, chloramphenicol resistant; Kanr, kanamycin resistant.
TABLE 3.
Primers used in this study
| Primer | Sequence (5′→3′)a |
|---|---|
| Primers for saeS deletion and complementation plasmid construction | |
| SaeS up BamHI (forward) | GGGGGATCCAACAACGACAACTAGCGGTAAAGA |
| SaeS up Sall (reverse) | AGTGTCGACACACCATTATCGGCTCCTTTCA |
| SaeS down EcoRI (forward) | CCCGAATTCTAGCCCATGATTTAAAAACACCTT |
| SaeS down Bglll (reverse) | CCCAGATCTTTCCTACATCTATATCACTGCTTACACTG |
| SaeRS Comp II KpnI For | TTTTTGGTACCGGTAAATTAATGCTTACTAACTACAA |
| SaeRS Comp II EcoRI Rev | CCCCCGAATTCTTATGTCGTAATGTCTAATTTGTG |
| Primers for sarA deletion and complementation plasmid construction | |
| SarA upstream For attB1 | GGGGACAAGTTTGTACAAAAAAGCAGGCTAGGTGCAGCATTAACAACACT |
| SarA upstream Revb | AATTGCCATGGTTAAAACCTC |
| SarA downstream Forb | GAACTATAATTTTGTTTAGCG |
| SarA downstream rev primer attB2 | GGGGACCACTTTGTACAAGAAAGCTGGGTTGAGGGAGGTGTCACAATGA |
| SarA complement KpnI For | TTTTTGGTACCTTTAACATTTAGCTTATCATTTTAA |
| SarA complement SacI Rev | CCCCCGAGCTCTTATAGTTCAATTTCGTTGTTTGCT |
| Primers for sequencing | |
| SaeRS screen For | CTGGGGGATATGTTTTACC |
| SaeRS screen Rev | GTCCCTATGCGTATTAAGGA |
| pMAD seq FP | GGGGAAGGCCATCCAGCCTCGCGTC |
| pMAD seq RP | AATCTAGCTAATGTTACGTTACACA |
| SarA flank For | TCTTATCATTAAAACTGCACTGGGA |
| SarA flank Rev | GCGGTGGCAATTCGTTCATT |
| pKOR1For | CAGATCCATATCCTTCTTTTCTGA |
| pKOR1 Rev | GTGTGGAATTGTGAGCGGATA |
| pALC2073 Seq For | GGTTGCATGCCTGCAGGTCGACGG |
| pALC2073 Seq Rev | CAGTCACGACGTTGTAAAACG |
| Primers for qRT-PCR analysis | |
| tst-For | CTGATGCTGCCATCTGTGTT |
| tst-Rev | GTAAGCCCTTTGTTGCTTGC |
| agrA-For (P2) | GTGAAATTCGTAAGCATGACCCAGTTG |
| agrA-Rev (P2) | TGTAAGCGTGTATGTGCAGTTTCTAAAC |
| rnaIII-For (P3) | TAGATCACAGAGATGTGA |
| rnaIII-Rev (P3) | CTGAGTCCAAGGAAACTAACTC |
| saeR-For | CCAAGGGAACTCGTTTTACG |
| saeR-Rev | ACGCATAGGGACTTCATGAC |
| sarA-For | TGTTTGCTTCAGTGATTCGTTT |
| sarA-Rev | CATCAGCGAAAACAAAGAGAAA |
| rpoB-For | TCCTGTTGAACGCGCATGTAA |
| rpoB-Rev | GCTGGTATGGCTCGTGATGGTA |
| Primers for EMSA experiments | |
| SaeP1-For-biotin | ATTAGTTAAGCGATATTTAAACGAAGTTAAGAATTAGTTAATGGCA |
| SaeP1-Rev | TGCCATTAACTAATTCTTAACTTCGTTTAAATATCGCTTAACTAAT |
| ptst-For-biotin | AAAGTGACTTTAAAGAATATAACTA |
| ptst-Rev | TTTTAATTCTCCTTCATTCAAATGT |
| Probe1-For-IRDye800 | GTAACAAACACTTTTTAATTAATATATATTTAAACAATAATTTAGA |
| Probe1-Rev | TCTAAATTATTGTTTAAATATATATTAATTAAAAAGTGTTTGTTAC |
| Probe 2-For-IRDye800 | TTTTTAATTAATATATATTTAAACAATAATTTAGAGATGGTTAATTGATT |
| Probe2-Rev | AATCAATTAACCATCTCTAAATTATTGTTTAAATATATATTAATTAAAAA |
| Probe3-For-IRDye800 | AATTTAGAGATGGTTAATTGATTCATTTAAATAATATTTATACATTCT |
| Probe3-Rev | AGAATGTATAAATATTATTTAAATGAATCAATTAACCATCTCTAAATT |
Restriction sites (indicated in the primer name) are underlined in the primer sequence, and attB sites are shown in boldface.
Primer contained a phosphate group for blunt-end ligation.
To generate a markerless deletion in the saeS gene, 610 bp of DNA (including the first 2 codons of saeS) was PCR amplified and cloned into the BamHI and SalI sites of the temperature-sensitive integration plasmid pMAD (21). Next, 616 bp of DNA downstream of and within saeS was PCR amplified and cloned into the EcoRI and BglII sites of this plasmid to create pMAD::ΔsaeS (Table 2). The saeS deletion was constructed in the S. aureus MN8 chromosome as described previously (21) to create a 381-bp deletion within the 5′ region of the saeS gene, which was replaced by a 31-bp “scar” (Fig. 1). The correct deletion was confirmed by PCR and DNA sequencing analysis.
FIG 1.

Schematic and PCR analysis of the saeS and sarA in-frame deletions in S. aureus MN8. (A) Scale schematic of the 381-bp deletion in saeS (top) and the 358-bp deletion in sarA (bottom) and location of the PCR products used for analysis and sequencing of the corresponding deletions. (B) DNA agarose gel analysis using PCR products indicated in panel A for the individual and double MN8 sarA and saeS deletions as indicated.
To generate a markerless deletion in the sarA gene, 1,122 bp of DNA (including the first 3 codons of sarA) and 1,063 bp of DNA downstream of sarA (including the last 3 codons) was PCR amplified from the chromosome of S. aureus MN8. These products were ligated together and cloned into pKOR1 using the Gateway BP Clonase II system (Life Technologies). The sarA deletion was constructed in the S. aureus MN8 chromosome as described previously (22) to create a 358-bp, in-frame deletion within the sarA coding region (Fig. 1). The correct deletion was confirmed by PCR and DNA sequencing analysis.
To generate a complementation plasmid for the saeS mutation, a 2,755-bp fragment containing full-length saeQ, saeR, and saeS genes was PCR amplified and directionally cloned using KpnI and EcoRI into pALC2073. To generate a complementation plasmid for the sarA mutation, a 1,321-bp fragment contain full-length sarA and upstream promoter region was PCR amplified and directionally cloned using KpnI and SacI into pALC2073. As pALC2073 contains a tetracycline-inducible promoter, the complementation genes were overexpressed in some experiments by the addition of increasing concentrations of anhydrotetracycline (Sigma-Aldrich), as previously described (23).
Analysis of exoprotein profiles and TSST-1 expression.
S. aureus MN8 and the various mutants were subcultured to a starting optical density at 600 nm (OD600) of 0.05 in BHI medium and grown for indicated time points at 37°C in a shaking incubator. Cells were centrifuged, and cell-free supernatants equivalent to 42 OD600 units (∼4 × 1010 CFU) of cultures were collected. Proteins were precipitated 30 min on ice using trichloroacetic acid (TCA [Fisher Scientific]) at a final concentration of 6%, washed with ice-cold acetone, and resuspended in 250 μl 8 M urea. Proteins were analyzed by 12% SDS-PAGE, and specific proteins of interest were “picked” from Coomassie brilliant blue R-250 (Bio-Rad)-stained gels using an Ettan Spot Picker (GE Healthcare), in-gel digested using a Waters MASSPrep automated digestor (PerkinElmer), and identified by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry at the University of Western Ontario MALDI Mass Spectrometry Facility.
For Western blot analysis of TSST-1 expression, protein samples were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore) at 100 V for 1 h. The membrane was blocked at room temperature for 2 h with phosphate-buffered saline (PBS) containing 5% skimmed milk, 10% normal horse serum (NHS [Life Technologies]), and 10% fetal calf serum (FCS) (Wisent, Inc.). After removal of the blocking solution, the membrane was incubated at 4°C overnight with rabbit polyclonal anti-TSST-1 antisera (kindly provided by Patrick Schlievert, University of Iowa, IA) diluted 1:1,500 in PBS containing 2.5% skimmed milk, 5% NHS, and 5% FCS. Membranes were washed 3 times with PBS containing 0.1% Tween 20 (PBST) (Fisher Scientific) followed by incubation with IRDye 800-conjugated donkey anti-rabbit IgG antibody (Rockland) diluted 1:10,000 in PBS containing 2.5% skimmed milk, 5% NHS, and 5% FCS at room temperature for 1 h in the dark. After the membrane had been washed 3 times with PBST, the membrane was imaged using an Odyssey imager (LI-COR Biosciences, Lincoln, NE).
TSST-1 ELISA.
A sandwich enzyme-linked immunosorbent assay (ELISA) procedure was used for quantification of TSST-1 in S. aureus culture supernatants. Affinity-purified rabbit IgG anti-TSST-1 antibody (Abcam) diluted to 10 μg/ml in 0.01 M carbonate buffer at pH 9.6 (coating buffer) was adsorbed to high-binding polystyrene 96-well plates (Corning) at 37°C for 18 h. Unbound anti-TSST-1 was removed by being washed 4 times with PBST. A 1% bovine serum albumin (Sigma) blocking solution prepared in coating buffer was added to the wells at 37°C for 1 h, and wells were washed with PBST. Filtered supernatant samples were diluted in 1% normal rabbit serum (Thermo Fisher) prepared in PBST. Each diluted supernatant was incubated at room temperature for 30 min to eliminate possible interference of protein A. Recombinant TSST-1 reference standards (serially diluted in PBST from 10 ng/ml to 0.039 ng/ml) and supernatants were added to the wells, and the mixture was incubated at 37°C for 2 h. The plate wells were washed, and incubated with horseradish peroxidase-conjugated affinity purified rabbit IgG anti-TSST-1 antibody (Abcam) diluted 1:300 in PBS at room temperature for 1 h. After the wells were washed, 3,3′,5,5′-tetramethylbenzidine (TMB) substrate solution (BD) was added, and plates were incubated at 37°C for 20 min. A 2 N H2SO4 solution was added to stop the enzymatic reaction. Optical density was read at 450 nm in a microplate reader (Biotek Synergy H4). A standard curve was generated using the data of the reference standards. The model R2 of linear regression analysis was ≥98%. Total amounts of TSST-1 are reported as nanograms per milliliter.
Luciferase reporter assays.
The tst gene promoter (Ptst) was previously cloned into the luciferase reporter plasmid pAmilux (18, 24). The pAmilux::Ptst plasmid was transformed into the different MN8 clones. Cells were grown in BHI medium and subcultured to a starting OD600 of 0.01 (200 μl/well) in half-BHI medium (1:2 dilution of BHI medium in Milli-Q water). Each strain was tested for expression of luminescence over time by growth in a Biotek Synergy H4 multimode plate reader at 37°C with shaking in a flat, clear-bottom 96-well Microfluor 2 White plate (Thermo) sealed using Thermo acetate plate sealers. OD600 and luminescence readings were taken every hour with individual clones grown separately in quadruplicate.
qRT-PCR.
For quantitative real-time PCR (qRT-PCR), S. aureus strains were grown in 5 ml of TSB medium for 18 h. Cells were then subcultured at an OD600 of 0.05 in 3 ml of medium and grown for 4 and 8 h. RNA extraction was carried out using the RNeasy minikit (Qiagen) with the addition of 250 μg/ml lysostaphin to the lysis solution. First-strand cDNA was synthesized using Super-Script II reverse transcriptase and random primers (Invitrogen) from 500 ng of total RNA. PCR mixtures with the primers listed in Table 3 were prepared using iQ SYBR green supermix (Bio-Rad) and performed with the Rotor-Gene real-time analyzer (Corbett Life Science). All RNA samples were prepared using four biological replicates and analyzed against the expression of the housekeeping rpoB gene (18).
Recombinant protein expression and purification.
The protein expression plasmids for full-length SaeR and the C-terminal effector domain of SaeS (SaeSc) (25) were kindly provided by Taeok Bae (Indiana University School of Medicine). Protein purification was carried out as previously described, with some modifications (25). Briefly, cells were grown at 37°C in LB medium to an OD600 of ∼0.6, and 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) was added. The culture for SaeR expression was further incubated at room temperature overnight, while the culture for SaeSC expression was induced at 16°C with shaking. Cells were resuspended in 10 mM Tris-HCl–500 mM NaCl (pH 7.5) and lysed at 25,000 lb/in2 using a cell disruptor (Constants Systems, Ltd.). The His6-tagged proteins were purified using nickel column chromatography using a previously described protocol (26). The purified SaeR and SaeSc were dialyzed in buffers consisting of 10 mM Tris-HCl, 138 mM NaCl, and 2.7 mM KCl (pH 7.5) and 50 mM Tris-HCl, 50 mM KCl, and 1 mM MgCl2 (pH 8.0), respectively (27), and concentrated up to 10-fold using Amicon centrifugal filter devices (Millipore). The protein concentration was determined by the bicinchoninic acid assay (Thermo Scientific), and purified proteins were stored in buffers containing 10% glycerol at −80°C until used.
EMSAs.
Electrophoretic mobility shift assays (EMSAs) were performed using double-stranded fluorescently labeled DNA probes. The DNA probes for the PsaeP1 and Ptst promoter sequences were PCR amplified with 5′-biotin end-labeled primers (Sigma-Aldrich), and IRDye 800-conjugated streptavidin (Rockland) was used (1:2500 dilution) to detect DNA binding. Custom 5′ IRDye 800 end-labeled oligonucleotides (for probes 1, 2, and 3) were purchased from Integrated DNA Technologies (IDT) (Table 3). Equal amounts of forward and reverse DNA oligonucleotides (20 μM) were heated to 100°C for 3 min and allowed to anneal. Phosphorylation of SaeR and the EMSA were done as previously described (25, 28), with some modifications. Proteins and DNA probes were incubated at room temperature in a 25-μl volume containing phosphorylation buffer (10 mM Tris-HCl [pH 7.4], 50 mM KCl, 5 mM MgCl2, 10% glycerol), 240 μg/ml of bovine serum albumin, and 12 μg/ml poly(dI-dC). First, SaeSc was preincubated with 1.6 mM ATP (Sigma-Aldrich) for 10 min. Increasing amounts of SaeR were added to the mixture, and the mixture was further incubated for 10 min. DNA probes (40 nM) were mixed with various amounts of test proteins and incubated for 20 min. Samples were separated in 8% nondenaturing polyacrylamide gels in TBE buffer (10 mM Tris, 89 mM Tris-borate, 2 mM EDTA [pH 8.3]) at 85 V for 85 min. Gels were visualized using an Odyssey imager (LI-COR Biosciences).
RESULTS
The Sae two-component system is a positive regulator of TSST-1 expression.
To investigate a role for the Sae regulatory system on TSST-1 expression, we generated a markerless deletion within the histidine kinase-encoding saeS gene in the prototypical menstrual TSS strain S. aureus MN8 (Fig. 1). Analysis of the exoprotein profile of S. aureus MN8 ΔsaeS resulted in a dramatic reduction of multiple proteins, including the effectual elimination of TSST-1, as determined by mass spectrometry and Western blot analysis (Fig. 2A). Also as identified by mass spectrometry, we saw a large reduction in both the pro- and mature forms of the glycerol ester hydrolase (pro-Geh and Geh, respectively) and a minor reduction in components of the gamma hemolysin (Hlg) (Fig. 2A). Using the inducible complementation plasmid pALC2073, in trans complementation of the sae operon (including saeQRS) restored production of TSST-1, although induction with 500 ng/ml anhydrotetracycline was necessary to fully restore pro-Geh expression. These experiments indicate that the Sae two-component system is a positive regulator of TSST-1 expression in S. aureus MN8.
FIG 2.

Sae is a positive regulator and SarA is a negative regulator of TSST-1 expression in S. aureus MN8. Shown are exoprotein profiles (top panels) and Western blot analysis (bottom panels) of TSST-1 for wild-type S. aureus MN8 and the corresponding (A) saeS and (B) sarA deletion mutant and complemented strains. Concentrated supernatants from the indicated strains grown in BHI medium for 18 h were loaded onto 12% SDS-PAGE gels. Increasing concentrations (0, 5, 50, and 500 ng/ml) of anhydrotetracycline (aTet) were used to induce the promoter in the complemented strains. Molecular mass markers were loaded on the left and labeled in kilodaltons, and purified recombinant TSST-1 was loaded on the right and is indicated by the solid arrowheads.
The SarA stand-alone transcription regulator is a repressor of TSST-1 expression in S. aureus MN8.
Previous work has shown that depending upon the genetic background, SarA may function as either a positive (13) or negative (15) regulator of TSST-1 expression. To investigate a role for TSST-1 regulation by SarA in S. aureus MN8, we generated a markerless deletion within the sarA gene (Fig. 1). Analysis of the exoprotein profile of S. aureus MN8 ΔsarA resulted in a dramatic increase in TSST-1, the aureolysin protease (Aur), and the staphopain A protease (ScpA), a minor increase in nuclease (Nuc), and a notable reduction in expression of pro-Geh (Fig. 2B). Using the inducible complementation plasmid pALC2073, in trans complementation of sarA restored production of TSST-1, although similar to the saeQRS complementation plasmid, induction with 500 ng/ml anhydrotetracycline was necessary to fully restore pro-Geh to wild-type levels. These experiments indicate that SarA is an important negative regulator of TSST-1 expression in S. aureus MN8.
Transcriptional control of TSST-1 by Sae, SarA, and Agr.
Given the striking, but qualitative, data in Fig. 2, we quantified TSST-1 production by ELISA after 18 h of growth (Fig. 3A). Compared with wild-type S. aureus MN8, TSST-1 production was reduced to barely detectable levels in the MN8 ΔsaeS mutant. In S. aureus MN8 Δagr, TSST-1 protein was significantly reduced but not abolished, whereas MN8 ΔsarA produced ∼2.5× the amount of TSST-1 relative to wild-type MN8.
FIG 3.

TSST-1 quantitation, growth curve analysis, and TSST-1 promoter activity of wild-type S. aureus MN8 and the corresponding saeS, sarA, and agr mutants. (A) S. aureus MN8 strains were grown for 18 h in BHI medium, and TSST-1 secretion was quantified by ELISA. The data represent the means ± standard errors of the means (SEM) from three biological replicates (**, P < 0.05 by one-way ANOVA with Tukey's multiple comparison test). (B and C) The indicated S. aureus strains were grown at 37°C in a Biotek Synergy H4 multimode plate reader, and OD600 and luminescence readings were taken every hour for 15 h. Results are expressed as OD600 units (B) and luminescence in relative light units (defined as counts per minute) (C). The data represent the means from three independent experiments, each done with quadruplicate technical replicates.
To evaluate if the changes in TSST-1 expression were mediated at the transcriptional level, we transformed the luciferase reporter plasmid pAmilux (24) containing the tst promoter (Ptst) (18) into each of the S. aureus strains. Although each strain containing pAmilux::Ptst grew equivalently (Fig. 3B), we noted significant differences in luciferase activity for each of the three regulatory mutants (Fig. 3C). Transcription of tst in wild-type MN8 peaked at the late exponential phase (∼7 h), and activity declined during the stationary phase. Deletion of the agr locus resulted in a shift to earlier expression, peaking by 4 h, corresponding to the early exponential phase, and expression was subsequently reduced to background levels when cells entered stationary phase. Consistent with the lack of TSST-1 protein expression (Fig. 2A and 3A), deletion of saeS resulted in a complete reduction of tst expression over the entire growth curve. However, and also consistent with enhanced TSST-1 protein expression (Fig. 2B and 3A), the sarA mutant demonstrated a temporally similar curve to wild-type MN8, although with a marked increase in luminescent activity from the mid- to late-exponential phase.
In order to confirm our findings with the luciferase reporter system, we conducted qRT-PCR experiments at both 4 and 8 h, representing the early and late exponential phases of growth, respectively. In this analysis, we included the tst gene, as well as saeR, sarA, agrA, and the RNAIII transcripts. The qRT-PCR experiments with tst largely confirmed the results of the luciferase assays (Fig. 4A), although qRT-PCR could detect transcripts prior to the detection of luminescence by ∼2 h, and the levels of tst transcripts were thus very low at the 8-h time point in wild-type MN8. Both the agr and saeS mutants demonstrated a clear reduction in tst transcript levels for both time points, while the sarA mutant demonstrated a clear increase in tst transcript levels. Importantly, for each of the regulatory mutants, transcription of the corresponding mutant gene was not detectable above background; however, none of the regulatory mutants had a significant impact on transcription of the other regulatory systems at either time point. Secretion of TSST-1 from the different regulatory mutants at 4 and 8 h (Fig. 4B) was consistent with the transcriptional analysis at these time points. These collective experiments demonstrate that the Sae system is a positive transcriptional regulator, while SarA is a negative transcriptional regulator, of tst expression in S. aureus MN8. These experiments also further confirm that the Agr system is a positive regulator of tst (10, 18).
FIG 4.

qRT-PCR analysis of tst transcripts and relevant regulators in wild-type S. aureus MN8 and the saeS, sarA, and agr mutants. (A) cDNA was prepared at the indicated time points, and copies were normalized to the housekeeping rpoB gene. Data represent the means ± SEM from four biological replicates (*, P < 0.05, **, P < 0.01, and ***, P < 0.001, by one-way ANOVA with Tukey's multiple comparison test). (B) Exoprotein profiles (top panel) and anti-TSST-1 Western blot (bottom panel) analysis of the indicated S. aureus strains at the 4- and 8-h time points. Molecular mass markers were loaded on the left (labeled in kilodaltons), and purified recombinant TSST-1 was loaded on the right (indicated by solid arrowheads).
SaeR binds to the TSST-1 promoter.
Although SarA is known to bind to Ptst (13), given the complete repression of TSST-1 in the saeS deletion background, we hypothesized that SaeR, the response regulatory of the Sae system, would directly regulate TSST-1 transcription. We first assessed the Ptst promoter for a potential SaeR binding motif (25, 29). We located two sequences that could theoretically represent SaeR binding motifs, highlighted by the TTAA(N7)TTAA motif (Fig. 5A). One of the potential SaeR motifs was located immediately upstream of the −35 site (13), which is a common feature of genes directly regulated by SaeR (25, 29). To test binding of SaeR to the Ptst promoter, we produced recombinant SaeR, as well as the cytoplasmic C-terminal domain of the Sae histidine kinase SaeSc (25). SaeSc was used to phosphorylate SaeR in vitro, and we first assessed the ability of phosphorylated SaeR to bind to a known target promoter, namely, the P1 promoter of the Sae operon (PsaeP1) (25). Phosphorylated SaeR (SaeR∼P) bound to PsaeP1 in a concentration-dependent manner (Fig. 5B). Removal of ATP from the in vitro phosphorylation reaction to preclude phosphorylation of SaeR prevented binding to PsaeP1 (Fig. 5B, lane 6). Next, we tested binding of SaeR∼P to an intergenic 271-bp fragment located immediately upstream of tst (Fig. 5C). This experiment demonstrated binding of SaeR∼P to the Ptst promoter region. Again, no binding was detected when ATP was omitted from the phosphorylation reaction. Competition experiments using excess, unlabeled Ptst inhibited binding to labeled Ptst in a concentration-dependent manner (Fig. 5C, right panel). Next, we generated three sets of smaller probes to contain each potential SaeR binding motif (probes 1 and 3) or one probe to contain both potential SaeR binding motifs (probe 2) (Fig. 5A). A very weak shift was detected with probe 1 (Fig. 5D), and probe 2 demonstrated two apparent shifts (Fig. 5E), while probe 3 demonstrated a single, weak shift similar to probe 1 (Fig. 5F). In each case, binding was dependent upon the phosphorylation status of SaeR. These collective experiments demonstrate that SaeR∼P binds directly to Ptst and that tandem sequences consistent with the SaeR consensus binding site appear to be important for optimum binding.
FIG 5.

DNA binding analysis of phosphorylated SaeR (P∼SaeR) to the PsaeP1 and Ptst promoters. (A) Schematic map and nucleotide sequence of Ptst in S. aureus MN8 (locus tag prefix HMPREF0769). The characterized transcriptional start site and −10 and −35 regions are labeled with boxes, and the characterized SarA binding sites is highlighted in blue (13). The proposed SaeR binding sequences are highlighted in green. Probes 1, 2, and 3 correspond to the EMSAs shown in panels D, E, and F, respectively. (C to F) DNA binding analysis of P∼SaeR to PsaeP1 (B), Ptst (C), probe 1 (D), probe 2 (E), and probe 3 (F). Panel C (right panel) also shows a competitive binding analysis using 0-, 5-, 10-, and 50-fold excess unlabeled Ptst (gradient denoted by the triangle). Unbound DNA probes are indicated by open arrowheads, and DNA shifts are indicated by solid arrowheads. Protein concentrations, and the presence of ATP in the reaction buffer, are indicated for each lane.
Sae is a dominant regulatory system that is essential for TSST-1 expression.
Given the data demonstrating that mutation of sarA dramatically increased TSST-1 expression (Fig. 2A, 3A and C, and 4), while mutation of saeS dramatically reduced TSST-1 expression (Fig. 2B, 3A and C, and 4), to investigate which regulatory system played a more dominant role, we generated a double mutation of both regulatory systems in S. aureus MN8 (Fig. 1). Despite the lack of the SarA repressor, the S. aureus ΔsaeS ΔsarA double mutant demonstrated a dramatic reduction in TSST-1 expression that essentially phenocopied the single saeS mutation (Fig. 6), while Aur and ScpA expression appeared to phenocopy the sarA mutation. These findings indicate that although both SarA and Sae are key and opposing regulators of TSST-1, expression of TSST-1 requires functional Sae, even in the absence of repression by SarA.
FIG 6.
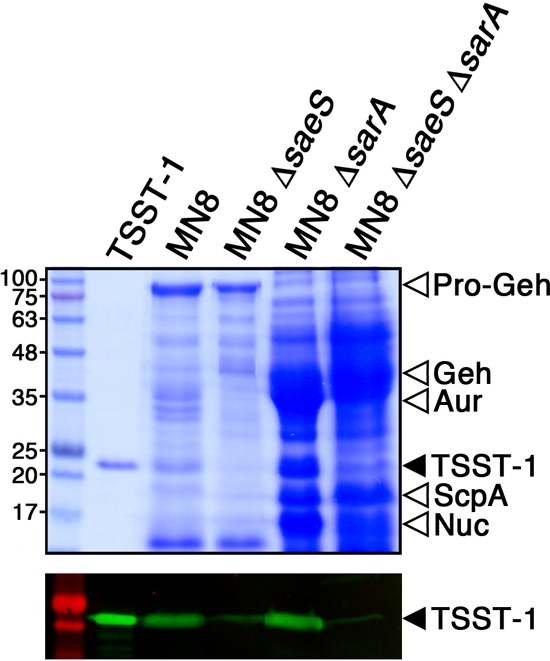
The Sae two-component regulator is necessary for TSST-1 expression in the absence of repression by SarA. Shown are exoprotein profiles (top panel) and Western blot (bottom panel) analysis of TSST-1 for wild-type S. aureus MN8 and the corresponding mutants. Concentrated supernatants from the indicated strains grown in BHI medium were loaded onto 12% SDS-PAGE gels. Molecular mass markers in kilodaltons are shown on the left. Purified recombinant TSST-1 is indicated by the solid arrowhead. The locations of pro-Geh, mature Geh, Aur, ScpA, and nuclease (Nuc) are indicated by open arrowheads.
DISCUSSION
Prior work from our laboratory discovered that particular strains of lactobacilli are able to produce small signaling cyclic dipeptide molecules that repressed the expression of the TSST-1 superantigen in S. aureus MN8 (18). In that work, repression of TSST-1 was clearly correlated to repression of agr, a well-established positive regulator of TSST-1 expression (10), although the precise details as to how TSST-1 is regulated by agr are not known. Nevertheless, mutation of agr does not completely abolish TSST-1 expression (10, 13, 15, 18), and quorum sensing inhibitors based on the agr autoinducing peptide structure also do not eliminate TSST-1 expression (30). Furthermore, in the agr mutant background, TSST-1 could be further repressed by the lactobacillus signals (18), indicating that repression of TSST-1 was not entirely dependent upon agr. Herein, this work confirms that although agr functions as an important positive regulator of TSST-1, the sae regulatory system is the principal regulator that directly controls TSST-1 production and is essential for expression of this important exotoxin.
During the kinetic analysis experiments, transcription of tst in wild-type MN8 was predictably initiated at the late exponential phase (Fig. 3) (10). In MN8 ΔsarA, tst transcription occurred at coinciding time points relative to wild-type MN8, but at much higher levels, which was entirely consistent with the protein expression levels (Fig. 2B, 3A, and 4B) and qRT-PCR analysis (Fig. 4A). Since SarA binds directly to the tst promoter (13), this implies that in the context of TSST-1 regulation in S. aureus MN8, the role of SarA is to compete with SaeR-mediated activation of tst transcription. This is also consistent with the overlapping binding sites of SarA (13) and the SaeR binding motifs localized by the EMSAs (Fig. 5). SarA has been reported to upregulate RNAIII in S. aureus, through enhancing transcription of the agr P2 promoter (31). With S. aureus MN8, however, there was no apparent cross talk between agr, sae, and sarA during the early (4-h) and late (8-h) exponential phases (Fig. 4A), consistent with other S. aureus strains (32). Thus, the reduced levels of TSST-1 in MN8 Δagr were likely, for the most part, independent of sae or sarA.
Earlier studies using a Ptst::lux reporter system integrated into the lipase gene of S. aureus 8325-4 (14) provided evidence that SarA may function as a transcriptional activator of tst expression (13). However, 8325-4 does not carry tst but as noted (15) is known to harbor a mutation in rsbU resulting in a sigma B defect (33). Deletion of the alternative sigma factor sigB gene in strain RN4282, which does carry tst, resulted in increased expression of tst, which also resulted in decreased levels of sarA transcript and increased levels of RNAIII (15). In S. aureus RN4282, SarA functioned to repress TSST-1 production (15). As the effects of sigB disruption on sarA have been previously noted (34), SigB likely acts indirectly, through the derepression of tst, due to decreased sarA. Combined with our work in S. aureus MN8, which is a prototypical menstrual TSS isolate (17), SarA is a TSST repressor in tst-containing S. aureus strains.
S. aureus MN8, similar to other contemporary clonal complex 30 isolates, contains a G55R mutation within AgrC that is known to result in decreased levels of RNAIII and reduced virulence in a mouse model of bacteremia (35). Consistent with this mutation, levels of RNAIII transcripts are reduced in S. aureus MN8 compared with other S. aureus agr groups (18). Yet, S. aureus MN8 Δagr consistently produced reduced, but detectable, levels of TSST-1. The shift to the early expression profile in MN8 Δagr is entirely consist with the protein analysis, where expression was not abolished but peaked at a relatively low cell density, accounting for the small quantities of TSST-1 produced. Evidence exists that agr can antagonize Sae (36), and thus in the absence of agr, the sae system may potentially function prior to 4 h, which could account for the temporal shift in transcription. In addition, activation of agr may function to relieve repression of TSST-1 by the repressor of toxins (Rot), which would no longer be repressed by RNAIII (37, 38).
There are two proposed DNA binding motifs for SaeR (25, 29). Both motifs indicate a highly conserved “primary” TTAAN7TTAA sequence that overlaps the 5′ end of the −35 promoter region. As noted previously, many Sae-regulated genes contain an additional but less conserved “secondary” TTAA repeat motif (25). Figure 7 shows an alignment of the SaeP1 promoter region, and the Sbi promoter region, both of which are highly regulated by the Sae system in S. aureus USA300 (29). In addition, the Geh promoter appears to be Sae regulated in S. aureus MN8, as well as the TSST-1 promoter. Mutation analysis of the SaeP1 promoter demonstrated that the presence of a G residue upstream of either primary TTAA sequence facilitates direct binding by SaeR (25). Of note, the Ptst “primary” motif sequence does not appear to be optimal, yet tst transcript is completely repressed in the absence of a functional Sae system. The evident weak binding of SaeR to Ptst may reflect, in part, the in vitro nature of the system, which lacks RNA polymerase known to enhance SaeR binding to PsaeP1 (39). However, based on these findings, we predict that an additional element may function to enhance SaeR binding to Ptst. Although Rot is generally considered an exotoxin repressor, Rot and SaeR both cooperate to enhance promoter activity for the staphylococcal superantigen-like genes (40). Since Rot is likely a repressor of tst transcription (15), we do not expect that Rot promotes SaeR binding to Ptst. The staphylococcal respiratory response (srr) two-component system has been shown to influence TSST-1 expression, primarily under microaerophilic conditions. SrrA has also been demonstrated to weakly bind Ptst, and thus it is possible that Sae cooperates with other systems, such as Srr, in the regulation of TSST-1. Nevertheless, the apparent weak binding of SaeR to Ptst in vitro is consistent with other studies in which Sae is a key regulator (e.g., nuc and ssl promoters) (40, 41).
FIG 7.

Alignment of promoter element nucleotide sequences of SaeR binding motifs located upstream of PsaeP1, Psbi, Pgeh, Ptst, and Pseb. The −35 promoter binding regions are underlined, and green highlights outline the primary and secondary SaeR binding motifs.
Apart from regulation of TSST-1 in S. aureus MN8, the reduction of pro-Geh expression in MN8 ΔsaeS (Fig. 2) is consistent with Sae functioning as a positive transcriptional regulator of geh in USA300 (29), as well as a potential SaeR binding site (25, 29) located upstream of geh (Fig. 7). In MN8 ΔsarA, as expected (42, 43), we noted a prominent increase in expression of the aureolysin (Aur) metalloprotease and staphopain A (ScpA) cysteine protease. Aur is responsible for maturation of pro-Geh to mature Geh (44), which explains the near complete lack of pro-Geh in the sarA mutant. Thus, although the regulatory role of SarA and Sae for these other important exoproteins was largely predicted, a dominant role for regulation by Sae is selective for particular exotoxins.
Consistent with a prior model as to how phosphorylated SaeR recruits RNA polymerase to Sae-regulated target promoters (25), we herein show that the Sae system is required for expression of TSST-1 and thus functions as a dominant transcriptional activator of TSST-1 expression. Although the regulatory queues that control Sae activation are complex, the specific signals that govern Sae control of TSST-1 expression during the context of menstrual TSS remain uncharacterized. Given that increased expression of saeRS from the inducible complementation system did not enhance TSST-1 expression (Fig. 2A), we would predict that host factors that alter SaeR phosphorylation, rather than autoinduction of the sae system, would play a more important role for enhanced TSST-1 expression. Apart from the role of TSST-1 in both menstrual and nonmenstrual forms of TSS, another major superantigen with a clear role in nonmenstrual TSS is staphylococcal enterotoxin B (SEB) (9). SEB expression is known to be controlled by Sae (32, 45), while this two-component regulator had no effect on transcription of sea or sel-k (45). Immediately upstream of the −35 promoter region of seb (46) is a potential SaeR consensus binding sequence (Fig. 7). In an in vivo rabbit model of non-menstrual TSS, agr activation was not necessary for the expression of SEB and the development of TSS (47). Thus, potentially, Sae may also be the dominant regulator of SEB expression in S. aureus. A picture has therefore emerged whereby the Sae system seems to have evolved to be a critical regulator of S. aureus “trademark” superantigen exotoxins involved in TSS, and thus Sae may represent an important target for the rational design of targeted therapeutics with a focus to disarm the exotoxin arsenal of S. aureus.
ACKNOWLEDGMENTS
We thank Taeok Bae (Indiana University) for the kind gift of the SaeR and SaeSc expression plasmids, Patrick Schlievert (University of Iowa) for the kind gift of anti-TSST-1 antisera, and Timothy Foster (Trinity College Dublin) for the kind gift of E. coli SA30B.
REFERENCES
- 1.Lowy FD. 1998. Staphylococcus aureus infections. N Engl J Med 339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 2.Dinges MM, Orwin PM, Schlievert PM. 2000. Exotoxins of Staphylococcus aureus. Clin Microbiol Rev 13:16–34. doi: 10.1128/CMR.13.1.16-34.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCormick JK, Yarwood JM, Schlievert PM. 2001. Toxic shock syndrome and bacterial superantigens: an update. Annu Rev Microbiol 55:77–104. doi: 10.1146/annurev.micro.55.1.77. [DOI] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention. 2011. Toxic shock syndrome; 2011 case definition. Centers for Disease Control and Prevention, Atlanta, GA. [Google Scholar]
- 5.DeVries AS, Lesher L, Schlievert PM, Rogers T, Villaume LG, Danila R, Lynfield R. 2011. Staphylococcal toxic shock syndrome 2000–2006: epidemiology, clinical features, and molecular characteristics. PLoS One 6:e22997. doi: 10.1371/journal.pone.0022997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis JP, Chesney PJ, Wand PJ, LaVenture M. 1980. Toxic-shock syndrome: epidemiologic features, recurrence, risk factors, and prevention. N Engl J Med 303:1429–1435. doi: 10.1056/NEJM198012183032501. [DOI] [PubMed] [Google Scholar]
- 7.Shands KN, Schmid GP, Dan BB, Blum D, Guidotti RJ, Hargrett NT, Anderson RL, Hill DL, Broome CV, Band JD, Fraser DW. 1980. Toxic-shock syndrome in menstruating women: association with tampon use and Staphylococcus aureus and clinical features in 52 cases. N Engl J Med 303:1436–1442. doi: 10.1056/NEJM198012183032502. [DOI] [PubMed] [Google Scholar]
- 8.Spaulding AR, Salgado-Pabón W, Kohler PL, Horswill AR, Leung DYM, Schlievert PM. 2013. Staphylococcal and streptococcal superantigen exotoxins. Clin Microbiol Rev 26:422–447. doi: 10.1128/CMR.00104-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schlievert PM. 1986. Staphylococcal enterotoxin B and toxic-shock syndrome toxin-1 are significantly associated with non-menstrual TSS. Lancet i:1149–1150. (Letter.) [DOI] [PubMed] [Google Scholar]
- 10.Recsei P, Kreiswirth B, O'Reilly M, Schlievert P, Gruss A, Novick RP. 1986. Regulation of exoprotein gene expression in Staphylococcus aureus by agr. Mol Gen Genet 202:58–61. doi: 10.1007/BF00330517. [DOI] [PubMed] [Google Scholar]
- 11.Yarwood JM, McCormick JK, Schlievert PM. 2001. Identification of a novel two-component regulatory system that acts in global regulation of virulence factors of Staphylococcus aureus. J Bacteriol 183:1113–1123. doi: 10.1128/JB.183.4.1113-1123.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagao M, Okamoto A, Yamada K, Hasegawa T, Hasegawa Y, Ohta M. 2009. Variations in amount of TSST-1 produced by clinical methicillin resistant Staphylococcus aureus (MRSA) isolates and allelic variation in accessory gene regulator (agr) locus. BMC Microbiol 9:52. doi: 10.1186/1471-2180-9-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andrey DO, Renzoni A, Monod A, Lew DP, Cheung AL, Kelley WL. 2010. Control of the Staphylococcus aureus toxic shock tst promoter by the global regulator SarA. J Bacteriol 192:6077–6085. doi: 10.1128/JB.00146-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan PF, Foster SJ. 1998. Role of SarA in virulence determinant production and environmental signal transduction in Staphylococcus aureus. J Bacteriol 180:6232–6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andrey DO, Jousselin A, Villanueva M, Renzoni A, Monod A, Barras C, Rodriguez N, Kelley WL. 2015. Impact of the regulators SigB, Rot, SarA and SarS on the toxic shock tst promoter and TSST-1 expression in Staphylococcus aureus. PLoS One 10:e0135579. doi: 10.1371/journal.pone.0135579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seidl K, Bischoff M, Berger-Bächi B. 2008. CcpA mediates the catabolite repression of tst in Staphylococcus aureus. Infect Immun 76:5093–5099. doi: 10.1128/IAI.00724-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schlievert PM, Blomster DA. 1983. Production of staphylococcal pyrogenic exotoxin type C: influence of physical and chemical factors. J Infect Dis 147:236–242. doi: 10.1093/infdis/147.2.236. [DOI] [PubMed] [Google Scholar]
- 18.Li J, Wang W, Xu SX, Magarvey NA, McCormick JK. 2011. Lactobacillus reuteri-produced cyclic dipeptides quench agr-mediated expression of toxic shock syndrome toxin-1 in staphylococci. Proc Natl Acad Sci U S A 108:3360–3365. doi: 10.1073/pnas.1017431108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Novick R. 1967. Properties of a cryptic high-frequency transducing phage in Staphylococcus aureus. Virology 33:155–166. doi: 10.1016/0042-6822(67)90105-5. [DOI] [PubMed] [Google Scholar]
- 20.Monk IR, Tree JJ, Howden BP, Stinear TP, Foster TJ. 2015. Complete bypass of restriction systems for major Staphylococcus aureus lineages. mBio 6:e00308-15. doi: 10.1128/mBio.00308-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arnaud M, Chastanet A, Debarbouille M. 2004. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, Gram-positive bacteria. Appl Environ Microbiol 70:6887–6891. doi: 10.1128/AEM.70.11.6887-6891.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bae T, Schneewind O. 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55:58–63. doi: 10.1016/j.plasmid.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 23.Bateman BT, Donegan NP, Jarry TM, Palma M, Cheung AL. 2001. Evaluation of a tetracycline-inducible promoter in Staphylococcus aureus in vitro and in vivo and its application in demonstrating the role of sigB in microcolony formation. Infect Immun 69:7851–7857. doi: 10.1128/IAI.69.12.7851-7857.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mesak LR, Yim G, Davies J. 2009. Improved lux reporters for use in Staphylococcus aureus. Plasmid 61:182–187. doi: 10.1016/j.plasmid.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 25.Sun F, Li C, Jeong D, Sohn C, He C, Bae T. 2010. In the Staphylococcus aureus two-component system sae, the response regulator SaeR binds to a direct repeat sequence and DNA binding requires phosphorylation by the sensor kinase SaeS. J Bacteriol 192:2111–2127. doi: 10.1128/JB.01524-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nur-ur Rahman AK, Bonsor DA, Herfst CA, Pollard F, Peirce M, Wyatt AW, Kasper KJ, Madrenas J, Sundberg EJ, McCormick JK. 2011. The T cell receptor beta-chain second complementarity determining region loop (CDR2beta) governs T cell activation and Vbeta specificity by bacterial superantigens. J Biol Chem 286:4871–4881. doi: 10.1074/jbc.M110.189068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Q, Cho H, Yeo W-S, Bae T. 2015. The extracytoplasmic linker peptide of the sensor protein SaeS tunes the kinase activity required for staphylococcal virulence in response to host signals. PLoS Pathog 11:e1004799. doi: 10.1371/journal.ppat.1004799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laakso HA, Marolda CL, Pinter TB, Stillman MJ, Heinrichs DE. 2016. A heme-responsive regulator controls synthesis of staphyloferrin B in Staphylococcus aureus. J Biol Chem 291:29–40. doi: 10.1074/jbc.M115.696625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nygaard TK, Pallister KB, Ruzevich P, Griffith S, Vuong C, Voyich JM. 2010. SaeR binds a consensus sequence within virulence gene promoters to advance USA300 pathogenesis. J Infect Dis 201:241–254. doi: 10.1086/649570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tal-Gan Y, Stacy DM, Foegen MK, Koenig DW, Blackwell HE. 2013. Highly potent inhibitors of quorum sensing in Staphylococcus aureus revealed through a systematic synthetic study of the group-III autoinducing peptide. J Am Chem Soc 135:7869–7882. doi: 10.1021/ja3112115. [DOI] [PubMed] [Google Scholar]
- 31.Reyes D, Andrey DO, Monod A, Kelley WL, Zhang G, Cheung AL. 2011. Coordinated regulation by AgrA, SarA, and SarR to control agr expression in Staphylococcus aureus. J Bacteriol 193:6020–6031. doi: 10.1128/JB.05436-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rogasch K, Rühmling V, Pané-Farré J, Höper D, Weinberg C, Fuchs S, Schmudde M, Bröker BM, Wolz C, Hecker M, Engelmann S. 2006. Influence of the two-component system SaeRS on global gene expression in two different Staphylococcus aureus strains. J Bacteriol 188:7742–7758. doi: 10.1128/JB.00555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giachino P, Engelmann S, Bischoff M. 2001. Sigma B activity depends on RsbU in Staphylococcus aureus. J Bacteriol 183:1843–1852. doi: 10.1128/JB.183.6.1843-1852.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bischoff M, Dunman P, Kormanec J, Macapagal D, Murphy E, Mounts W, Berger-Bächi B, Projan S. 2004. Microarray-based analysis of the Staphylococcus aureus sigmaB regulon. J Bacteriol 186:4085–4099. doi: 10.1128/JB.186.13.4085-4099.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeLeo FR, Kennedy AD, Chen L, Bubeck Wardenburg J, Kobayashi SD, Mathema B, Braughton KR, Whitney AR, Villaruz AE, Martens CA, Porcella SF, McGavin MJ, Otto M, Musser JM, Kreiswirth BN. 2011. Molecular differentiation of historic phage-type 80/81 and contemporary epidemic Staphylococcus aureus. Proc Natl Acad Sci U S A 108:18091–18096. doi: 10.1073/pnas.1111084108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giraudo AT, Cheung AL, Nagel R. 1997. The sae locus of Staphylococcus aureus controls exoprotein synthesis at the transcriptional level. Arch Microbiol 168:53–58. doi: 10.1007/s002030050469. [DOI] [PubMed] [Google Scholar]
- 37.Geisinger E, Adhikari RP, Jin R, Ross HF, Novick RP. 2006. Inhibition of rot translation by RNAIII, a key feature of agr function. Mol Microbiol 61:1038–1048. doi: 10.1111/j.1365-2958.2006.05292.x. [DOI] [PubMed] [Google Scholar]
- 38.Boisset S, Geissmann T, Huntzinger E, Fechter P, Bendridi N, Possedko M, Chevalier C, Helfer AC, Benito Y, Jacquier A, Gaspin C, Vandenesch F, Romby P. 2007. Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev 21:1353–1366. doi: 10.1101/gad.423507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cho H, Jeong D-W, Li C, Bae T. 2012. Organizational requirements of the SaeR binding sites for a functional P1 promoter of the sae operon in Staphylococcus aureus. J Bacteriol 194:2865–2876. doi: 10.1128/JB.06771-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benson MA, Lilo S, Nygaard T, Voyich JM, Torres VJ. 2012. Rot and SaeRS cooperate to activate expression of the staphylococcal superantigen-like exoproteins. J Bacteriol 194:4355–4365. doi: 10.1128/JB.00706-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Olson ME, Nygaard TK, Ackermann L, Watkins RL, Zurek OW, Pallister KB, Griffith S, Kiedrowski MR, Flack CE, Kavanaugh JS, Kreiswirth BN, Horswill AR, Voyich JM. 2013. Staphylococcus aureus nuclease is an SaeRS-dependent virulence factor. Infect Immun 81:1316–1324. doi: 10.1128/IAI.01242-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ziebandt AK, Weber H, Rudolph J, Schmid R, Höper D, Engelmann S, Hecker M. 2001. Extracellular proteins of Staphylococcus aureus and the role of SarA and sigma B. Proteomics 1:480–493. [DOI] [PubMed] [Google Scholar]
- 43.Jones RC, Deck J, Edmondson RD, Hart ME. 2008. Relative quantitative comparisons of the extracellular protein profiles of Staphylococcus aureus UAMS-1 and its sarA, agr, and sarA agr regulatory mutants using one-dimensional polyacrylamide gel electrophoresis and nanocapillary liquid chromatography coupled with tandem mass spectrometry. J Bacteriol 190:5265–5278. doi: 10.1128/JB.00383-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cadieux B, Vijayakumaran V, Bernards MA, McGavin MJ, Heinrichs DE. 2014. Role of lipase from community-associated methicillin-resistant Staphylococcus aureus strain USA300 in hydrolyzing triglycerides into growth-inhibitory free fatty acids. J Bacteriol 196:4044–4056. doi: 10.1128/JB.02044-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kusch K, Hanke K, Holtfreter S, Schmudde M, Kohler C, Erck C, Wehland J, Hecker M, Ohlsen K, Bröker B, Engelmann S. 2011. The influence of SaeRS and σ(B) on the expression of superantigens in different Staphylococcus aureus isolates. Int J Med Microbiol 301:488–499. doi: 10.1016/j.ijmm.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 46.Mahmood R, Khan SA. 1990. Role of upstream sequences in the expression of the staphylococcal enterotoxin B gene. J Biol Chem 265:4652–4656. [PubMed] [Google Scholar]
- 47.Yarwood JM, McCormick JK, Paustian ML, Kapur V, Schlievert PM. 2002. Repression of the Staphylococcus aureus accessory gene regulator in serum and in vivo. J Bacteriol 184:1095–1101. doi: 10.1128/jb.184.4.1095-1101.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]