

Abstract Abstract
The application of complex data sources to pulmonary vascular diseases is an emerging and promising area of investigation. The use of -omics platforms, in silico modeling of gene networks, and linkage of large human cohorts with DNA biobanks are beginning to bear biologic insight into pulmonary hypertension. These approaches to high-throughput molecular phenotyping offer the possibility of discovering new therapeutic targets and identifying variability in response to therapy that can be leveraged to improve clinical care. Optimizing the methods for analyzing complex data sources and accruing large, well-phenotyped human cohorts linked to biologic data remain significant challenges. Here, we discuss two specific types of complex data sources—gene regulatory networks and DNA-linked electronic medical record cohorts—that illustrate the promise, challenges, and current limitations of these approaches to understanding and managing pulmonary vascular disease.
Keywords: pulmonary vascular disease, systems biology, computational network modeling, electronic medical record, biorepository, genomics
The emergence of large, complex sources of biologic data is presenting new opportunities and challenges for discovery in modern medicine. Technical improvements in “-omics” tools (e.g., genomics, metabolomics, proteomics) now allow relatively cost-efficient high-throughput molecular profiling of biologic samples. Concurrent advances in computational strategies for modeling biologic networks in silico are also contributing new insights to our understanding of disease pathophysiology. Integration of these tools and linkage of biologic data with well-phenotyped human cohorts is a promising approach to discovering new targets of therapeutic interest and markers of disease susceptibility and to understanding variability in phenotypic expression and therapeutic response. A significant challenge for investigators using these tools is determining the optimal methods for distilling meaningful information from vast amounts of data.
The application of complex data sources to biologic discovery in pulmonary vascular diseases such as pulmonary hypertension (PH) is evolving rapidly. For example, in silico modeling and functional validation of complex gene regulatory networks has identified microRNAs (miRNAs) as important mediators of pulmonary vascular disease.1 In addition, transcriptional profiling of peripheral blood in humans has been shown to noninvasively discriminate patients with vasodilator-responsive pulmonary arterial hypertension (PAH).1,2 The National Institutes of Health has recognized the importance of using a “big-data” approach to understanding pulmonary vascular disease, as evident by the creation of the Pulmonary Vascular Disease–omics (PVDomics) Consortium.3 The PVDomics Consortium comprises six institutions tasked with applying deep clinical and molecular phenotyping approaches to better understand and treat PH subtypes.
This article describes in detail the advantages and challenges of using two specific complex data sources as they pertain to pulmonary vascular disease. These topics and the data presented in this review were presented at the 2015 Grover Conference on Pulmonary Vascular Disease. First, we discuss the current limitations of systems-biology approaches in PH and new methods for elucidating the role of gene regulatory networks. Next, we describe the potential for gene and biomarker discovery using electronic medical record cohorts linked to biorepositories.
Part 1: Leveraging network biology to consider the molecular complexity of pulmonary vascular disease
Computational network modeling can be a powerful and emerging tool for the identification of novel disease regulators in pulmonary vascular disease by gleaning biological information from in silico analysis of gene network architecture and kinetics. However, for pulmonary vascular disease, it has been limited by the lack of available molecular profiling data from relevant diseased tissue and optimized computational strategies for assessing the integrated effects of the complex interactions among disease genes.
Limitations of reductionist biology for research in complex diseases such as PH
For more than a century, traditional reductionist discovery methods applied to human diseases such as PH have been supported by an assumption that single biological factors operate in simple mechanistic association to control disease pathobiology. While useful in an earlier era, such oversimplification has limited our full understanding of complex human disease. Data have been accumulating from vast endeavors of high-throughput -omics profiling in general (e.g., the ENCODE project,4-7 which has sought to identify all functional elements in the human genome). In this postgenomic era, we also appreciate that PH is driven by a complex and interconnected network of molecular processes, but these networks are only just beginning to be defined and catalogued in this disease.8 Deciphering these networks, however, has been challenging, given the dearth of computational methods able to model these complex systems in such a way that biological fidelity is retained. If we can find a way to build upon and harness such high-throughput data, we could gain unique insight into the integrative nature of PH and human disease in general. Hints of such potential for systems-wide molecular discovery in PH have recently been reported,1,9-11 but otherwise, systems biology in PH has remained relatively unexplored. Additional advances in this realm could allow for novel predictions of disease molecules, the interconnected actions of those molecules in pathogenesis, the molecular relationships among distinct disease conditions, and the concerted response to therapeutic interventions. As a result, new avenues will be open to explore individualized nuances in disease expression, susceptibility states, responses to targeted therapies, and preclinical disease manifestations—ideas that go far beyond the static genomic predictions emphasized now by personalized-medicine programs. An entirely new framework of human disease classification may even be revealed through network analysis, leading to discovery of critical relationships among seemingly disparate diseases12 and PH.13 Thus, there exists a pressing need to distill critical insight from the volumes of molecular data derived from human tissue for application to clinical and therapeutic use.
Current systems approaches in PH and other complex human diseases
To better understand disease complexities, studies of simple disease networks have been performed, but their direct translation to human pathogenesis has been limited.14 These studies have been driven by the unproven assumption that random isolated changes in any disease gene could drastically alter the overall state of the associated disease network and thus affect disease phenotype. According to that logic, an understanding of disease network kinetics and dynamics is impossible without a complete network map. To this end, molecular atlases continue to be updated with biochemical, genomic, gene regulatory, protein-protein, and epigenetic data (as reviewed by Barabási et al.15), the compilation of which contains all known molecular connections affecting human health and disease. Architectural networks of this kind, however, while providing a comprehensive view of the molecular landscape of disease, lack critical information about the relative strength, tissue specificity, and at times even the directionality of the catalogued interactions. It is likely not the case that all disease genes are created equal, even in a multifactorial disease context, and without these additional features it becomes difficult to identify those genes and interactions that would most benefit from therapeutic intervention.
Clearly, a comprehensive quantitative characterization of all molecular interactions is a worthwhile goal. Yet such a characterization is still in its early stages. Disease-specific high-throughput profiling may take decades because of the relative anatomic inaccessibility of PH-relevant tissue in vivo and the financial constraints of extensive measurements.16 As such, current high-throughput data sets relevant to PH are growing but still sparse, either in animal models of PH17 or in humans.18 Concerted efforts are ongoing from both federal and private funding initiatives to increase the richness of these molecular profiling data sets across populations of PH patients. However, data collection will not be immediate. Furthermore, it is not obvious how such data would be parsed and analyzed once collected. While the modeling of static network topology in other contexts has been accomplished (i.e., the ENCODE project9,19), studies that have modeled biological network dynamics have been limited to single-cell contexts amenable to high-throughput profiling but insufficient to reflect more complex illnesses (as we have reviewed14,20). Less compelling reports, while often mathematically eloquent, have been otherwise too far removed from basic biology or computationally too complex to validate experimentally. Thus, while advancing, systems approaches to understanding PH are nonetheless currently limited by (1) the lack of crucial quantitative information in the most widely available comprehensive gene atlases and networks, (2) the high financial cost of obtaining dynamic and kinetic network data on an as-needed basis, and (3) the difficulty of producing complex models that are nonetheless faithful to biology and testable in real-world models.
Modeling miRNA networks in PH by coupling computational network theory with experimental biology
Even with these current limitations, it is still possible to make advances in deciphering the network activity in PH biology. Our group has been studying the PH-relevant regulatory actions of miRNAs, which are small, noncoding RNA molecules that rely on antisense sequence complementarity to engage messenger transcripts, leading to negative regulation of gene expression. Such posttranscriptional processes of gene regulation carry pervasive functions throughout plant and animal cells. However, the extreme pleiotropy of such molecules makes it difficult to determine which miRNAs may have the most robust effects on an entire network of genes and, consequently, on a resultant disease pathophenotype. Our group has endeavored to utilize computational network theory to analyze PH disease gene network architecture in order to identify miRNAs that carry broad systems-level control over the PH disease state.
To do so, we developed an in silico18,21,22 approach to rank miRNAs with systems-wide effects on known downstream PH-relevant genes and their first-degree interactors (“the PH network,” mapped with databases cataloguing transcriptome/proteome interactions). Specifically, we constructed a comprehensive network of PH-relevant genes and analyzed its pathway enrichment (color coding denotes functional pathway) and architectural structure (circles denote architectural clusters; Fig. 1A). We ranked miRNAs (“miRNA spanning score”) on the basis of the size and intercluster spread of their target pools and identified miR-130/301 as a highly influential regulatory factor.1 We ranked genes in the miR-130/301 target pool in silico by “hubness” and other centrality metrics and found top-ranking genes to form the outline of two known proliferative pathways in the lung (Fig. 1B). Subsequently, we verified these findings in vivo and in vitro, demonstrating that miR-130/301 was upregulated in the pulmonary vasculature of PH patients (multiple etiologies; Fig. 1C) and that it modulated vascular proliferation in pulmonary arterial endothelial cells (data not shown), as well as in pulmonary arterial smooth muscle cells, via the STAT3-miR-204-SRC axis (Fig. 1D). Separately, on the basis of the remaining PH-relevant targets, we defined a role for miR-130/301 in controlling pulmonary vasomotor tone.23,24 Finally, on the basis of an observed overlap between a fibrosis network and the PH network, accompanied by a highly ranked miRNA spanning score in both networks, we delineated a self-sustaining feedback loop driven by a central mechanosensitive YAP/TAZ-miR-130/301 axis, controlling extracellular matrix remodeling and vascular stiffness in PH.9,25 In sum, as predicted by our network-based algorithms, a model was confirmed, defining the global influence of miR-130/301 on vascular proliferation (Fig. 1E), vasomotor tone, and vascular stiffness in response to a variety of PH triggers. Thus, by combining such bioinformatics with unique reagents derived from animal models and humans, systems-level discovery in PH is possible and has distinct advantages, as compared with traditional scientific approaches.
Figure 1.

Prediction of microRNA (miR-)130/301-PPARγ (peroxisome proliferator–activated receptor γ; also PPARG) signaling axis in pulmonary hypertension (PH) via in silico network modeling. A, As we previously reported,1 we constructed a comprehensive network of pulmonary hypertension (PH)–relevant genes and analyzed its pathway enrichment (color coding denotes functional pathway) and architectural structure (circles denote architectural clusters). B, We ranked microRNAs (miRNAs/miRs) on the basis of the size and intercluster spread of their target pools and identified miR-130/301 as a highly influential regulatory factor. We ranked genes in the miR-130/301 target pool in silico by “hubness” and other centrality metrics and found top-ranking genes to form the outline of two known proliferative pathways in the lung. C, D, We verified these findings in vivo and in vitro, demonstrating that miR-130/301 was upregulated in the pulmonary vasculature of PH patients (multiple etiologies; C) and that it modulated vascular proliferation in PAECs (data not shown) and PASMCs by the STAT3-miR-204-SRC axis (D). E, As predicted by our network-based algorithms, a model was confirmed of the global influence of miR-130/301 on vascular proliferation in response to a variety of PH triggers. α-SMA: α smooth muscle actin; APLN: apelin; BMPR2: bone morphogenetic protein receptor 2; CAV1: caveolin 1; FGF2: fibroblast growth factor 2; HIF-2α: hypoxia-inducible factor 2α; IL: interleukin; OCT4: octamer-binding transcription factor 4; PAEC: pulmonary arterial endothelial cell; PAH: pulmonary arterial hypertension; PASMC: pulmonary arterial smooth muscle cell; STAT3: signal transducer and activator of transcription 3. Images adapted from Bertero et al.1 and reprinted with permission.
Future directions
Clearly, we are just at the beginning of optimally considering complex gene network architecture and kinetics in the development of PH. While our approach has attempted to leverage the pleiotropy of miRNAs in constructing our discovery platform, an obvious next step would include an overlay of our expanding appreciation of the genomic architectural landscape and genetic associations relevant to PH.20,26 Alternatively, or perhaps in combination, the emerging renaissance of information regarding mammalian metabolic networks could lead to an ample foundation for systems-based discovery, particularly in the pulmonary vasculature.18,22,25 New technologies to procure diseased PAH tissue from living PH patients23,27-29 will also figure prominently in further building our repertoire of high-throughput molecular profiling in this disease. Once more complete data sets are assembled, advents in “big-data” analysis, such as high-scale dynamic network modeling, would considerably advance our abilities to trace the molecular relationships between inception and end-stage PAH—far beyond the capabilities offered through the study of static gene network architecture alone. However, such large-scale dynamic modeling, which attempts to account for temporal separation between distinct cellular processes, would require extensive time-course data for accurate inference and thus would carry a high financial cost, at least by current standards. Yet the combination of such network-based theory with biological experimentation in PH, when possible, would have transformative implications. Results could guide the identification of the currently perplexing molecular origins of PH, the development of new diagnostic tools to identify populations at risk, and the generation of more powerful drugs that carry systems-level control over disease manifestation. As a result, this work could launch an entirely new direction in “network medicine”—applicable not only to PH research but also other complex human diseases.
Part 2: Electronic medical records linked to biobanks: opportunities for insight into pulmonary vascular disease
The role of electronic medical records in clinical research
Electronic medical records (EMRs) are a pervasive part of contemporary medical care, particularly at academic institutions.25 In addition to their impact on clinical care, the widespread adoption of EMRs has accelerated clinical research. Using a combination of common data fields (e.g., vital signs, laboratory values) and natural-language processing, EMRs permit the identification of large cohorts of subjects with a specific phenotype (and relevant control subjects). This allows investigators to make observations that would not be practical or possible in prospectively enrolled cohorts. This approach is particularly attractive for the study of rare diseases, in which the aggregation of larger cohorts is challenging.
EMR-linked biorepositories
Using an EMR alone, observations are limited to epidemiologic descriptions of clinical features and outcomes. Over the past decade, there have been coordinated efforts to link EMRs with biologic data (with a particular focus on genomic data) to allow insight into disease etiology and pathophysiology. These efforts are exemplified by the Electronic Medical Records and Genomic (eMERGE) network, initiated in 2007 and sponsored by the National Human Genome Research Institute.26 Krishnamoorthy et al.25 provide an in-depth review of the eMERGE network’s development and activities. The eMERGE network is tasked with developing best practices for identifying phenotypes within EMRs and integrating EMRs with genetic data. Algorithms for identifying specific phenotypes (e.g., resistant hypertension and asthma) are developed and tested across eMERGE network sites for validation.
Each of the 9 eMERGE sites maintains a prospective DNA biorepository linked to their EMRs, which allows the discovery and validation of genotype-phenotype associations. Examples of discoveries using EMR-linked DNA biobanks include variants associated with PR segment duration on electrocardiography and variants associated with white blood cell count.27-29Most eMERGE sites have employed an “opt-in” approach to DNA collection, in which patients with targeted phenotypes are consented for blood collection. An advantage of this approach is that patient identifiers can be maintained, which allows recontact of patients (and potentially family members) for future studies. Until recently, the Vanderbilt University BioVU program used an “opt-out” approach. At the time of consent for treatment, all inpatients and outpatients were given an option to decline to have their DNA collected in BioVU. Under BioVU, if blood is collected for clinical indications and excess sample remains after clinical testing, DNA is extracted and linked to the patient’s medical record. Unlike the other eMERGE network biorepositories, BioVU links DNA to a deidentified version of Vanderbilt’s EMR known as the “Synthetic Derivative.” BioVU now employs an opt-in approach at the time of consent for treatment. Therefore, patients cannot be contacted for future research, and familial studies are not possible. In addition to DNA, EMRs are also being linked to prospective plasma collection, which will facilitate biomarker studies (e.g., proteomics, metabolomics) to identify intermediate links between genotype and phenotype.30 The design and implementation of Vanderbilt’s EMR-linked DNA repository have been described previously.31 Key components of the initiation of the BioVU program include active community engagement and education to inform ethical considerations, robust EMR and sample deidentification mechanisms, and a user-friendly web interface to allow case review.
EMR-based cohorts versus traditional prospective cohorts
Traditional prospective cohort studies face several challenges. Accruing cohorts large enough for adequately powered evaluations of clinical outcomes (and subgroup analyses) can be extremely costly and time-consuming, particularly with respect to rare diseases. Moreover, many cohort studies and registries target patients with specific diseases while excluding subjects who may be highly relevant controls with similar comorbidity profiles and environmental exposures. Long-term follow-up in cohort studies adds cost because it requires continued investment in infrastructure to maintain contact with subjects.
EMR-based cohorts overcome a number of the limitations of traditional cohort studies (Table 1). The combination of a high-volume clinical center and established EMRs allows investigators to identify large cohorts for most clinical phenotypes. Bowton et al.32 recently showed that the median cohort size for 28 validated phenotypes in Vanderbilt’s Synthetic Derivative was 1,123 (interquartile range [IQR]: 492–4,158) subjects, compared with 623 (IQR: 273–2,095) subjects in similar cohort studies funded by the National Institutes of Health. Moreover, the median time to construct the EMR-based cohorts was 3 months, compared with 3 years for NIH-funded cohorts. In addition to identification of “cases” with a phenotype of interest, EMRs allow concurrent identification of relevant healthy and disease controls, which is particularly important for genetic-association studies. An additional cost-effective advantage of EMR cohorts is the potential to examine multiple phenotypes within one cohort. For example, in the study by Bowton et al.,32 90% of the cases or controls for the 28 phenotypes investigated were reused as a case or control for another phenotype.
Table 1.
Advantages and limitations of EMR cohorts linked to DNA biobanks
| Advantages | Limitations |
|---|---|
| Identify large cohorts for most phenotypes, including many rare diseases; Efficient creation of cohorts compared with prospective studies; Extraction of relevant controls (e.g., subjects referred for RHC found not to have PH); Able to examine multiple phenotypes within one cohort (cost-effective); Multisite EMRs can be combined to generate larger cohorts; EMRs can be linked to external data sources (Medicare, state registries, etc.); Genetic association discovery and replication can be performed in single or multisite cohorts |
Phenotype specificity depends on accuracy of case definition; Temporality can be challenging to ascertain (e.g., between risk factors and development of disease); No truly “healthy” control subjects when using hemodynamic diagnosis of PH; Available data limited to clinically relevant testing; Unable to prespecify timing of longitudinal follow-up; Merging genotyping platforms can be challenging; Specific to deidentified cohorts: subjects lost to follow-up cannot be contacted; cannot perform family studies; imaging/hemodynamic strips may not be available for review |
EMR: electronic medical record; PH: pulmonary hypertension; RHC: right heart catheterization.
There are several challenges and limitations to working with EMR-based cohorts (Table 1). The clinical data (laboratory tests, imaging, etc.) available are limited to what the subject’s physician(s) deemed relevant to their care. Although most data relevant to a particular phenotype are generally present (e.g., right heart catheterization [RHC] data in patients with PH), comparisons may be limited if similar studies are not clinically indicated in controls. Unlike many prospective cohort studies, no predefined longitudinal follow-up can be obtained in an EMR cohort. Longitudinal data can be limited or missing in patients who receive care at more than one hospital system. EMRs can be linked to the Social Security Death Index, but capture is not 100%; for survival analyses, survivors are commonly censored at the date of last medical contact, which underestimates true survival time. Studies involving EMR cohorts are often granted waivers of consent, prohibiting investigators from contacting patients. Patient contact is impossible by design in deidentified EMRs.
Development of PH cohorts
Most large, published PH cohorts are single- or multicenter registries targeting patients with PAH. There is a paucity of published data from EMR-based PH cohorts, although at least two have been developed recently. Maron et al.33 recently described the hemodynamic profile in a very large cohort (n = 21,915) of patients undergoing RHC in the Veterans Affairs hospital system, using data extracted from the Veteran’s Affairs Clinical Assessment Reporting and Tracking program. Hemodynamic data in this cohort are complemented by demographics, comorbidity profiles, and survival status. Examination of this cohort will undoubtedly lead to important observations regarding the prognostic importance and risk factors for the development of PH. Given the demographics of veterans in the United States, this cohort is inherently enriched in men and will contain relatively few cases of PAH.
Assad and colleagues34,35 recently presented data from a cohort of patients extracted from Vanderbilt’s Synthetic Derivative. The remainder of this section describes the creation of the Vanderbilt cohort as an illustration of the advantages, challenges, and limitations of EMR-based cohorts. An inherent requirement for discriminating PH phenotypes is invasive hemodynamic measurements by RHC to determine pulmonary capillary wedge pressure and pulmonary vascular resistance (PVR). Hemodynamic data are semistructured within catheterization reports in the Synthetic Derivative, which required code to be written to extract each individual value (Fig. 2). Catheterization reports in Vanderbilt’s EMRs have undergone four formatting iterations since 1998. Therefore, a unique algorithm was written and tested for each report; moreover, data were extracted for the baseline resting condition as well as for values after provocation with nitric oxide and/or fluid challenge (when performed). After data were extracted from each report, a random sample of 50 charts was reviewed to identify systematic errors. After necessary modifications to the extraction algorithm, this process was repeated until data accuracy was greater than 95%. The next step involved evaluating the distribution of each continuous variable for potential outliers and data entry errors. A previously published data set of manually reviewed RHC tracings (including a variety of PH phenotypes) was used to set physiologically plausible limits for hemodynamic values and vital signs. Values that fell outside these limits, obvious data entry errors, or nonphysiologic values (e.g., oxygen saturation of 110%) were removed and later imputed. The indication for RHC was also extracted from each report.
Figure 2.

Illustration of hemodynamic data extraction from Vanderbilt’s Synthetic Derivative. This represents a typical deidentified right heart catheterization report in Vanderbilt’s Synthetic Derivative demonstrating resting baseline values and repeat values after nitric oxide inhalation. A code is programmed for each individual hemodynamic data point and exported to a text file.
In addition to hemodynamic data, echocardiographic data were extracted. Echocardiographic data in the Synthetic Derivative were previously curated by Wells et al.36 Data were extracted from the echocardiogram closest in time to the RHC, including chamber dimensions, valve Doppler gradients, and semiquantitative valve and ventricular function. Approximately 85% of all subjects in the cohort had an echocardiogram on record, with a median interval from RHC of 2 days (IQR: −19 days to 1 day). Relevant comorbidities (e.g., coronary artery disease, heart failure, chronic obstructive pulmonary disease [COPD], and connective-tissue disease, among many others) were extracted using a combination of multiple instances of an ICD-9 (International Classification of Diseases, Ninth Revision) code and/or validated eMERGE algorithms. All major classes of cardiovascular medications (antihypertensives, diuretics, statins, etc.), all approved pulmonary vasodilators, and all approved diabetes mellitus treatments were recorded for the time periods before and up to 6 months after RHC. Therefore, medications initiated as a result of the RHC data were captured. Common cardiovascular, metabolic, and hematologic laboratory results closest in time (before or after) to RHC were also recorded. Finally, the Synthetic Derivative was linked to the Social Security Death Index to provide information on vital status; survivors were censored at the date of last clinical encounter (e.g., last clinic visit, discharge date, prescription refill).
In total, data from 9,982 RHCs were extracted, including 5,797 first-time, unique cases (Fig. 3). Patients with minimal hemodynamic data and those with “extreme physiology” (severe shock or near death based on vital signs) were excluded (n = 367). Subjects who had an acute myocardial infarction, prior heart or lung transplantation, chronic thromboembolic PH, or complex congenital heart disease (combined n = 795) were also excluded. After these exclusions, 1,898 subjects had a mean PA pressure of <25 mmHg, and 2,737 subjects met hemodynamic criteria for PH. On the basis of consensus guidelines, the majority of subjects with PH (n = 1,766; 65%) were classified as having pulmonary venous hypertension (PVH). Among 971 subjects with precapillary PH, 558 (57%) had elevated PVR without evidence of COPD or interstitial lung disease, thus meeting a nominal definition of PAH. A diagnosis of PAH requires exclusion of other potential causes of PH; therefore, deidentified medical records within the Synthetic Derivative were manually reviewed to verify a clinical diagnosis of PAH. Because PAH is a rare disease, manual verification of the diagnosis in the deidentified EMRs is not prohibitive. Some phenotypes of interest, however, number in the tens of thousands (e.g., diabetes, heart failure), making manual chart review impractical. For these cases, the accuracy of case identification is extrapolated from manual review of a random sample of the identified cases.
Figure 3.
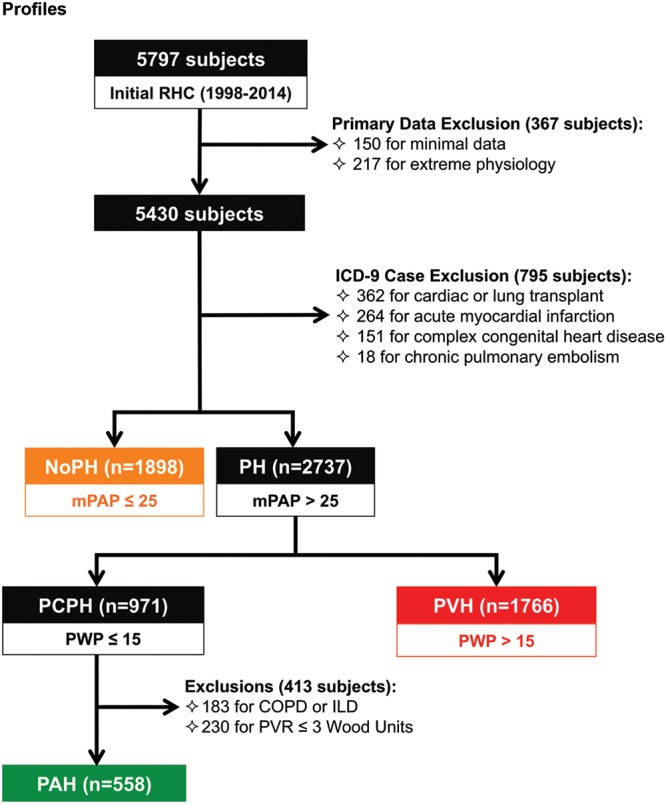
Diagnostic flow diagram of Vanderbilt cohort according to hemodynamic profiles. After extraction of all first-time unique right heart catheterizations (RHCs), a number of exclusions were applied, including subjects with minimal data or extreme physiology, subjects with prior heart or lung transplant, and subjects with chronic thromboembolic pulmonary hypertension (PH) or complex congenital heart disease. Subjects were subsequently stratified by hemodynamic profile into absence of PH (noPH), precapillary PH (PCPH), and pulmonary venous hypertension (PVH). PCPH was further subdivided into pulmonary arterial hypertension (PAH) on the basis of elevated pulmonary vascular resistance (PVR) and absence of parenchymal lung disease. COPD: chronic obstructive pulmonary disease; ICD-9: International Classification of Diseases, Ninth Revision; ILD: interstitial lung disease; mPAP: mean pulmonary artery pressure; PWP: pulmonary wedge pressure;
There are important limitations when considering observations made from EMR-based PH cohorts. First, hemodynamic tracings may not be available for review (as in the Vanderbilt cohort). Therefore, it is possible that subjects are misclassified because of errors in the computer-generated mean. We have previously shown that this error is modest on a population level (approximately 4% in our cohort34), but this does not exclude the likelihood of diagnostic misclassification on a patient level. Next, the sensitivity and specificity of using ICD-9 codes to identify comorbidities are not optimal and vary between medical records. Specificity can be increased (at the expense of sensitivity) by requiring multiple instances of an ICD-9 code and/or by requiring additional circumstantial evidence for a disease (e.g., presence of a disease-specific medication). Finally, because subjects in large cohorts do not always undergo a comprehensive workup, World Health Organization group classification must be inferred from a constellation of circumstantial evidence. For example, group III PH could be inferred from PH with a normal wedge pressure on RHC, multiple ICD-9 codes for COPD, low FEV1 (forced expiratory volume in 1 s) on pulmonary function tests, and use of chronic bronchodilators. Manual review of thousands of charts for dozens of data points is simply not practical; readers should keep these limitations in mind when interpreting results from EMR-based cohorts. Nonetheless, advances in data extraction from complex, long-standing EMRs have the potential to provide epidemiologic and biologic insights that are not possible with retrospective chart review or prospective cohort studies.
Approximately 20% of the final Vanderbilt cohort has DNA banked in the BioVU program, including 684 subjects with PVH and 239 with PAH. Of these, 254 have been genotyped on the Illumina Exome BeadChip platform. Although small compared with traditional genome-wide association studies, these samples provide an opportunity to perform hypothesis-generating genetic analysis in PH subphenotypes in which no biologic data have previously been published, such as combined pre- and postcapillary PH. The existence of longitudinal clinical and hemodynamic data in EMRs will also allow examination of a potential genetic influence on vasodilator response in PAH subjects. For example, Benza et al.37 recently studied the pharmacogenomics of the clinical response to endothelin receptor antagonist therapy and identified single-nucleotide polymorphisms (SNPs) in the endothelin 1 pathway that are associated with clinical efficacy.
An innovative application of a linked DNA biobank and medical record is the development of the phenome-wide association scan (PheWAS) technique by Denny et al.38,39 PheWAS is a method for agnostically examining the relationship between individual SNPs and clinical phenotypes. Denny et al.38 collapsed all ICD-9 codes into a smaller number of nonredundant PheWAS codes that represent all the possible clinical diagnoses in the medical record. The prevalence of an SNP of interest on the Exome BeadChip is compared between cases and controls for each clinical diagnosis (PheWAS code). In nearly 14,000 subjects in BioVU with exome chip data, Denny et al.39 validated previously existing genotype-phenotype relationships (e.g., Alzheimer’s [APOE], diabetes type 1 [HLA-DQB1], and rheumatoid arthritis [HLA-DRB1]).
Of relevance to pulmonary vascular disease, Mosely et al.40 recently used the PheWAS technique to identify clinical diagnoses associated with SNPs in genes previously associated with PAH. A total of 65 genes previously associated with PAH in prior genome-wide association studies were present on the exome chip, representing 311 SNPs in or within 25,000 base pairs of those genes. SNP variation in those 65 genes was significantly related to disorders of iron metabolism (n = 111 cases, P = 4.8 × 10−9). This finding reinforces several recent findings related to abnormal iron metabolism in PAH41-44 and suggests a possible genetic correlation, while also identifying specific genes of potential experimental and therapeutic interest.
EMR-linked biobanks offer great promise for epidemiologic and biologic insights into pulmonary vascular disease. Investigators and the scientific community should be aware of the inherent limitations of EMR-based cohorts when designing studies and interpreting findings from these cohorts. Long-standing EMRs subtending a large clinical population are particularly valuable for the study of rarer pulmonary vascular phenotypes, such as PAH and combined pre- and postcapillary PH. Findings from such resources require replication and functional validation, but EMR-linked biobanks offer opportunities for scientific discovery that are impractical (or impossible) in prospective cohorts.
Conclusions
Molecular and genetic profiling techniques are beginning to add to our understanding of the pathophysiology of pulmonary vascular disease. Computational gene network modeling is a particularly attractive approach to diseases influenced by a complex and interconnected network of molecular processes such as PH. The use of EMR-based cohorts linked to biologic data offers the unique advantage of accruing large numbers of patients across many pulmonary vascular phenotypes. Despite current limitations, our ability to harness and extract information from these and other complex data sources continues to improve. As a result, within the next decade, we expect that such endeavors will result in major advances in our understanding of the pathogenesis of PH and in our clinical care of these historically neglected patients.
Source of Support: American Heart Association Fellow to Faculty grant 13FTF16070002 and Actelion Entelligence Young Investigator Award (EB), National Institutes of Health (NIH) awards (1P0 HL108800-01A1 [EB], 1U01 HL125212 [EB], HL096834 [SC], and HL124021 [SC]), and the Pulmonary Hypertension Association (EB and SC). Data presented here were obtained from Vanderbilt University Medical Center’s BioVU, which is supported by institutional funding, the 1S10RR025141-01 instrumentation award (National Center for Research Resources), and the Vanderbilt Clinical and Translational Science Award grant UL1TR000445 from the National Center for Advancing Translational Sciences/NIH.
Conflict of Interest: None declared.
References
- 1.Bertero T, Lu Y, Annis S, Hale A, Bhat B, Saggar R, Saggar R, et al. Systems-level regulation of microRNA networks by miR-130/301 promotes pulmonary hypertension. J Clin Invest 2014;124(8):3514–3528. [DOI] [PMC free article] [PubMed]
- 2.Hemnes AR, Trammell AW, Archer SL, Rich S, Yu C, Nian H, Penner N, et al. A peripheral blood signature of vasodilator-responsive pulmonary arterial hypertension. Circulation 2015;131(4):401–409. [DOI] [PMC free article] [PubMed]
- 3.National Institutes of Health. Redefining pulmonary hypertension through pulmonary vascular disease phenomics: clinical centers (CC) (U01). http://grants.nih.gov/grants/guide/rfa-files/RFA-HL-14-027.html. Published November 25, 2013. Accessed October 3, 2015.
- 4.ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007;447(7146):799–816. [DOI] [PMC free article] [PubMed]
- 5.Margulies EH, Cooper GM, Asimenos G, Thomas DJ, Dewey CN, Siepel A, Birney E, et al. Analyses of deep mammalian sequence alignments and constraint predictions for 1% of the human genome. Genome Res 2007;17(6):760–774. [DOI] [PMC free article] [PubMed]
- 6.Affymetrix/Cold Spring Harbor Laboratory ENCODE Transcriptome Project. Post-transcriptional processing generates a diversity of 5ʹ-modified long and short RNAs. Nature 2009;457(7232):1028–1032. [DOI] [PMC free article] [PubMed]
- 7.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489(7414):57–74. [DOI] [PMC free article] [PubMed]
- 8.Zhao M, Austin ED, Hemnes AR, Loyd JE, Zhao Z. An evidence-based knowledgebase of pulmonary arterial hypertension to identify genes and pathways relevant to pathogenesis. Mol Biosyst 2014;10(4):732–740. [DOI] [PMC free article] [PubMed]
- 9.Bertero T, Cottrill KA, Lu Y, Haeger CM, Dieffenbach P, Annis S, Hale A, et al. Matrix remodeling promotes pulmonary hypertension through feedback mechanoactivation of the YAP/TAZ-miR-130/301 circuit. Cell Rep 2015;13(5):1016–1032. [DOI] [PMC free article] [PubMed]
- 10.Hemnes AR, Zhao M, West J, Newman JH, Rich S, Archer SL, Robbins IM, et al. Critical genomic networks and vasoreactive variants in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2016. doi:10.1164/rccm.201508-1678OC. [DOI] [PMC free article] [PubMed]
- 11.Rhodes CJ, Im H, Cao A, Hennigs JK, Wang L, Sa S, Chen P-I, et al. RNA sequencing analysis detection of a novel pathway of endothelial dysfunction in pulmonary arterial hypertension. Am J Respir Crit Care Med 2015;192(3):356–366. [DOI] [PMC free article] [PubMed]
- 12.Loscalzo J, Kohane I, Barabási A-L. Human disease classification in the postgenomic era: a complex systems approach to human pathobiology. Mol Syst Biol 2007;3:124. doi:10.1038/msb4100163. [DOI] [PMC free article] [PubMed]
- 13.Bertero T, Cottrill KA, Annis S, Bhat B, Gochuico BR, Osorio JC, Rosas I, et al. A YAP/TAZ-miR-130/301 molecular circuit exerts systems-level control of fibrosis in a network of human diseases and physiologic conditions. Sci Rep 2015;5:18277. doi:10.1038/srep18277. [DOI] [PMC free article] [PubMed]
- 14.Chan SY, Loscalzo J. The emerging paradigm of network medicine in the study of human disease. Circ Res 2012;111(3):359–374. [DOI] [PMC free article] [PubMed]
- 15.Barabási A-L, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet 2011;12(1):56–68. [DOI] [PMC free article] [PubMed]
- 16.Chan SY, Loscalzo J. Pathogenic mechanisms of pulmonary arterial hypertension. J Mol Cell Cardiol 2008;44(1):14–30. [DOI] [PMC free article] [PubMed]
- 17.Wu W, Dave NB, Yu G, Strollo PJ, Kovkarova-Naumovski E, Ryter SW, Reeves SR, et al. Network analysis of temporal effects of intermittent and sustained hypoxia on rat lungs. Physiol Genomics 2008;36(1):24–34. [DOI] [PMC free article] [PubMed]
- 18.Zhao Y, Peng J, Lu C, Hsin M, Mura M, Wu L, Chu L, et al. Metabolomic heterogeneity of pulmonary arterial hypertension. PLoS ONE 2014;9(2):e88727. doi:10.1371/journal.pone.0088727. [DOI] [PMC free article] [PubMed]
- 19.Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan K-K, Cheng C, Mu XJ, et al. Architecture of the human regulatory network derived from ENCODE data. Nature 2012;489(7414):91–100. [DOI] [PMC free article] [PubMed]
- 20.Soubrier F, Chung WK, Machado R, Grünig E, Aldred M, Geraci M, Loyd JE, et al. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol 2013;62(25 suppl.):D13–D21. [DOI] [PubMed]
- 21.Parikh VN, Jin RC, Rabello S, Gulbahce N, White K, Hale A, Cottrill KA, et al. MicroRNA-21 integrates pathogenic signaling to control pulmonary hypertension: results of a network bioinformatics approach. Circulation 2012;125(12):1520–1532. [DOI] [PMC free article] [PubMed]
- 22.Fessel JP, Hamid R, Wittmann BM, Robinson LJ, Blackwell T, Tada Y, Tanabe N, Tatsumi K, Hemnes AR, West JD. Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulm Circ 2012;2(2):201–213. [DOI] [PMC free article] [PubMed]
- 23.Pollett JB, Benza RL, Murali S, Shields KJ, Passineau MJ. Harvest of pulmonary artery endothelial cells from patients undergoing right heart catheterization. J Heart Lung Transplant 2013;32(7):746–749. [DOI] [PubMed]
- 24.Bertero T, Cottrill K, Krauszman A, Lu Y, Annis S, Hale A, Bhat B, et al. The microRNA-130/301 family controls vasoconstriction in pulmonary hypertension. J Biol Chem 2015;290(4):2069–2085. [DOI] [PMC free article] [PubMed]
- 25.Krishnamoorthy P, Gupta D, Chatterjee S, Huston J, Ryan JJ. A review of the role of electronic health record in genomic research. J Cardiovasc Transl Res 2014;7:(8)692–700. [DOI] [PubMed]
- 26.Gottesman O, Kuivaniemi H, Tromp G, Faucett WA, Li R, Manolio TA, Sanderson SC, et al. The Electronic Medical Records and Genomics (eMERGE) network: past, present, and future. Genet Med 2013;15(10):761–771. [DOI] [PMC free article] [PubMed]
- 27.Denny JC, Ritchie MD, Crawford DC, Schildcrout JS, Ramirez AH, Pulley JM, Basford MA, Masys DR, Haines JL, Roden DM. Identification of genomic predictors of atrioventricular conduction: using electronic medical records as a tool for genome science. Circulation 2010;122(20):2016–2021. [DOI] [PMC free article] [PubMed]
- 28.Denny JC, Crawford DC, Ritchie MD, Bielinski SJ, Basford MA, Bradford Y, Chai HS, et al. Variants near FOXE1 are associated with hypothyroidism and other thyroid conditions: using electronic medical records for genome- and phenome-wide studies. Am J Hum Genet 2011;89(4):529–542. [DOI] [PMC free article] [PubMed]
- 29.Crosslin DR, McDavid A, Weston N, Nelson SC, Zheng X, Hart E, de Andrade M, et al. Genetic variants associated with the white blood cell count in 13,923 subjects in the eMERGE network. Hum Genet 2012;131(4):639–652. [DOI] [PMC free article] [PubMed]
- 30.Bowton EA, Collier SP, Wang X, Sutcliffe CB, Van Driest SL, Couch LJ, Herrera M, et al. Phenotype-driven plasma biobanking strategies and methods. J Pers Med 2015;5(2):140–152. [DOI] [PMC free article] [PubMed]
- 31.Roden DM, Pulley JM, Basford MA, Bernard GR, Clayton EW, Balser JR, Masys DR. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther 2008;84(3):362–369. [DOI] [PMC free article] [PubMed]
- 32.Bowton E, Field JR, Wang S, Schildcrout JS, Van Driest SL, Delaney JT, Cowan J, et al. Biobanks and electronic medical records: enabling cost-effective research. Sci Transl Med 2014;6:234cm3. doi:10.1126/scitranslmed.3008604. [DOI] [PMC free article] [PubMed]
- 33.Maron BA, Hess E, Maddox TM, Opotowsky AR, Tedford RJ, Lahm T, Joynt KE, et al. Association of borderline pulmonary hypertension with mortality and hospitalization in a large patient cohort: insights from the Veterans Affairs Clinical Assessment, Reporting, and Tracking program. Circulation 2016;133(13):1240–1248. [DOI] [PMC free article] [PubMed]
- 34.Assad TR, Hemnes AR, Doss LN, Wells QS, Farber-Eger E, Brittain EL. Determining the clinical and hemodynamic profile of combined pulmonary hypertension in a large de-identified database. Am J Respir Crit Care Med 2015;191(MeetingAbstracts):A5522.
- 35.Assad TR, Brittain EL, Halliday SJ, Wells QS, Farber-Eger E, Robbins IM, Pugh ME, Newman JH, Hemnes AR. Hemodynamic evidence of vascular remodeling in combined pulmonary hypertension. Am J Respir Crit Care Med 2015;191(MeetingAbstracts):A3827.
- 36.Wells QS, Farber-Eger E, Crawford DC. Extraction of echocardiographic data from the electronic medical record is a rapid and efficient method for study of cardiac structure and function. J Clin Bioinf 2014;4:12. doi:10.1186/2043-9113-4-12. [DOI] [PMC free article] [PubMed]
- 37.Benza RL, Gomberg-Maitland M, Demarco T, Frost AE, Torbicki A, Langleben D, Pulido T, et al. Endothelin-1 pathway polymorphisms affect outcome in pulmonary arterial hypertension. Am J Respir Crit Care Med 2015;192(11):1345–1354. [DOI] [PMC free article] [PubMed]
- 38.Denny JC, Ritchie MD, Basford MA, Pulley JM, Bastarache L, Brown-Gentry K, Wang D, Masys DR, Roden DM, Crawford DC. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 2010;26(9):1205–1210. [DOI] [PMC free article] [PubMed]
- 39.Denny JC, Bastarache L, Ritchie MD, Carroll RJ, Zink R, Mosley JD, Field JR, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol 2013;31(12):1102–1110. [DOI] [PMC free article] [PubMed]
- 40.Mosley JD, Brittain EL, Loyd JE, Denny JC, Austin ED, Larkin EK. Letter by Mosley regarding article, “Iron Homeostasis and Pulmonary Hypertension: Iron Deficiency Leads to Pulmonary Vascular Remodeling in the Rat.” Circ Res 2015;117(6):e56–e57. [DOI] [PMC free article] [PubMed]
- 41.Rhodes CJ, Howard LS, Busbridge M, Ashby D, Kondili E, Gibbs JSR, Wharton J, Wilkins MR. Iron deficiency and raised hepcidin in idiopathic pulmonary arterial hypertension: clinical prevalence, outcomes, and mechanistic insights. J Am Coll Cardiol 2011;58(3):300–309. [DOI] [PubMed]
- 42.Rhodes CJ, Wharton J, Howard L, Gibbs JSR, Vonk-Noordegraaf A, Wilkins MR. Iron deficiency in pulmonary arterial hypertension: a potential therapeutic target. Eur Respir J 2011;38(6):1453–1460. [DOI] [PubMed]
- 43.Ruiter G, Lankhorst S, Boonstra A, Postmus PE, Zweegman S, Westerhof N, van der Laarse WJ, Vonk-Noordegraaf A. Iron deficiency is common in idiopathic pulmonary arterial hypertension. Eur Respir J 2011;37(6):1386–1391. [DOI] [PubMed]
- 44.Cotroneo E, Ashek A, Wang L, Wharton J, Dubois O, Bozorgi S, Busbridge M, Alavian KN, Wilkins MR, Zhao L. Iron homeostasis and pulmonary hypertension: iron deficiency leads to pulmonary vascular remodeling in the rat. Circ Res 2015;116(10):1680–1690. [DOI] [PubMed]