

Abstract Abstract
Pulmonary arterial hypertension (PAH) is a multifactorial disease characterized by interplay of many cellular, molecular, and genetic events that lead to excessive proliferation of pulmonary cells, including smooth muscle and endothelial cells; inflammation; and extracellular matrix remodeling. Abnormal vascular changes and structural remodeling associated with PAH culminate in vasoconstriction and obstruction of pulmonary arteries, contributing to increased pulmonary vascular resistance, pulmonary hypertension, and right ventricular failure. The complex molecular mechanisms involved in the pathobiology of PAH are the limiting factors in the development of potential therapeutic interventions for PAH. Over the years, our group and others have demonstrated the critical implication of lipids in the pathogenesis of PAH. This review specifically focuses on the current understanding of the role of oxidized lipids, lipid metabolism, peroxidation, and oxidative stress in the progression of PAH. This review also discusses the relevance of apolipoprotein A-I mimetic peptides and microRNA-193, which are known to regulate the levels of oxidized lipids, as potential therapeutics in PAH.
Keywords: metabolism, oxidative stress, pulmonary hypertension
Pulmonary arterial hypertension (PAH) is a rare but fatal disease, characterized by persistent elevation in pulmonary artery pressures.1,2 PAH is also a serious complication of several connective-tissue diseases, including systemic lupus erythematosus, progressive systemic sclerosis, and rheumatoid arthritis.3,4 PAH could also be associated with pulmonary thromboembolism, portal hypertension, HIV infection, hepatitis C infection, intravenous drug abuse, and various other pulmonary disorders.5-9 Many cellular and genetic events are involved in the pathogenesis of PAH. PAH is associated with marked vascular injury caused by endothelial dysfunction of small pulmonary arteries, promoting vasoconstriction. In addition, structural abnormalities, excessive hypertrophy of smooth muscle cells lining the arterioles, endothelial cell proliferation resulting in plexiform lesions, extracellular matrix remodeling leading to fibrosis, and activation of inflammatory cells vastly contribute to the pathogenesis of the disease. All these vascular changes increase the afterload on the right ventricle, leading to right ventricular (RV) hypertrophy, decompensation, and failure.1 There are many complex processes and events that culminate in the development and progression of PAH. This has resulted in the advancement of many therapies targeting the endothelin 1, phosphodiesterase, nitric oxide, and prostacyclin pathways to slow the progression of the disease.10-14 This review specifically focuses on the role of oxidative stress, lipid oxidation, and peroxidation as contributing factors in PAH. Finally, this review discusses the emerging potential of high-density lipoprotein (HDL) mimetic peptides, which bind to oxidized lipids with high affinity, as well as microRNA-193, which targets oxidized lipids, as novel therapeutics.
Lipid and lipoprotein metabolism in PAH
PAH is associated with an increase in oxidative stress participating in the oxidation of lipids. Oxidized lipids participate in many pathophysiological hallmarks of PAH, including smooth muscle cell (SMC) proliferation, endothelial cell (EC) apoptosis, and inflammation (Table 1).53-55
Table 1.
Role of oxidized lipids and lipid metabolism in pulmonary hypertension
| PAH hallmarks | Effect | References |
|---|---|---|
| HETEs and HODEs | ||
| Vascular remodeling | Activation of PDGF/15-LOX/15-HETE axis | 15, 16 |
| PASMC proliferation | 12-LOX/12-HETE and 15-LOX/15-HETE axis activates ERK1/2 pathway | 17, 18 |
| Resistance to apoptosis | 15-HETE activates ERK1/2, PI3K/Akt, and ROCK/iNOS pathways | 19–22 |
| Vasoconstriction | 15-HETE inhibits expression of Kv1.5, Kv2.1, and Kv3.4 and activates Rho/ROCK signaling | 23–27 |
| Angiogenesis | 15-HETE activates ROCK pathways promoting angiogenesis | 16 |
| Fibrosis | 15-HETE activates TGF-β/Smad2/3 axis | 28 |
| Inflammation | Increased LDL/HDL inflammatory index | 29 |
| Leukotrienes | ||
| PASMC proliferation | LTB4 activates BLT1 receptor | 30 |
| Inflammation | Macrophages LTB activates LOX enzyme | 31, 32 |
| Epoxyeicosatrienoic acid (EET) | 8,9-, 11,12-, 14,15-EET promote JNK1/2 activation in PAECs | 33 |
| Resistance to apoptosis | ||
| Endothelial dysfunction | ||
| Plexiform lesions | ||
| Lipids metabolism | ||
| Vascular remodeling | Impaired PPARγ signaling | 34–38 |
| Mitochondrial dysfunction | Inhibition of fatty acid oxidation promotes mitochondria hyperpolarization | 39–46 |
| Vasoconstriction | 16, 47–51 | |
| Endothelial dysfunction | Oxidized LDL activates NF-κB pathways | 52 |
ERK: extracellular signal–regulated kinase; HDL: high-density lipoprotein; HETE: hydroxyeicosatetraenoic acid; HODE: hydroxyoctadecadienoic acid; iNOS: nitric oxide synthase; JNK: c-Jun N-terminal kinase; LDL: low-density lipoprotein; LOX: lipoxygenase; LTB: leukotriene B; PAECs: pulmonary artery endothelial cells; PAH: pulmonary arterial hypertension; PASMC: pulmonary artery smooth muscle cell; PDGF: platelet-derived growth factor; PI3K: phosphoinositide 3-kinase; PPARγ: peroxisome proliferator–activated receptor γ; ROCK: Rho-associated protein kinase; TGF: transforming growth factor.
Role of oxidized fatty acids produced from arachidonic acid via the lipoxygenase pathway in PAH
In this section, we focus on oxidized lipids in PAH generated from linoleic acid (LA) and arachidonic acid (AA). LA can be oxidized by 5-lipoxygenase (5-LOX) and 15-LOX to form 9-hydroxyoctadecadienoic acid (9-HODE) and 13-HODE, respectively (Fig. 1A). Involvement of these oxidized products as contributing factors in oxidative stress in PAH has recently been demonstrated.16,28,29,56,57 LA is also the precursor of AA, which by enzymatic oxidation gives rise to different oxidized lipids, including 5-hydroxyeicosatetraenoic acid (5-HETE), 12-HETE, and 15-HETE (Fig. 1A). These three oxidized metabolites are found to be elevated in the lung tissue samples obtained from patients with primary pulmonary hypertension (PH).58 The 12-HETE levels are increased in the lung SMCs cultured from hypoxia-treated rats, and exogenous 12-HETE treatment stimulated proliferation of SMCs.17 Similarly, 15-HETE plays an important role in hypoxic PAH.59 Increased activity of 15-LOX in pulmonary arteries upon exposure to hypoxia catalyzes and enhances 15-HETE production.57 Vascular remodeling in hypoxic PAH is in part mediated by a positive feedback regulatory loop between 15-HETE and hypoxia-inducible factor 1α (HIF-1α), a critical oxygen-sensing transcription factor in PAH.60 Increased levels of 15-HETE stimulate proliferation of pulmonary artery SMCs (PASMCs);18 pulmonary arterial vasoconstriction via K+ channels and the protein kinase C (PKC) signal transduction pathway;23-26 the Rho/Rho-associated protein kinase (Rho/ROCK) signaling pathway;27 inhibition of apoptosis in PASMCs mediated via several signaling pathways, such as ERK1/2 (extracellular signal–regulated kinases),19 PI3K (phosphatidylinositol 3-kinase)/Akt,20 the ROCK pathway,21 and the nitric oxide synthase (iNOS) pathway;22 and pulmonary vascular remodeling mediated via platelet-derived growth factor.15 In addition, 15-LOX/15-HETE induces the p38 MAPK (mitogen-activated protein kinase)–dependent transforming growth factor (TGF) β1/Smad2/3 intracellular signaling pathways to mediate vascular fibrosis in the adventitia of the pulmonary arterial wall, resulting in pulmonary artery remodeling.28 Also, 15-HETE has been shown to mediate vascular medial hypertrophy and EC migration and angiogenesis contributing to hypoxic PH.16 A mutual positive regulatory mechanism exists between telomerase reverse transcriptase and the 15-LOX/15-HETE pathway that could mediate migration, proliferation, and cell cycle distribution of PASMCs in hypoxia-induced pulmonary vascular remodeling.61 Inhibition of 5-LOX by diethlycarbamazine, an enzyme responsible for 5-HETE and leukotriene synthesis (Fig 1B), has been demonstrated to improve PAH in the Sugen/hypoxia rat model.62 Indeed, inhibition of 5-LOX improves RV function by decreasing RV systolic pressure (RVSP) and hypertrophy. Al-Husseini et al.62 demonstrated that this improvement is mediated by a decrease in inflammation and pulmonary vascular wall thickness. Furthermore, we recently demonstrated that PH is associated with elevated plasma levels of 5-HETE, 12-HETE, 15-HETE, 9-HODE, and 13-HODE in the monocrotaline (MCT)-induced PH model in rodents.56 We also reported significantly higher levels of plasma HETEs and HODEs in PAH patients (idiopathic and associated PAH secondary to connective-tissue disease).29 Conversely, we showed that increasing oxidized lipids in vivo by feeding mice with 15-HETE leads to PH, establishing a strong connection between the levels of oxidized lipids and the pathophysiology of PH.56 Collectively, these studies reinforce a major role of oxidized lipids in the development of PH (Fig. 2).
Figure 1.
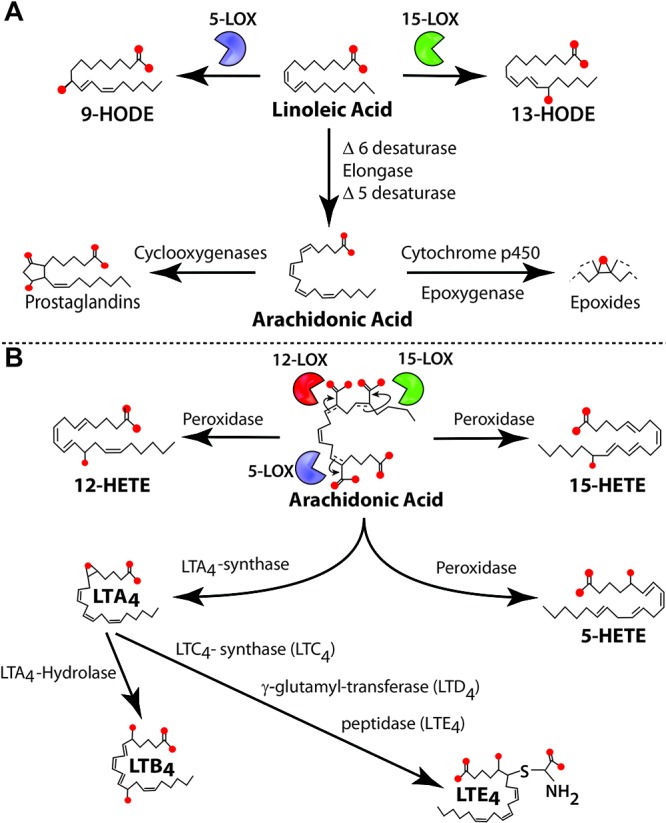
Pathways involved in linoleic and arachidonic acid metabolism. A, Linoleic acid is metabolized by 5- and 15-lipoxygenase (LOX) to form 9-hydroxyoctadecadienoic acid (9-HODE) and 13-HODE, respectively. Linoleic acid can also be metabolized to arachidonic acid. In turn, arachidonic acid is used to form prostaglandins (by cyclooxygenases) and epoxides (by the cytochrome p450 epoxygenase pathway). B, Arachidonic acid can also be metabolized to 5-hydroxyeicosatetraenoic acid (5-HETE), 12-HETE, and 15-HETE by LOXs and to leukotrienes (LTs) such as LTA4 (by LTA4-synthase), LTB4 (by LTA4-hydrolase), LTC4 (by LTC4-synthase), LTD4 (by γ-glutamyl-transferase), and LTE4 (by peptidase).
Figure 2.

Role of oxidized lipids, oxidative stress, and lipid metabolism in promoting pulmonary arterial hypertension (PAH). A, Many altered pathways and abnormalities are involved in the progression and development of PAH. PASMC: pulmonary artery smooth muscle cell. B, Oxidized lipids (hydroxyeicosatetraenoic acids [HETE]s and hydroxyoctadecadienoic acids [HODEs]), leukotrienes (LTs), epoxyeicosatrienoic acids (EETs), and lipid metabolism contribute only to a selective subset of pathways (highlighted); pathways that are not involved in the pathogenesis of PAH are shaded (See Table 1 for references).
Role of other metabolites of AA in PAH
Many by-products of AA play an important role as mediators in PAH. For instance, epoxyeicosatrienoic acid (EET), which is derived from AA by cytochrome p450 epoxygenase (Fig. 1A), plays an essential role in vasoconstriction and the modulation of proliferative and angiogenic properties in pulmonary artery ECs (PAECs) in PH and other diseases.63,64 It has been shown that the EET and JNK/c-Jun pathways are involved in pulmonary vascular remodeling caused by proliferation of ECs, inhibition of apoptosis, and stimulation of angiogenesis, culminating in pulmonary artery endothelial plexiform lesions (Fig. 2B).33 Kandhi et al.65 demonstrated that jugular administration of EET led to an increase in RVSP in a dose-dependent manner. Thus, the effect of EET on RVSP seems synergic with hypoxia. Wang et al.66 have clearly demonstrated that hypoxic pulmonary vasoconstriction response, which is impaired in PH, is modulated by endothelium calcium signaling via activation of EETs, further supporting the essential role of EETs in PH.
Leukotrienes (LTs) are another class of lipid mediators derived from the 5-LOX pathway of AA metabolism (Fig. 1B).67,68 They trigger immune response by recruitment and activation of leukocytes and play an essential role as mediators in pulmonary inflammation.31,32 Several studies have shown that leukotriene B4 (LTB4) is involved in PAH pathogenesis.69-71 Rodent models of PH, including MCT- and SU5416 (a VEGFR2 [vascular endothelial growth factor receptor 2] inhibitor)-treated rat models, have shown elevated levels of LTB4.30,72 LTB4 induces apoptosis of PAECs, proliferation of PASMCs, and fibroblast activation, three major pathologic events leading to PAH.30,73 We have recently demonstrated that plasma levels of LTB4, but not those of LTC4 and LTE4, are increased in the rat model of MCT-induced PH (Fig. 2B).56 Taken together, these data strengthen a critical role of oxidized lipids in the pathophysiological mechanism of PH (Table 1).
Plasma HDL and PAH
HDL cholesterol (HDL-C) is protective in coronary artery disease because of its antioxidant and anti-inflammatory properties.74,75 Apolipoprotein A-I (ApoA-I), the major protein component of HDL-C, is present at lower levels in PAH, which correlates with increased endothelial dysfunction.76 Metabolic syndrome and insulin resistance are associated with low circulating HDL-C levels and may predispose to the development of pulmonary vascular disease.77,78 Indeed, insulin resistance and RV hypertrophy with pulmonary vascular remodeling are observed in an apolipoprotein E–deficient mouse model.79 In addition, depressed circulating levels of HDL-C are associated with worse clinical outcomes in PAH patients and are independent of other cardiovascular risk factors.80 This observation is further supported by another independent study showing that circulating HDL cholesterol levels are depressed in a cohort of patients with idiopathic PAH (IPAH) and are associated with worse clinical outcomes.81 On the whole, these data suggest a potential role of circulating HDL in metabolic syndrome and PH and are supported by the prevalence of subclinical PH in patients with metabolic syndrome.82
Role of lysophosphatidic acid
Studies have investigated the effects of lysophosphatidic acid (LPA) signaling and metabolism on vascular SMCs and ECs, indicating that LPA may also have implications for the remodeling of pulmonary vasculature.83,84 LPA is a bioactive lipid molecule produced by the plasma lysophospholipase D enzyme autotoxin.85,86 LPA has been shown to stimulate migration and proliferation of SMCs and to alter EC function; consequently, it plays a critical role in vascular development.87-89 Mouse models with loss-of-function mutations in genes required for LPA production and signaling have been used to investigate the pathophysiological role of LPA metabolism and signaling in diverse settings, including pulmonary inflammation and hypoxia-induced vascular remodeling.90,91
PAH is associated with oxidative stress and lipid peroxidation
PAH is associated with increased oxidative stress. This leads to tissue damage by oxidation of many important biological molecules, including DNA damage and lipid peroxidation.58 Increased reactive oxygen species (ROS) production is involved in the pathogenesis of PH in various animal models. Chronic hypoxia–induced PH in mice is associated with increased intrapulmonary superoxide levels and other pathophysiological changes, which are abolished by the antioxidant xanthine oxidase inhibitor allopurinol.92,93 Elevated RV superoxide levels are also observed in the MCT rat model of PH. Antioxidant therapies with intratracheal administration of antioxidant superoxide dismutase or resveratrol suppressed the progression of PH.94-97 Tissue hypoxia and an increase of inflammatory cytokines in the lungs of animal models of PH lead to elevated levels of ROSs.58,97 PAH is associated with oxidative stress arising from the accumulation of ROSs, including superoxide and peroxide. Reaction of these highly reactive molecules with functional groups in membrane lipids and proteins can produce harmful oxidative-breakdown products.98 One such class of metabolites is the chemically stable bioactive lipid peroxidation product of AA known as “isoprostanes.” Levels of isoprostanes are elevated in many pulmonary vascular diseases, including PAH.99-104 Isoprostanes not only serve as biomarkers of the disease but also act as signal transduction molecules exerting multiple biological effects through prostanoid receptors and other signaling pathways.47,105-107 They can exert their effects on pulmonary vasculature in many ways, including pulmonary vasoconstriction,48,108,109 induction of pulmonary endothelium to release endothelin 1, vasoconstriction in general,49,110 and nonspecific effects on smooth muscle, such as hyperresponsiveness and hypertrophy,50,51 thereby serving as important mediators in many lung pathologies including PAH.
Lipid peroxidation products of AA, including isoprostanes as well as other end products, such as malondialdehyde, are increased in patients with PAH.103,111,112 Lungs of transgenic mice with bone morphogenetic protein receptor II (BMPR2) mutation showed an increase in isoprostanes, indicative of a rise in oxidative stress that was of mitochondrial origin and specific to vasculature.113 Chronic hypoxic exposure of rats results in the production of ROSs, including phosphatidylcholine hydroperoxide, a primary peroxidation product of phosphatidylcholine, and serves as a contributing factor for pulmonary vascular thickening and development of PH.93 A reduction in the levels of antioxidants, including β-carotene and α-tocopherol, is observed in patients with IPAH compared to control subjects.114 Studies have evaluated the role of NADPH oxidase in the development of PH. In a lamb model of PH of the newborn, it has been shown that the NADPH oxidase enzyme complex may contribute to proliferation of SMCs by producing increasing superoxide levels.115,116 Moreover, the proliferative effect of endothelin 1 on fetal PASMCs is a consequence of increased generation of ROSs.117 Regulation of the growth of PASMCs by the transcription factor GATA4 is inhibited by antioxidant serotonin via suppression of NADPH oxidase or monoamine oxidase A, further reinforcing the critical role of ROSs in altering signaling pathways involved in PH.118,119 Several other studies have suggested that antioxidant therapy can suppress the progression of PH. Many compounds with antioxidant properties, including the superoxide dismutase mimetic tempol and resveratrol, are effective in preventing the development of PH in various animal models.97,120 In addition, cell culture studies, transgenic mouse models, and human samples confirm that the heritable form of PAH caused by mutations in the BMPR2 pathway is also associated with increased oxidative stress that is very vascular specific and most likely of mitochondrial origin. Lectin-like oxidized low-density lipoprotein (oxLDLs) is involved in endothelial dysfunction and injury upon stimulation by many factors, including inflammation and shear stress;52 oxLDL plays an important role in cardiovascular diseases such as myocardial infarction and atherosclerosis.121,122 Overexpression of lectin-like oxidized low-density lipoprotein receptor 1 (LOX-1), an endothelial receptor of oxLDL in the lungs of transgenic mice, promotes oxidative stress by ROS generation and induces PH in chronic hypoxia.123 Thus, oxidative stress and lipid peroxidation could make a major contribution to the pathogenesis of PAH (Fig. 2; Table 1).
Oxidative stress and impaired mitochondrial function in the pathogenesis of PAH and associated RV failure
There is a growing interest in the potential involvement of abnormal cellular metabolism and impaired mitochondrial function in the pathogenesis of PAH and associated RV failure. These changes may participate in the factors involved in the resistance to apoptosis and increased vascular cell proliferation, which are characteristics of PAH.39 RV failure is associated with many metabolic transformations at the cellular and molecular levels affecting glucose and fatty acid metabolism. Glycolytic shift is observed in the right ventricles (RVs) of both humans with PAH40 and rat models of PH induced by MCT or RV pressure overload by pulmonary artery banding.41 Previous studies have suggested that limitation of the energy supply due to a mitochondrial metabolic switch from the energy-rich oxidative metabolism of glucose to glycolysis, arising from pathological pyruvate dehydrogenase kinase (PDK) activation, leads to RV failure.41,42 Involvement of dysregulated fat metabolism in the failing RV during the progression of PAH has also been highlighted. In a transgenic rodent model of BMPR2 mutation, dysfunctional BMPR2 signaling in the RV results in triglyceride and ceramide deposition and potential fat toxicity.43,44 Indeed, mutations in the gene for BMPR2 were identified to cause familial primary PH.113,124,125 Fessel et al.126 have provided an extensive analysis of widespread metabolomic and transcriptomic changes affected by BMPR2 mutations in the pathogenesis of PH. The role of fatty acid oxidation in PAH is further emphasized in a study showing that mice lacking the gene for the metabolic enzyme malonyl-coenzyme A decarboxylase (MCD), an enzyme involved in fatty acid oxidation, do not develop PAH during chronic hypoxia.45 Studies have shown that serum levels of secreted glycoproteins involved in lipid metabolism and angiogenesis, such as angiopoietin-like protein 3 (ANGPTL3), are positively correlated with RVSPs and could contribute to PAH in systemic sclerosis.46 In summary, abnormal cellular metabolism and impaired mitochondrial function could play an important role in the pathogenesis of PAH (Fig. 2).
Ventricular dysfunction associated with pulmonary artery atherosclerosis
It has been observed that patients with obstructive sleep apnea, which is characterized by episodes of hypoxia and hypercapnia during sleep, are susceptible to atherosclerotic disease in the pulmonary vasculature. This was demonstrated in LDL receptor–deficient mice by exposing them to intermittent hypoxia/hypercapnia for periods of 8 or 16 weeks. Intermittent hypoxia/hypercapnia resulted in marked increase in atherosclerotic lesions in the pulmonary artery, accompanied by RV and left ventricular dysfunction.127 In addition, pulmonary artery atheroscelerosis is accelerated in patients with hypertensive pulmonary disease and shows significant correlation with RV dilation and hypertrophy.128 It has been reported that pulmonary artery atherosclerosis is also characterized by increased lipid peroxidation in pulmonary artery lesions.129
Role of oxidized lipids in PAH inflammation
Recruitment of inflammatory cells and an increase in inflammatory mediators are hallmarks of PAH.130 The pathogenesis of PH includes an inflammatory response, resulting in a higher circulating levels of monocyte chemoattractant protein 1 (MCP-1), interleukin (IL) 6, IL-8, and tumor necrosis factor α (TNF-α) in patients with IPAH and chronic thromboembolic PH than in healthy controls.131-134 Oxidized lipids are known to promote inflammatory processes in many diseases such as atherosclerosis.135 For example, oxidized LDL has been demonstrated to promote MCP-1 expression (Fig. 3).136 Growing evidence demonstrates the implication of oxidized lipids derived from LA and AA in the inflammatory mechanism in PAH. Indeed, 5-HETE promotes neutrophil recruitment.137 Interestingly, 9-HODE and 13-HODE are capable of exerting both pro- and anti-inflammatory reactions by regulation of monocyte/macrophage activation.138 Enzymatic synthesis of 9-HODE and 13-HODE leads to the production of 9-(S)-HODE and 13-(S)-HODE. On the one hand, these two enantiomers are known to have anti-inflammatory properties by virtue of binding and activating peroxisome proliferator–activated receptor γ (PPARγ), leading to downregulation of inflammatory mediators such as IL-12, interferon α, and TNF-α and thus inhibiting inflammatory cell activation.138-140 On the other hand, nonenzymatic production of HODEs in the case of oxidative stress, as observed in PAH, leads to the synthesis of 9-/13-(S)-HODEs and 9-/13-(R)-HODEs in the same proportion.141 Recently, it was demonstrated that the production of 9-/13-HODEs by nonenzymatic pathways leads to monocyte/macrophage activation and inflammation in atherosclerosis disease.141 In addition to an enhancement of inflammation arising from oxidative stress in PAH, downregulation of PPARγ in PAH impairs the anti-inflammatory effect of oxidized lipids. Nonetheless, more studies are required to fully understand the precise role of HODEs in PAH pathophysiology with respect to inflammation. Furthermore, oxidative stress can also promote inflammation by leading to the synthesis of compounds, such as isoprostanes, known to promote inflammatory cell recruitment.142 Finally, LTs produced from AA also exert a proinflammatory effect. Among these, LTB4 has been demonstrated to be a chemoattractive compound for neutrophil and to promote expression of the ICAM-1 protein by ECs, leading to leukocyte recruitment.69 Moreover, LTC4 and LTE4 could also promote inflammation by activating the expression of TGF-β1.143-145 Taken together, these data give evidence of major involvement of oxidized lipids in inflammation observed in PAH and make them attractive pharmacological targets to counteract PAH pathology.
Figure 3.

Hypothetical scheme of production and effects of oxidized fatty acids. In the oxidized fatty acid pathway (purple arrows) linoleic acid (LA) and arachidonic acid (AA) in the cytoplasm (shown with an asterisk to indicate the starting point) are enzymatically cleaved through lipoxygenases (LOXs) into hydroperoxyeicosatetraenoid acids (HPETEs) and hydroperoxyoctadecadienoic acids (HPODEs) that are further oxidized into hydroxyeicosatetraenoic acids (HETEs) and hydroxyoctadecadienoic acids (HODEs). The apolipoprotein A-I mimetic peptide 4F inhibits production of HETEs and HODEs. HETEs and HODEs in the blood bind to G protein–coupled receptors (GPCRs) and induce intracellular pathways, leading to the activation of transcription repressor retinoid X receptor alpha (RXR-α), which inhibits microRNA-193 (miR193) expression. Inhibition of miR193 increases HETE and HODE production by targeting the LOX pathway; miR193 is also secreted in the blood. In the inflammation pathway (red arrows), HPETEs and HPODEs, the reactive intermediates of HETEs and HODEs, oxidize LDL, which initiates the inflammatory response, including initiating the transcription of monocyte chemoattractant protein 1 (MCP-1), monocyte migration, and aggravation of pulmonary arterial hypertension (PAH) symptoms. EC: endothelial cells; PASMC: pulmonary artery smooth muscle cells.
Novel therapeutic strategies in PAH
Role of HDL (ApoA-I) mimetic peptides
HDL is a major lipid carrier in the bloodstream and plays a critical role in vascular disease. It is known that HDL protects against atherosclerosis through several mechanisms, including the ability to extract cholesterol and phospholipids from peripheral cells and transfer them to the liver for excretion. Moreover, HDL also protects against lipid oxidation and inflammation. However, under certain pathological conditions, as in PAH, these antioxidant and anti-inflammatory properties of HDL decrease, accompanied by a drastic increase in the levels of oxidized lipids. Therefore, HDL can act as both an anti- and a proinflammatory molecule, depending on the context and environment. Indeed, we have determined the “inflammatory indices” of HDL and LDL in IPAH and associated PAH (APAH) patients.29 We found that LDL inflammatory indices were significantly higher in IPAH and APAH patients than in controls. Furthermore, HDL was proinflammatory in both IPAH and APAH.29
The major component of HDL in plasma is ApoA-I, which possesses anti-atherosclerotic, anti-inflammatory, and antioxidant properties.146 The mechanistic relationship between ApoA-I and pulmonary function was highlighted by genetic deletion of ApoA-I in mice. ApoA-I-null (ApoA-I−/−) mice show an increase in proinflammatory HDL, indicative of high oxidative stress and increased airway hyperresponsiveness as well as impaired pulmonary vascular function.147 PH in patients with sickle cell disease is associated with altered expression of ApoA-I, contributing to sickle cell disease–associated vasculopathy.148 ApoA-I concentrations were decreased in the lungs of idiopathic pulmonary fibrosis patients and in an experimental bleomycin-induced fibrosis model.149 The local treatment with ApoA-I has been shown to be very effective against the development of experimental lung injury and fibrosis.149 ApoA-I appears to be a promising therapeutic molecule, considering its therapeutic potential in reducing inflammation and fibrosis in the animal model of bleomycin-induced pulmonary fibrosis.150
Several HDL mimetic peptides, which mimic the lipid-binding properties of ApoA-I, have been engineered to mimic the anti-inflammatory and antioxidant properties of HDL. Among these, the 4F peptide (18 amino acids with 4 phenylalanines, at positions 3, 6, 14, and 18) has received the most attention over the past decade. The 4F peptide is highly effective in improving vascular dysfunction implicated in the pathogenesis of many diseases and disorders, including atherosclerosis, diabetes, hypercholesterolemia, and sickle cell disease.151-153 In the context of lung disease, 4F decreases airway hyperresponsiveness, inflammation, and oxidative stress in a murine model of asthma.154 We have recently demonstrated that the levels of oxidized lipids are elevated in the plasma of PH rats,56 as well as in PAH patients,29 and may contribute to the inflammatory response and vascular changes involved in the progression of PH. Therapy with 4F has been shown to be very effective in restoring the levels of oxidized lipids and rescue of preexisting PH in animal models.56 We have examined the effect of 4F on HDL and LDL inflammatory indices in an arterial wall model and a monocyte migration assay in IPAH and APAH patients.29 HDL, as well as LDL, inflammatory indices were decreased significantly after ex vivo treatment with 4F to levels comparable to healthy controls.29
Role of the microRNA 193–oxidized lipids axis in PH
MicroRNAs (miRNAs) are small, regulatory, noncoding, single-stranded RNA molecules involved in the regulation of several physiological pathways, including apoptosis, cell migration, vascular development, and cell proliferation, via modulation of target genes.155,156 Altered expression of miRNAs could result in a dysregulated expression of their target genes, consequently causing or exacerbating several pathological conditions, including cardiovascular diseases and PH.157,158 Several miRNAs, including miR-21, miR-204, and miR-328, have been reported to regulate pathogenic signaling in the development and progression of PH.159-161 We have recently demonstrated that PH is associated with increased plasma levels of oxidized lipids in rodents as well as in IPAH patients.29 Therapy with 4F was very effective in restoring their levels and led to rescue of preexisting PH in models of both hypoxia and MCT. Mechanistically, we identified the miRNA miR-193-3p (miR193) as a downstream effector molecule whose expression was significantly downregulated in the lungs in two experimental animal models of PH. The 4F therapy fully restored expression of miR193 to its level in the control group.56 Overexpression of miR193 in the lungs rescued preexisting PH induced by either MCT or hypoxia in animal models.56 We also found that overexpression of miR193 in SMCs isolated from small pulmonary arteries of PAH patients (confirmed by right catheterization) reduces proliferation, whereas knockdown of miR193 in SMCs isolated from small pulmonary arteries of control subjects with no PAH (discarded nondonor lungs) increases proliferation.56 Our data highlight the therapeutic role of miR193 in reversing pulmonary vascular remodeling.
We further showed that oxidized lipids regulate expression of miR193 through the transcriptional factor retinoid X receptor alpha (RXR-α).56 Oxidized lipids induce the expression of RXR-α in PASMCs. This induction results in an increased binding of RXR-α on the promoter of miR193, thus causing its subsequent downregulation and a net increase in the expression of lipoxygenases, the enzymes responsible for the production of oxidized lipids (Fig. 3). However, 4F can decrease the overall content and binding of RXR-α to an miR193 promoter by sequestering oxidized lipids, ultimately leading to miR193 induction.56 This study explored an essential aspect of oxidized lipid–induced pathology of PH, wherein miRNA (i.e., miR193) modulation is involved in the molecular and functional outcome. Future studies on the role of miRNAs in the oxidized lipid–mediated induction of PH with insight into novel targets are warranted to develop a multiple-miRNA therapeutic approach to tackle the disease effectively.
Oxidized lipids and miR193 as potential biomarkers of PH
Several tests are currently used in clinical practice for evaluation of PAH, including 6-minute walk distance (6MWD); hemodynamic parameters, such as pulmonary artery pressure and cardiac output/cardiac index; B-type natriuretic peptide (BNP) and N-terminal-pro-BNP (NT-proBNP); and New York Heart Association functional class. Unfortunately, all of these parameters have significant limitations, since they are either invasive (pulmonary artery pressure using direct catheterization) or not very specific, as BNP/NT-proBNP can be influenced by left heart dysfunction and/or renal impairment and 6MWD by arthritis or myositis. Hence, there is an urgent need for discovering new, reliable, specific biomarkers that can predict disease progression and survival in PAH. Our recent work shows that oxidized lipids are significantly elevated in the plasma of rodents with PH. In the same setting, miR193 expression is significantly downregulated. Similarly, downregulation of miR193 was also observed in plasma samples obtained from PAH patients.56 These data raise the possibility that a combination approach using both oxidized lipids and miR193 expression may be exploited as a potential biomarker panel in PAH to assess the disease severity or response to therapy in PAH patients (Fig. 3). Analysis of a large cohort of human samples is needed to affirm the reliability and development of oxidized lipids and miR193 as a biomarker panel for PAH.
Summary
PAH is a multifactorial and heterogeneous disease associated with dysregulation of many molecular mechanisms contributing to the pathogenesis of the disease. The underlying causes of PAH include structural changes such as vascular remodeling, induction of proproliferative pathways, increased inflammation and oxidative stress, altered metabolic signaling, and genetic mutations. In this review, we have mainly focused on the involvement of oxidized lipids, lipid peroxidation, and impaired cellular mechanisms, including metabolism and oxidative stress, in the pathophysiology of PAH (Fig. 2). We have also elucidated the emerging role of HDL in the context of PH and the downstream mechanisms, including miR193, involved in the therapeutic potential of the ApoA-I mimetic peptide 4F in PH (Fig. 3). Given the heterogeneity of PAH, it is important to explore in depth the molecular mechanisms involved in the cause and consequence of the disease. Understanding the involvement of oxidized lipids in the pathophysiology of PAH may help in the development of more effective therapeutics and would increase the existing repertoire of potential therapeutics for this rare disease.
Source of Support: This work was supported in part by American Heart Association grants AHA17240020 (SS) and HL119886 (ME) and Clinical and Translational Science Institute grant UL1TR000124 (ME).
Conflict of Interest: STR is a principal in Bruin Pharma. All other authors: none declared.
References
- 1.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 2012;122(12):4306–4313. [DOI] [PMC free article] [PubMed]
- 2.Fallah F. Recent strategies in treatment of pulmonary arterial hypertension, a review. Glob J Health Sci 2015;7(4):307–322. [DOI] [PMC free article] [PubMed]
- 3.Shariff N, Kumar A, Narang R, Malhotra A, Mukhopadhyaya S, Sharma SK. A study of pulmonary arterial hypertension in patients with rheumatoid arthritis. Int J Cardiol 2007;115(1):75–76. [DOI] [PubMed]
- 4.Sung YK, Chung L. Connective tissue disease-associated pulmonary arterial hypertension. Rheum Dis Clin N Am 2015;41(2):295–313. [DOI] [PubMed]
- 5.Jaffe RB, Koschmann EB. Intravenous drug abuse: pulmonary, cardiac, and vascular complications. Am J Roentgenol Radium Ther Nucl Med 1970;109(1):107–120. [PubMed]
- 6.Saleemi S. Portopulmonary hypertension. Ann Thorac Med 2010;5(1):5–9. [DOI] [PMC free article] [PubMed]
- 7.Cicalini S, Almodovar S, Grilli E, Flores S. Pulmonary hypertension and human immunodeficiency virus infection: epidemiology, pathogenesis, and clinical approach. Clin Microbiol Infect 2011;17(1):25–33. [DOI] [PubMed]
- 8.Cool CD, Voelkel NF, Bull T. Viral infection and pulmonary hypertension: is there an association? Expert Rev Respir Med 2011;5(2):207–216. [DOI] [PubMed]
- 9.Adir Y, Harari S. Pulmonary hypertension associated with chronic obstructive lung disease and idiopathic pulmonary fibrosis. Curr Opin Pulm Med 2014;20(5):414–420. [DOI] [PubMed]
- 10.Kovalchin JP, Mott AR, Rosen KL, Feltes TF. Nitric oxide for the evaluation and treatment of pulmonary hypertension in congenital heart disease. Tex Heart Inst J 1997;24(4):308–316. [PMC free article] [PubMed]
- 11.Lee SH, Channick RN. Endothelin antagonism in pulmonary arterial hypertension. Semin Respir Crit Care Med 2005;26(4):402–408. [DOI] [PubMed]
- 12.Cannon BC, Feltes TF, Fraley JK, Grifka RG, Riddle EM, Kovalchin JP. Nitric oxide in the evaluation of congenital heart disease with pulmonary hypertension: factors related to nitric oxide response. Pediatr Cardiol 2005;26(5):565–569. [DOI] [PubMed]
- 13.Gomberg-Maitland M, Olschewski H. Prostacyclin therapies for the treatment of pulmonary arterial hypertension. Eur Respir J 2008;31(4):891–901. [DOI] [PubMed]
- 14.Wilkins MR, Wharton J, Grimminger F, Ghofrani HA. Phosphodiesterase inhibitors for the treatment of pulmonary hypertension. Eur Respir J 2008;32(1):198–209. [DOI] [PubMed]
- 15.Zhang L, Ma J, Shen T, Wang S, Ma C, Liu Y, Ran Y, Wang L, Liu L, Zhu D. Platelet-derived growth factor (PDGF) induces pulmonary vascular remodeling through 15-LO/15-HETE pathway under hypoxic condition. Cell Signal 2012;24(10):1931–1939. [DOI] [PubMed]
- 16.Ma C, Li Y, Ma J, Liu Y, Li Q, Niu S, Shen Z, Zhang L, Pan Z, Zhu D. Key role of 15-lipoxygenase/15-hydroxyeicosatetraenoic acid in pulmonary vascular remodeling and vascular angiogenesis associated with hypoxic pulmonary hypertension. Hypertension 2011;58(4):679–688. [DOI] [PubMed]
- 17.Preston IR, Hill NS, Warburton RR, Fanburg BL. Role of 12-lipoxygenase in hypoxia-induced rat pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol 2006;290(2):L367–L374. [DOI] [PubMed]
- 18.Shan R, Chen L, Li X, Wu H, Liang Q, Tang X. Hypoxia promotes rabbit pulmonary artery smooth muscle cells proliferation through a 15-LOX-2 product 15(S)-hydroxyeicosatetraenoic acid. Prostaglandins Leukot Essent Fatty Acids 2012;86(1–2):85–90. [DOI] [PubMed]
- 19.Jiang J, Wang S, Wang Z, Ma J, Liu S, Li W, Zhu D. The role of ERK1/2 in 15-HETE-inhibited apoptosis in pulmonary arterial smooth muscle cells. J Recept Signal Transduct Res 2011;31(1):45–52. [DOI] [PubMed]
- 20.Wang S, Wang Y, Jiang J, Wang R, Li L, Qiu Z, Wu H, Zhu D. 15-HETE protects rat pulmonary arterial smooth muscle cells from apoptosis via the PI3K/Akt pathway. Prostaglandins Other Lipid Mediat 2010;91(1–2):51–60. [DOI] [PubMed]
- 21.Ma J, Liang S, Wang Z, Zhang L, Jiang J, Zheng J, Yu L, Zheng X, Wang R, Zhu D. ROCK pathway participates in the processes that 15-hydroxyeicosatetraenoic acid (15-HETE) mediated the pulmonary vascular remodeling induced by hypoxia in rat. J Cell Physiol 2010;222(1):82–94. [DOI] [PubMed]
- 22.Nie X, Song S, Zhang L, Qiu Z, Shi S, Liu Y, Yao L, Zhu D. 15-Hydroxyeicosatetraenoic acid (15-HETE) protects pulmonary artery smooth muscle cells from apoptosis via inducible nitric oxide synthase (iNOS) pathway. Prostaglandins Other Lipid Mediat 2012;97(1–2):50–59. [DOI] [PubMed]
- 23.Li Y, Li Q, Wang Z, Liang D, Liang S, Tang X, Guo L, Zhang R, Zhu D. 15-HETE suppresses K+ channel activity and inhibits apoptosis in pulmonary artery smooth muscle cells. Apoptosis 2009;14:42–51. [DOI] [PubMed]
- 24.Guo L, Tang X, Chu X, Sun L, Zhang L, Qiu Z, Chen S, Li Y, Zheng X, Zhu D. Role of protein kinase C in 15-HETE-induced hypoxic pulmonary vasoconstriction. Prostaglandins Leukot Essent Fatty Acids 2009;80(2–3):115–123. [DOI] [PubMed]
- 25.Li Q, Bi HR, Zhang R, Zhu DL. Kv3.4 channel is involved in rat pulmonary vasoconstriction induced by 15-hydroxyeicosatetraenoic acid [in Chinese with English abstract]. Sheng Li Xue Bao 2006;58:77–82. [PubMed]
- 26.Chu X, Tang X, Guo L, Bao H, Zhang S, Zhang J, Zhu D. Hypoxia suppresses KV1.5 channel expression through endogenous 15-HETE in rat pulmonary artery. Prostaglandins Other Lipid Mediat 2009;88(1–2):42–50. [DOI] [PubMed]
- 27.Wang Y, Liang D, Wang S, Qiu Z, Chu X, Chen S, Li L, et al. Role of the G-protein and tyrosine kinase–Rho/ROK pathways in 15-hydroxyeicosatetraenoic acid induced pulmonary vasoconstriction in hypoxic rats. J Biochem 2010;147(5):751–764. [DOI] [PubMed]
- 28.Zhang L, Li Y, Chen M, Su X, Yi D, Lu P, Zhu D. 15-LO/15-HETE mediated vascular adventitia fibrosis via p38 MAPK-dependent TGF-β. J Cell Physiol 2014;229(2):245–257. [DOI] [PubMed]
- 29.Ross DJ, Hough G, Hama S, Aboulhosn J, Belperio JA, Saggar R, Van Lenten BJ, et al. Proinflammatory high-density lipoprotein results from oxidized lipid mediators in the pathogenesis of both idiopathic and associated types of pulmonary arterial hypertension. Pulm Circ 2015;5(4):640–648. [DOI] [PMC free article] [PubMed]
- 30.Tian W, Jiang X, Tamosiuniene R, Sung YK, Qian J, Dhillon G, Gera L, et al. Blocking macrophage leukotriene B4 prevents endothelial injury and reverses pulmonary hypertension. Sci Transl Med 2013;5:200ra117. doi:10.1126/scitranslmed.3006674. [DOI] [PMC free article] [PubMed]
- 31.Malmsten CL, Palmblad J, Udén AM, Rådmark O, Engstedt L, Samuelsson B. Leukotriene B4: a highly potent and stereospecific factor stimulating migration of polymorphonuclear leukocytes. Acta Physiol Scand 1980;110(4):449–451. [DOI] [PubMed]
- 32.Doig MV, Ford-Hutchinson AW. The production and characterisation of products of the lipoxygenase enzyme system released by rat peritoneal macrophages. Prostaglandins 1980;20(6):1007–1019. [DOI] [PubMed]
- 33.Ma J, Zhang L, Han W, Shen T, Ma C, Liu Y, Nie X, Liu M, Ran Y, Zhu D. Activation of JNK/c-Jun is required for the proliferation, survival, and angiogenesis induced by EET in pulmonary artery endothelial cells. J Lipid Res 2012;53(6):1093–1105. [DOI] [PMC free article] [PubMed]
- 34.Ameshima S, Golpon H, Cool CD, Chan D, Vandivier RW, Gardai SJ, Wick M, Nemenoff RA, Geraci MW, Voelkel NF. Peroxisome proliferator-activated receptor gamma (PPARγ) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ Res 2003;92(10):1162–1169. [DOI] [PubMed]
- 35.Guignabert C, Alvira CM, Alastalo TP, Sawada H, Hansmann G, Zhao M, Wang L, El-Bizri N, Rabinovitch M. Tie2-mediated loss of peroxisome proliferator-activated receptor-γ in mice causes PDGF receptor-β-dependent pulmonary arterial muscularization. Am J Physiol Lung Cell Mol Physiol 2009;297(6):L1082–L1090. [DOI] [PMC free article] [PubMed]
- 36.Hansmann G, de Jesus Perez VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, Schellong S, et al. An antiproliferative BMP-2/PPARγ/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest 2008;118(5):1846–1857. [DOI] [PMC free article] [PubMed]
- 37.Liu G, Li X, Li Y, Tang X, Xu J, Li R, Hao P, Sun Y. PPARδ agonist GW501516 inhibits PDGF-stimulated pulmonary arterial smooth muscle cell function related to pathological vascular remodeling. Biomed Res Int 2013;2013:903947. doi:10.1155/2013/903947. [DOI] [PMC free article] [PubMed]
- 38.Nisbet RE, Bland JM, Kleinhenz DJ, Mitchell PO, Walp ER, Sutliff RL, Hart CM. Rosiglitazone attenuates chronic hypoxia-induced pulmonary hypertension in a mouse model. Am J Respir Cell Mol Biol 2010;42(4):482–490. [DOI] [PMC free article] [PubMed]
- 39.Tuder RM, Davis LA, Graham BB. Targeting energetic metabolism: a new frontier in the pathogenesis and treatment of pulmonary hypertension. Am J Respir Crit Care Med 2012;185(3):260–266. [DOI] [PMC free article] [PubMed]
- 40.Rich S, Pogoriler J, Husain AN, Toth PT, Gomberg-Maitland M, Archer SL. Long-term effects of epoprostenol on the pulmonary vasculature in idiopathic pulmonary arterial hypertension. Chest 2010;138(5):1234–1239. [DOI] [PMC free article] [PubMed]
- 41.Piao L, Fang YH, Cadete VJ, Wietholt C, Urboniene D, Toth PT, Marsboom G, et al. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. J Mol Med (Berl) 2010;88(1):47–60. [DOI] [PMC free article] [PubMed]
- 42.Piao L, Marsboom G, Archer SL. Mitochondrial metabolic adaptation in right ventricular hypertrophy and failure. J Mol Med (Berl) 2010;88(10):1011–1020. [DOI] [PMC free article] [PubMed]
- 43.Hemnes AR, Brittain EL, Trammell AW, Fessel JP, Austin ED, Penner N, Maynard KB, et al. Evidence for right ventricular lipotoxicity in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med 2014;189(3):325–334. [DOI] [PMC free article] [PubMed]
- 44.Tuder RM, Robinson JC, Graham BB. Fat and cardiotoxicity in hereditary pulmonary hypertension. Am J Respir Crit Care Med 2014;189(3):247–249. [DOI] [PMC free article] [PubMed]
- 45.Sutendra G, Bonnet S, Rochefort G, Haromy A, Folmes KD, Lopaschuk GD, Dyck JR, Michelakis ED. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci Transl Med 2010;2:44ra58. doi:10.1126/scitranslmed.3001327. [DOI] [PubMed]
- 46.Ichimura Y, Asano Y, Akamata K, Aozasa N, Noda S, Taniguchi T, Takahashi T, et al. Serum angiopoietin-like protein 3 levels: possible correlation with progressive skin sclerosis, digital ulcers and pulmonary vascular involvement in patients with systemic sclerosis. Acta Derm Venereol 2014;94(2):157–162. [DOI] [PubMed]
- 47.Liu C, Tazzeo T, Janssen LJ. Isoprostane-induced airway hyperresponsiveness is dependent on internal Ca2+ handling and Rho/ROCK signaling. Am J Physiol Lung Cell Mol Physiol 2006;291(6):L1177–L1184. [DOI] [PubMed]
- 48.Janssen LJ, Premji M, Netherton S, Coruzzi J, Lu-Chao H, Cox PG. Vasoconstrictor actions of isoprostanes via tyrosine kinase and Rho kinase in human and canine pulmonary vascular smooth muscles. Br J Pharmacol 2001;132(1):127–134. [DOI] [PMC free article] [PubMed]
- 49.Yi SL, Kantores C, Belcastro R, Cabacungan J, Tanswell AK, Jankov RP. 8-Isoprostane-induced endothelin-1 production by infant rat pulmonary artery smooth muscle cells is mediated by Rho-kinase. Free Radic Biol Med 2006;41(6):942–949. [DOI] [PubMed]
- 50.Katsuyama M, Fan C, Yabe-Nishimura C. NADPH oxidase is involved in prostaglandin F2α-induced hypertrophy of vascular smooth muscle cells: induction of NOX1 by PGF2α. J Biol Chem 2002;277(16):13438–13442. [DOI] [PubMed]
- 51.Janssen LJ. Isoprostanes: an overview and putative roles in pulmonary pathophysiology. Am J Physiol Lung Cell Mol Physiol 2001;280(6):L1067–L1082. [DOI] [PubMed]
- 52.Mehta JL, Li D. Identification, regulation and function of a novel lectin-like oxidized low-density lipoprotein receptor. J Am Coll Cardiol 2002;39(9):1429–1435. [DOI] [PubMed]
- 53.Deigner HP, Hermetter A. Oxidized phospholipids: emerging lipid mediators in pathophysiology. Curr Opin Lipidol 2008;19(3):289–294. [DOI] [PubMed]
- 54.Mehta JL, Chen J, Hermonat PL, Romeo F, Novelli G. Lectin-like, oxidized low-density lipoprotein receptor-1 (LOX-1): a critical player in the development of atherosclerosis and related disorders. Cardiovasc Res 2006;69(1):36–45. [DOI] [PubMed]
- 55.Lähteenmäki TA, Seppo L, Laakso J, Korpela R, Vanhanen H, Tikkanen MJ, Vapaatalo H. Oxidized LDL from subjects with different dietary habits modifies atherogenic processes in endothelial and smooth muscle cells. Life Sci 2000;66(5):455–465. [DOI] [PubMed]
- 56.Sharma S, Umar S, Potus F, Iorga A, Wong G, Meriwether D, Breuils-Bonnet S, et al. Apolipoprotein A-I mimetic peptide 4F rescues pulmonary hypertension by inducing microRNA-193-3p. Circulation 2014;130(9):776–785. [DOI] [PMC free article] [PubMed]
- 57.Zhu D, Medhora M, Campbell WB, Spitzbarth N, Baker JE, Jacobs ER. Chronic hypoxia activates lung 15-lipoxygenase, which catalyzes production of 15-HETE and enhances constriction in neonatal rabbit pulmonary arteries. Circ Res 2003;92(9):992–1000. [DOI] [PubMed]
- 58.Bowers R, Cool C, Murphy RC, Tuder RM, Hopken MW, Flores SC, Voelkel NF. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med 2004;169(6):764–769. [DOI] [PubMed]
- 59.Moreno JJ. New aspects of the role of hydroxyeicosatetraenoic acids in cell growth and cancer development. Biochem Pharmacol 2009;77(1):1–10. [DOI] [PubMed]
- 60.Yao L, Nie X, Shi S, Song S, Hao X, Li S, Zhu D. Reciprocal regulation of HIF-1α and 15-LO/15-HETE promotes anti-apoptosis process in pulmonary artery smooth muscle cells during hypoxia. Prostaglandins Other Lipid Mediat 2012;99(3–4):96–106. [DOI] [PubMed]
- 61.Shen T, Ma J, Zhang L, Yu X, Liu M, Hou Y, Wang Y, Ma C, Li S, Zhu D. Positive feedback-loop of telomerase reverse transcriptase and 15-lipoxygenase-2 promotes pulmonary hypertension. PLoS ONE 2013;8(12):e83132. doi:10.1371/journal.pone.0083132. [DOI] [PMC free article] [PubMed]
- 62.Al-Husseini A, Wijesinghe DS, Farkas L, Kraskauskas D, Drake JI, Van Tassel B, Abbate A, Chalfant CE, Voelkel NF. Increased eicosanoid levels in the Sugen/chronic hypoxia model of severe pulmonary hypertension. PLoS ONE 2015;10(3):e0120157. doi:10.1371/journal.pone.0120157. [DOI] [PMC free article] [PubMed]
- 63.Xu X, Zhang XA, Wang DW. The roles of CYP450 epoxygenases and metabolites, epoxyeicosatrienoic acids, in cardiovascular and malignant diseases. Adv Drug Deliv Rev 2011;63(8):597–609. [DOI] [PubMed]
- 64.Keserü B, Barbosa-Sicard E, Popp R, Fisslthaler B, Dietrich A, Gudermann T, Hammock BD, et al. Epoxyeicosatrienoic acids and the soluble epoxide hydrolase are determinants of pulmonary artery pressure and the acute hypoxic pulmonary vasoconstrictor response. FASEB J 2008;22(12):4306–4315. [DOI] [PMC free article] [PubMed]
- 65.Kandhi S, Froogh G, Qin J, Luo M, Wolin MS, Huang A, Sun D. EETs elicit direct increases in pulmonary arterial pressure in mice. Am J Hypertens 2016;29(5):598–604. [DOI] [PMC free article] [PubMed]
- 66.Wang L, Yin J, Nickles HT, Ranke H, Tabuchi A, Hoffmann J, Tabeling C, et al. Hypoxic pulmonary vasoconstriction requires connexin 40-mediated endothelial signal conduction. J Clin Invest 2012;122(11):4218–4230. [DOI] [PMC free article] [PubMed]
- 67.Yokomizo T, Uozumi N, Takahashi T, Kume K, Izumi T, Shimizu T. Leukotriene A4 hydrolase and leukotriene B4 metabolism. J Lipid Mediat Cell Signal 1995;12(2–3):321–332. [DOI] [PubMed]
- 68.Capraro V. Intestinal absorption [in Italian]. Arch Fisiol 1979;71(1–4):67–78. [PubMed]
- 69.Tian W, Jiang X, Sung YK, Qian J, Yuan K, Nicolls MR. Leukotrienes in pulmonary arterial hypertension. Immunol Res 2014;58(2–3):387–393. [DOI] [PMC free article] [PubMed]
- 70.Voelkel NF, Tuder RM, Wade K, Hoper M, Lepley RA, Goulet JL, Koller BH, Fitzpatrick F. Inhibition of 5-lipoxygenase-activating protein (FLAP) reduces pulmonary vascular reactivity and pulmonary hypertension in hypoxic rats. J Clin Invest 1996;97(11):2491–2498. [DOI] [PMC free article] [PubMed]
- 71.Wright L, Tuder RM, Wang J, Cool CD, Lepley RA, Voelkel NF. 5-Lipoxygenase and 5-lipoxygenase activating protein (FLAP) immunoreactivity in lungs from patients with primary pulmonary hypertension. Am J Respir Crit Care Med 1998;157(1):219–229. [DOI] [PubMed]
- 72.Tabata T, Ono S, Song C, Noda M, Suzuki S, Tanita T, Fujimura S. Role of leukotriene B4 in monocrotaline-induced pulmonary hypertension [in Japanese]. Nihon Kyobu Shikkan Gakkai Zasshi 1997;35(2):160–166. [PubMed]
- 73.Qian J, Tian W, Jiang X, Tamosiuniene R, Sung YK, Shuffle EM, Tu AB, et al. Leukotriene B4 activates pulmonary artery adventitial fibroblasts in pulmonary hypertension. Hypertension 2015;66(6):1227–1239. [DOI] [PMC free article] [PubMed]
- 74.Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease: the Framingham Study. Am J Med 1977;62(5):707–714. [DOI] [PubMed]
- 75.Ansell BJ, Navab M, Hama S, Kamranpour N, Fonarow G, Hough G, Rahmani S, et al. Inflammatory/antiinflammatory properties of high-density lipoprotein distinguish patients from control subjects better than high-density lipoprotein cholesterol levels and are favorably affected by simvastatin treatment. Circulation 2003;108(22):2751–2756. [DOI] [PubMed]
- 76.Toikka JO, Ahotupa M, Viikari JS, Niinikoski H, Taskinen M, Irjala K, Hartiala JJ, Raitakari OT. Constantly low HDL-cholesterol concentration relates to endothelial dysfunction and increased in vivo LDL-oxidation in healthy young men. Atherosclerosis 1999;147(1):133–138. [DOI] [PubMed]
- 77.Gordon SC, Polson DJ, Shirkhoda A. Budd-Chiari syndrome complicating pre-eclampsia: diagnosis by magnetic resonance imaging. J Clin Gastroenterol 1991;13(4):460–462. [DOI] [PubMed]
- 78.Zamanian RT, Hansmann G, Snook S, Lilienfeld D, Rappaport KM, Reaven GM, Rabinovitch M, Doyle RL. Insulin resistance in pulmonary arterial hypertension. Eur Respir J 2009;33(2):318–324. [DOI] [PMC free article] [PubMed]
- 79.Hansmann G, Wagner RA, Schellong S, Perez VA, Urashima T, Wang L, Sheikh AY, Suen RS, Stewart DJ, Rabinovitch M. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-γ activation. Circulation 2007;115(10):1275–1284. [DOI] [PubMed]
- 80.Heresi GA, Aytekin M, Newman J, DiDonato J, Dweik RA. Plasma levels of high-density lipoprotein cholesterol and outcomes in pulmonary arterial hypertension. Am J Respir Crit Care Med 2010;182(5):661–668. [DOI] [PMC free article] [PubMed]
- 81.Zhao QH, Peng FH, Wei H, He J, Chen FD, Di RM, Jiang X, et al. Serum high-density lipoprotein cholesterol levels as a prognostic indicator in patients with idiopathic pulmonary arterial hypertension. Am J Cardiol 2012;110(3):433–439. [DOI] [PubMed]
- 82.Gopal DM, Santhanakrishnan R, Wang YC, Ayalon N, Donohue C, Rahban Y, Perez AJ, et al. Impaired right ventricular hemodynamics indicate preclinical pulmonary hypertension in patients with metabolic syndrome. J Am Heart Assoc 2015;4(3):e001597. doi:10.1161/JAHA.114.001597. [DOI] [PMC free article] [PubMed]
- 83.Hayashi K, Takahashi M, Nishida W, Yoshida K, Ohkawa Y, Kitabatake A, Aoki J, Arai H, Sobue K. Phenotypic modulation of vascular smooth muscle cells induced by unsaturated lysophosphatidic acids. Circ Res 2001;89(3):251–258. [DOI] [PubMed]
- 84.Lee H, Goetzl EJ, An S. Lysophosphatidic acid and sphingosine 1-phosphate stimulate endothelial cell wound healing. Am J Physiol Cell Physiol 2000;278(3):C612–C618. [DOI] [PubMed]
- 85.Moolenaar WH. Lysophosphatidic acid, a multifunctional phospholipid messenger. J Biol Chem 1995;270(22):12949–12952. [DOI] [PubMed]
- 86.Smyth SS, Cheng HY, Miriyala S, Panchatcharam M, Morris AJ. Roles of lysophosphatidic acid in cardiovascular physiology and disease. Biochim Biophys Acta Mol Cell Biol Lipids 2008;1781(9):563–570. [DOI] [PMC free article] [PubMed]
- 87.Gennero I, Xuereb JM, Simon MF, Girolami JP, Bascands JL, Chap H, Boneu B, Sié P. Effects of lysophosphatidic acid on proliferation and cytosolic Ca++ of human adult vascular smooth muscle cells in culture. Thromb Res 1999;94(5):317–326. [DOI] [PubMed]
- 88.van Meeteren LA, Ruurs P, Stortelers C, Bouwman P, van Rooijen MA, Pradère JP, Pettit TR, et al. Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol Cell Biol 2006;26(13):5015–5022. [DOI] [PMC free article] [PubMed]
- 89.Chua CC, Hamdy RC, Chua BH. Upregulation of endothelin-1 production by lysophosphatidic acid in rat aortic endothelial cells. Biochim Biophys Acta Mol Cell Res 1998;1405(1–3):29–34. [DOI] [PubMed]
- 90.Choi JW, Lee CW, Chun J. Biological roles of lysophospholipid receptors revealed by genetic null mice: an update. Biochim Biophys Acta Mol Cell Biol Lipids 2008;1781(9):531–539. [DOI] [PMC free article] [PubMed]
- 91.Cheng HY, Dong A, Panchatcharam M, Mueller P, Yang F, Li Z, Mills G, Chun J, Morris AJ, Smyth SS. Lysophosphatidic acid signaling protects pulmonary vasculature from hypoxia-induced remodeling. Arterioscler Thromb Vasc Biol 2012;32(1):24–32. [DOI] [PMC free article] [PubMed]
- 92.Liu JQ, Zelko IN, Erbynn EM, Sham JS, Folz RJ. Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91phox). Am J Physiol Lung Cell Mol Physiol 2006;290(1):L2–L10. [DOI] [PubMed]
- 93.Hoshikawa Y, Ono S, Suzuki S, Tanita T, Chida M, Song C, Noda M, Tabata T, Voelkel NF, Fujimura S. Generation of oxidative stress contributes to the development of pulmonary hypertension induced by hypoxia. J Appl Physiol 2001;90(4):1299–1306. [DOI] [PubMed]
- 94.DeMarco VG, Whaley-Connell AT, Sowers JR, Habibi J, Dellsperger KC. Contribution of oxidative stress to pulmonary arterial hypertension. World J Cardiol 2010;2(10):316–324. [DOI] [PMC free article] [PubMed]
- 95.Pichardo J, Palace V, Farahmand F, Singal PK. Myocardial oxidative stress changes during compensated right heart failure in rats. Mol Cell Biochem 1999;196(1–2):51–57. [PubMed]
- 96.Kamezaki F, Tasaki H, Yamashita K, Tsutsui M, Koide S, Nakata S, Tanimoto A, et al. Gene transfer of extracellular superoxide dismutase ameliorates pulmonary hypertension in rats. Am J Respir Crit Care Med 2008;177(2):219–226. [DOI] [PubMed]
- 97.Csiszar A, Labinskyy N, Olson S, Pinto JT, Gupte S, Wu JM, Hu F, et al. Resveratrol prevents monocrotaline-induced pulmonary hypertension in rats. Hypertension 2009;54(3):668–675. [DOI] [PMC free article] [PubMed]
- 98.Janssen LJ. Isoprostanes and lung vascular pathology. Am J Respir Cell Mol Biol 2008;39(4):383–389. [DOI] [PubMed]
- 99.Carpenter CT, Price PV, Christman BW. Exhaled breath condensate isoprostanes are elevated in patients with acute lung injury or ARDS. Chest 1998;114(6):1653–1659. [DOI] [PubMed]
- 100.Montuschi P, Corradi M, Ciabattoni G, Nightingale J, Kharitonov SA, Barnes PJ. Increased 8-isoprostane, a marker of oxidative stress, in exhaled condensate of asthma patients. Am J Respir Crit Care Med 1999;160(1):216–220. [DOI] [PubMed]
- 101.Montuschi P, Collins JV, Ciabattoni G, Lazzeri N, Corradi M, Kharitonov SA, Barnes PJ. Exhaled 8-isoprostane as an in vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am J Respir Crit Care Med 2000;162(3):1175–1177. [DOI] [PubMed]
- 102.Montuschi P, Ciabattoni G, Paredi P, Pantelidis P, du Bois RM, Kharitonov SA, Barnes PJ. 8-Isoprostane as a biomarker of oxidative stress in interstitial lung diseases. Am J Respir Crit Care Med 1998;158(5):1524–1527. [DOI] [PubMed]
- 103.Cracowski JL, Cracowski C, Bessard G, Pépin JL, Bessard J, Schwebel C, Stanke-Labesque F, Pison C. Increased lipid peroxidation in patients with pulmonary hypertension. Am J Respir Crit Care Med 2001;164(6):1038–1042. [DOI] [PubMed]
- 104.Jankov RP, Luo X, Cabacungan J, Belcastro R, Frndova H, Lye SJ, Tanswell AK. Endothelin-1 and O2-mediated pulmonary hypertension in neonatal rats: a role for products of lipid peroxidation. Pediatr Res 2000;48(3):289–298. [DOI] [PubMed]
- 105.Elmhurst JL, Betti PA, Rangachari PK. Intestinal effects of isoprostanes: evidence for the involvement of prostanoid EP and TP receptors. J Pharmacol Exp Ther 1997;282(3):1198–1205. [PubMed]
- 106.Catalli A, Zhang D, Janssen LJ. Receptors and signaling pathway underlying relaxations to isoprostanes in canine and porcine airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 2002;283(5):L1151–L1159. [DOI] [PubMed]
- 107.Clarke DL, Belvisi MG, Hardaker E, Newton R, Giembycz MA. E-ring 8-isoprostanes are agonists at EP2- and EP4-prostanoid receptors on human airway smooth muscle cells and regulate the release of colony-stimulating factors by activating cAMP-dependent protein kinase. Mol Pharmacol 2005;67(2):383–393. [DOI] [PubMed]
- 108.Liu C, Tazzeo T, Guy A, Durand T, Janssen LJ. Pharmacological actions of isoprostane metabolites and phytoprostanes in human and bovine pulmonary smooth muscles. Prostaglandins Leukot Essent Fatty Acids 2007;76(1):57–64. [DOI] [PubMed]
- 109.Tazzeo T, Miller J, Janssen LJ. Vasoconstrictor responses, and underlying mechanisms, to isoprostanes in human and porcine bronchial arterial smooth muscle. Br J Pharmacol 2003;140(4):759–763. [DOI] [PMC free article] [PubMed]
- 110.Ruef J, Moser M, Kübler W, Bode C. Induction of endothelin-1 expression by oxidative stress in vascular smooth muscle cells. Cardiovasc Pathol 2001;10(6):311–315. [DOI] [PubMed]
- 111.Irodova NL, Lankin VZ, Konovalova GK, Kochetov AG, Chazova IE. Oxidative stress in patients with primary pulmonary hypertension. Bull Exp Biol Med 2002;133(6):580–582. [DOI] [PubMed]
- 112.Odhiambo A, Perlman DH, Huang H, Costello CE, Farber HW, Steinberg MH, McComb ME, Klings ES. Identification of oxidative post-translational modification of serum albumin in patients with idiopathic pulmonary arterial hypertension and pulmonary hypertension of sickle cell anemia. Rapid Commun Mass Spectrom 2007;21(14):2195–2203. [DOI] [PubMed]
- 113.Lane KL, Talati M, Austin E, Hemnes AR, Johnson JA, Fessel JP, Blackwell T, et al. Oxidative injury is a common consequence of BMPR2 mutations. Pulm Circ 2011;1(1):72–83. [DOI] [PMC free article] [PubMed]
- 114.Preston IR, Tang G, Tilan JU, Hill NS, Suzuki YJ. Retinoids and pulmonary hypertension. Circulation 2005;111(6):782–790. [DOI] [PubMed]
- 115.Brennan LA, Steinhorn RH, Wedgwood S, Mata-Greenwood E, Roark EA, Russell JA, Black SM. Increased superoxide generation is associated with pulmonary hypertension in fetal lambs: a role for NADPH oxidase. Circ Res 2003;92(6):683–691. [DOI] [PubMed]
- 116.Grobe AC, Wells SM, Benavidez E, Oishi P, Azakie A, Fineman JR, Black SM. Increased oxidative stress in lambs with increased pulmonary blood flow and pulmonary hypertension: role of NADPH oxidase and endothelial NO synthase. Am J Physiol Lung Cell Mol Physiol 2006;290(6):L1069–L1077. [DOI] [PubMed]
- 117.Wedgwood S, Dettman RW, Black SM. ET-1 stimulates pulmonary arterial smooth muscle cell proliferation via induction of reactive oxygen species. Am J Physiol Lung Cell Mol Physiol 2001;281(5):L1058–L1067. [DOI] [PubMed]
- 118.Liu Y, Suzuki YJ, Day RM, Fanburg BL. Rho kinase-induced nuclear translocation of ERK1/ERK2 in smooth muscle cell mitogenesis caused by serotonin. Circ Res 2004;95(6):579–586. [DOI] [PubMed]
- 119.Lawrie A, Spiekerkoetter E, Martinez EC, Ambartsumian N, Sheward WJ, MacLean MR, Harmar AJ, Schmidt AM, Lukanidin E, Rabinovitch M. Interdependent serotonin transporter and receptor pathways regulate S100A4/Mts1, a gene associated with pulmonary vascular disease. Circ Res 2005;97(3):227–235. [DOI] [PubMed]
- 120.Elmedal B, de Dam MY, Mulvany MJ, Simonsen U. The superoxide dismutase mimetic, tempol, blunts right ventricular hypertrophy in chronic hypoxic rats. Br J Pharmacol 2004;141(1):105–113. [DOI] [PMC free article] [PubMed]
- 121.Hu C, Dandapat A, Chen J, Fujita Y, Inoue N, Kawase Y, Jishage K, Suzuki H, Sawamura T, Mehta JL. LOX-1 deletion alters signals of myocardial remodeling immediately after ischemia-reperfusion. Cardiovasc Res 2007;76(2):292–302. [DOI] [PubMed]
- 122.Chen H, Li D, Sawamura T, Inoue K, Mehta JL. Upregulation of LOX-1 expression in aorta of hypercholesterolemic rabbits: modulation by losartan. Biochem Biophys Res Commun 2000;276(3):1100–1104. [DOI] [PubMed]
- 123.Ogura S, Shimosawa T, Mu S, Sonobe T, Kawakami-Mori F, Wang H, Uetake Y, et al. Oxidative stress augments pulmonary hypertension in chronically hypoxic mice overexpressing the oxidized LDL receptor. Am J Physiol Heart Circ Physiol 2013;305(2):H155–H162. [DOI] [PubMed]
- 124.Austin ED, Loyd JE. The genetics of pulmonary arterial hypertension. Circ Res 2014;115(1):189–202. [DOI] [PMC free article] [PubMed]
- 125.Newman JH, Wheeler L, Lane KB, Loyd E, Gaddipati R, Phillips JA III, Loyd JE. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med 2001;345(5):319–324. [DOI] [PubMed]
- 126.Fessel JP, Hamid R, Wittmann BM, Robinson LJ, Blackwell T, Tada Y, Tanabe N, Tatsumi K, Hemnes AR, West JD. Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulm Circ 2012;2(2):201–213. [DOI] [PMC free article] [PubMed]
- 127.Douglas RM, Bowden K, Pattison J, Peterson AB, Juliano J, Dalton ND, Gu Y, et al. Intermittent hypoxia and hypercapnia induce pulmonary artery atherosclerosis and ventricular dysfunction in low density lipoprotein receptor deficient mice. J Appl Physiol 2013;115(11):1694–1704. [DOI] [PMC free article] [PubMed]
- 128.Moore GW, Smith RR, Hutchins GM. Pulmonary artery atherosclerosis: correlation with systemic atherosclerosis and hypertensive pulmonary vascular disease. Arch Pathol Lab Med 1982;106(8):378–380. [PubMed]
- 129.Carpenter KL, Taylor SE, van der Veen C, Mitchinson MJ. Evidence of lipid oxidation in pulmonary artery atherosclerosis. Atherosclerosis 1995;118(1):169–172. [DOI] [PubMed]
- 130.Price LC, Wort SJ, Perros F, Dorfmüller P, Huertas A, Montani D, Cohen-Kaminsky S, Humbert M. Inflammation in pulmonary arterial hypertension. Chest 2012;141(1):210–221. [DOI] [PubMed]
- 131.Kimura H, Okada O, Tanabe N, Tanaka Y, Terai M, Takiguchi Y, Masuda M, et al. Plasma monocyte chemoattractant protein-1 and pulmonary vascular resistance in chronic thromboembolic pulmonary hypertension. Am J Respir Crit Care Med 2001;164(2):319–324. [DOI] [PubMed]
- 132.Itoh T, Nagaya N, Ishibashi-Ueda H, Kyotani S, Oya H, Sakamaki F, Kimura H, Nakanishi N. Increased plasma monocyte chemoattractant protein-1 level in idiopathic pulmonary arterial hypertension. Respirology 2006;11(2):158–163. [DOI] [PubMed]
- 133.Sanchez O, Marcos E, Perros F, Fadel E, Tu L, Humbert M, Dartevelle P, Simonneau G, Adnot S, Eddahibi S. Role of endothelium-derived CC chemokine ligand 2 in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2007;176(10):1041–1047. [DOI] [PubMed]
- 134.Zabini D, Heinemann A, Foris V, Nagaraj C, Nierlich P, Bálint Z, Kwapiszewska G, et al. Comprehensive analysis of inflammatory markers in chronic thromboembolic pulmonary hypertension patients. Eur Respir J 2014;44(4):951–962. [DOI] [PubMed]
- 135.Mori TA, Beilin LJ. Omega-3 fatty acids and inflammation. Curr Atheroscler Rep 2004;6(6):461–467. [DOI] [PubMed]
- 136.Terkeltaub R, Banka CL, Solan J, Santoro D, Brand K, Curtiss LK. Oxidized LDL induces monocytic cell expression of interleukin-8, a chemokine with T-lymphocyte chemotactic activity. Arterioscler Thromb 1994;14(1):47–53. [DOI] [PubMed]
- 137.Bittleman DB, Casale TB. 5-Hydroxyeicosatetraenoic acid (HETE)-induced neutrophil transcellular migration is dependent upon enantiomeric structure. Am J Respir Cell Mol Biol 1995;12(3):260–267. [DOI] [PubMed]
- 138.Vangaveti V, Baune BT, Kennedy RL. Hydroxyoctadecadienoic acids: novel regulators of macrophage differentiation and atherogenesis. Ther Adv Endocrinol Metab 2010;1(2):51–60. [DOI] [PMC free article] [PubMed]
- 139.Bochkov VN, Leitinger N. Anti-inflammatory properties of lipid oxidation products. J Mol Med (Berl) 2003;81(10):613–626. [DOI] [PubMed]
- 140.Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol 2002;2(10):748–759. [DOI] [PubMed]
- 141.Cabral M, Martín-Venegas R, Moreno JJ. Differential cell growth/apoptosis behavior of 13-hydroxyoctadecadienoic acid enantiomers in a colorectal cancer cell line. Am J Physiol Gastrointest Liver Physiol 2014;307(6):G664–G671. [DOI] [PubMed]
- 142.Janssen LJ, Catalli A, Helli P. The pulmonary biology of isoprostanes. Antioxid Redox Signal 2005;7(1–2):244–255. [DOI] [PubMed]
- 143.Stenmark KR, Morganroth ML, Remigio LK, Voelkel NF, Murphy RC, Henson PM, Mathias MM, Reeves JT. Alveolar inflammation and arachidonate metabolism in monocrotaline-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol 1985;248(6):H859–H866. [DOI] [PubMed]
- 144.Hirata H, Arima M, Fukushima Y, Sugiyama K, Tokuhisa T, Fukuda T. Leukotriene C4 aggravates bleomycin-induced pulmonary fibrosis in mice. Respirology 2013;18(4):674–681. [DOI] [PubMed]
- 145.Kondeti V, Al-Azzam N, Duah E, Thodeti CK, Boyce JA, Paruchuri S. Leukotriene D4 and prostaglandin E2 signals synergize and potentiate vascular inflammation in a mast cell–dependent manner through cysteinyl leukotriene receptor 1 and E-prostanoid receptor 3. J Allergy Clin Immunol 2016;137(1):289–298. [DOI] [PMC free article] [PubMed]
- 146.Meyer P, Nigam A, Marcil M, Tardif JC. The therapeutic potential of high-density lipoprotein mimetic agents in coronary artery disease. Curr Atheroscler Rep 2009;11(5):329–333. [DOI] [PubMed]
- 147.Wang W, Xu H, Shi Y, Nandedkar S, Zhang H, Gao H, Feroah T, et al. Genetic deletion of apolipoprotein A-I increases airway hyperresponsiveness, inflammation, and collagen deposition in the lung. J Lipid Res 2010;51(9):2560–2570. [DOI] [PMC free article] [PubMed]
- 148.Yuditskaya S, Tumblin A, Hoehn GT, Wang G, Drake SK, Xu X, Ying S, et al. Proteomic identification of altered apolipoprotein patterns in pulmonary hypertension and vasculopathy of sickle cell disease. Blood 2009;113(5):1122–1128. [DOI] [PMC free article] [PubMed]
- 149.Kim TH, Lee YH, Kim KH, Lee SH, Cha JY, Shin EK, Jung S, et al. Role of lung apolipoprotein A-I in idiopathic pulmonary fibrosis: antiinflammatory and antifibrotic effect on experimental lung injury and fibrosis. Am J Respir Crit Care Med 2010;182(5):633–642. [DOI] [PubMed]
- 150.Lämmer D. Test results in 1008 patients with contact allergy [in German]. Z Hautkr 1979;54:571–579. [PubMed]
- 151.Chiesa G, Sirtori CR. Use of recombinant apolipoproteins in vascular diseases: the case of apoA-I. Curr Opin Investig Drugs 2002;3(3):420–426. [PubMed]
- 152.Peterson SJ, Drummond G, Kim DH, Li M, Kruger AL, Ikehara S, Abraham NG. L-4F treatment reduces adiposity, increases adiponectin levels, and improves insulin sensitivity in obese mice. J Lipid Res 2008;49(8):1658–1669. [DOI] [PMC free article] [PubMed]
- 153.Ou J, Ou Z, Jones DW, Holzhauer S, Hatoum OA, Ackerman AW, Weihrauch DW, et al. L-4F, an apolipoprotein A-1 mimetic, dramatically improves vasodilation in hypercholesterolemia and sickle cell disease. Circulation 2003;107(18):2337–2341. [DOI] [PubMed]
- 154.Nandedkar SD, Weihrauch D, Xu H, Shi Y, Feroah T, Hutchins W, Rickaby DA, et al. D-4F, an apoA-1 mimetic, decreases airway hyperresponsiveness, inflammation, and oxidative stress in a murine model of asthma. J Lipid Res 2011;52(3):499–508. [DOI] [PMC free article] [PubMed]
- 155.Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science 2001;294(5543):853–858. [DOI] [PubMed]
- 156.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004;116(2):281–297. [DOI] [PubMed]
- 157.Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med 2007;13(5):613–618. [DOI] [PubMed]
- 158.Joshi SR, McLendon JM, Comer BS, Gerthoffer WT. MicroRNAs-control of essential genes: implications for pulmonary vascular disease. Pulm Circ 2011;1(3):357–364. [DOI] [PMC free article] [PubMed]
- 159.Courboulin A, Paulin R, Giguère NJ, Saksouk N, Perreault T, Meloche J, Paquet ER, et al. Role for miR-204 in human pulmonary arterial hypertension. J Exp Med 2011;208(3):535–548. [DOI] [PMC free article] [PubMed]
- 160.Guo L, Qiu Z, Wei L, Yu X, Gao X, Jiang S, Tian H, Jiang C, Zhu D. The microRNA-328 regulates hypoxic pulmonary hypertension by targeting at insulin growth factor 1 receptor and L-type calcium channel-α1C. Hypertension 2012;59(5):1006–1013. [DOI] [PubMed]
- 161.Parikh VN, Jin RC, Rabello S, Gulbahce N, White K, Hale A, Cottrill KA, et al. MicroRNA-21 integrates pathogenic signaling to control pulmonary hypertension: results of a network bioinformatics approach. Circulation 2012;125(12):1520–1532. [DOI] [PMC free article] [PubMed]