

Abstract Abstract
The proliferative endothelial and smooth muscle cell phenotype, inflammation, and pulmonary vascular remodeling are prominent features of pulmonary arterial hypertension (PAH). Mutations in bone morphogenetic protein type 2 receptor (BMPR2) have been identified as the most common genetic cause of PAH and females with BMPR2 mutations are 2.5 times as likely to develop heritable forms of PAH than males. Higher levels of estrogen have also been observed in males with PAH, implicating sex hormones in PAH pathogenesis. Recently, the estrogen metabolite 16α-OHE1 (hydroxyestrone) was implicated in the regulation of miR29, a microRNA involved in modulating energy metabolism. In females, decreased miR96 enhances serotonin’s effect by upregulating the 5-hydroxytryptamine 1B (5HT1B) receptor. Because PAH is characterized as a quasi-malignant disease, likely due to BMPR2 loss of function, altered signaling pathways that sustain this cancer-like phenotype are being explored. Extracellular signal–regulated kinases 1 and 2 and p38 mitogen-activated protein kinases (MAPKs) play a critical role in proliferation and cell motility, and dysregulated MAPK signaling is observed in various experimental models of PAH. Wnt signaling pathways preserve pulmonary vascular homeostasis, and dysregulation of this pathway could contribute to limited vascular regeneration in response to injury. In this review, we take a closer look at sex, sex hormones, and the interplay between sex hormones and microRNA regulation. We also focus on MAPK and Wnt signaling pathways in the emergence of a proproliferative, antiapoptotic endothelial phenotype, which then orchestrates an angioproliferative process of vascular remodeling, with the hope of developing novel therapies that could reverse the phenotype.
Keywords: vascular remodeling, sex hormones, microRNA, mitogen-activated protein kinase, Wnt
Pulmonary arterial hypertension (PAH) is a rare disorder characterized by endothelial cell proliferation, pulmonary vascular remodeling, pruning of distal vessels, and increased pulmonary resistance, culminating in right heart failure and death.1 Although the initial mechanisms responsible for the development of idiopathic PAH (IPAH) and other forms of PAH remain unknown, loss of normal bone morphogenetic protein type 2 receptor (BMPR2) function has been implicated in the formation of plexogenic lesions.2 These plexogenic lesions are believed to be a result of dysregulated angiogenesis, with proliferation of endothelial cells, vascular smooth muscle cells, myofibroblasts, and inflammatory cells in and around occluded pulmonary arteries.3
PAH develops more predominately in women than in men; survival is, however, poorer in men. These sex differences are incompletely understood. Female predominance suggests that sex hormones contributes to pulmonary vascular remodeling, in part through the ability of estrogen (E2) to reduce BMPR2 expression.4 However, in some experimental models of PAH (monocrotaline and chronic hypoxia), E2 appears to protect against PAH,5 while others have reported that endogenous E2 results in the development of experimental PAH.6,7 It is thought that survival is poorer in men because men may either respond less well to therapy or have poorer right ventricular compensation in the face of elevated pulmonary pressures than women, although neither theory has been proven. PAH is now more frequently diagnosed in elderly patients, resulting in a mean age at diagnosis between 50 ± 14 and 65 ± 15 years in current registries.8 Female predominance is quite variable among registries and may also not be present in elderly patients.9 A clearer understanding, however, of how the levels of E2, E2 metabolites, and E2 receptors play a role in the pathogenesis of PAH is needed.
PAH phenotypically is characterized as a quasi-malignant disease, and there have been many studies identifying aberrant signaling pathways in the development of PAH, including mitogen-activated protein kinase (MAPK) and Wnt signaling. There is increasing evidence that the activation of MAPKs controls the hyperproliferation and increased motility of endothelial cells and smooth muscle cells (SMCs) and targeted inhibition of MAPKs reverses the phenotype. However, some receptor tyrosine kinase (RTK) inhibitors have paradoxically resulted in the development of PAH, so safer and more targeted therapies must be developed. The Wnt signaling pathways, Wnt/β-catenin and Wnt/PCP (planar cell polarity), play a crucial role in cell fate determination and vascular regeneration in response to injury, and constitutive activation of these signaling pathways in PAH could promote cell survival and protect against vascular remodeling.10 Here we review the role of sex and dysregulated signaling pathways in PAH, with the hope of better understanding the complexity of this disease.
The Role of E2 and E2 Metabolites in PAH
IPAH and heritable forms of PAH (hPAH) have a mean age at diagnosis of 35 years and a median survival of 2.8 years untreated. Survival is not greatly improved by treatment, and there is an unmet clinical need for effective therapeutic approaches. Epidemiological studies report a greater incidence of the disease in females; the female-to-male ratio can be as great as 4∶1.8,11 The reasons for this increased frequency in females are unclear. Pulmonary artery smooth muscle cells (PASMCs) normally exhibit low rates of proliferation, migration, and apoptosis to maintain a low-resistance pulmonary circulation. However, alterations in signaling pathways can lead to abnormal proliferation, apoptosis, and migration, and the most important signaling pathway to this effect is the BMPR2 pathway. The primary genetic defect of hPAH (present in at least 70% of cases of hPAH) is a mutation in the gene encoding BMPR2.12,13 Females with mutations are about 2.5 times as likely to develop hPAH as males.14 Heritable PAH transmits as an autosomal dominant trait that exhibits genetic anticipation but also markedly reduced penetrance (20%–40%).14 The cause of the reduced penetrance is likely to be related to the need for a “second hit” caused by environmental and/or genetic modifiers. BMPR2 is a member of the transforming growth factor beta (TGF-β) superfamily. Signaling by bone morphogenetic protein (BMP) receptors involves heterodimerization of two transmembrane serine/threonine kinases: the constitutively active type 2 receptor BMPR2 and a corresponding type 1 receptor, BMPR1A or BMPR1B. Activated BMPR1 receptors phosphorylate a set of BMP-restricted Smad proteins (Smad1, 5, and 8), which then complex with the common partner Smad, Smad4 (Co-Smad), and translocate into the nucleus to regulate transcription of target genes in a tissue- and cell-specific manner. The proteins in the inhibitor of DNA binding (Id) family (Id1–Id4) are transcriptional targets of the BMP signaling. These proteins bind with high affinity to the E-protein subfamily of basic-loop-helix family of transcription factors and inhibit their binding to target DNA, regulating gene expression and cellular differentiation. Dysfunctional Smad signaling leads to abnormal cell proliferation associated with pulmonary vascular disease.15 Recently, higher levels of E2 were associated with PAH in men, and it is thought that sex-based differences in sex hormone processing and signaling may contribute to unique phenotypes in pulmonary vascular disease.16
E2 synthesis and metabolism
In premenopausal women, E2 synthesis occurs mainly in the ovarian follicles and corpus luteum; in postmenopausal women and men, adipose tissue is a major source of E2 synthesis.17 Aromatase (CYP19A1) is a member of the cytochrome P450 (CYP) superfamily and synthesizes E2 through the aromatization of androgens, specifically testosterone and androstenedione. Further metabolism is mediated by several CYP enzymes. In particular, CYP1B1 catalyzes the oxidation of estrone (E1)/17β-estradiol (E2) to 2- and 4-OHE2 (2- and 4-hydroxyestradiol). E2 hydroxylation can occur by CYP enzymes (including CYP1B1), resulting in 6, 7, 15, and 16(α- and β-)hydroxyestradiol. E2 can also be converted to (1) E1 by 17β-HSD (hydroxysteroid dehydrogenase) and hence via oxidation to 16α-OHE1 (hydroxyestrone), which is mitogenic, and (2) 2-OHE2 by CYP1A1/2, CYP1B1, and CYP3A4. Catechol-O-methyltransferase (COMT) catalyzes the methylation of catechol E2s to methoxy E2s (MEs), which simultaneously lowers the potential for DNA damage and increases the concentration of 2ME, an antiproliferative metabolite (Fig. 1).
Figure 1.
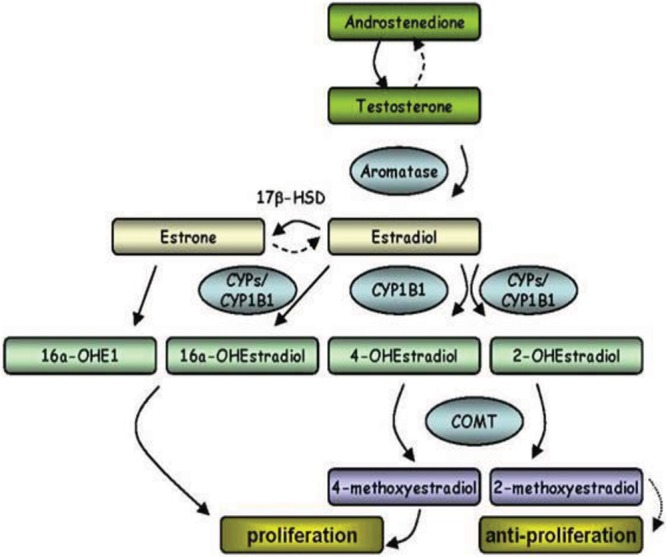
Summary of estrogen (E2) synthesis and metabolic pathways of key interest. See text for details. COMT: catechol-O-methyltransferase; CYP: cytochrome P450; HSD: hydroxysteroid dehydrogenase; OHE1: hydroxyestrone.
E2 receptors and BMPR2 signaling
The effects of E2 are primarily mediated by activation of the E2 receptors ERα and ERβ, and these effects can be both genomic and nongenomic. Very rapid, nongenomic effects of E2 have also been described through activation of the G-protein coupled E2 receptor (GPER). All three receptor types are expressed in human pulmonary arteries, and it is thought that E2 may suppress BMPR2 expression via the receptor ERα.4 ERα is highly expressed in female human PASMCs (hPASMCs) from PAH patients and mediates E2-induced proliferation of hPASMCs via MAPK and Akt signaling.18 The ERα antagonist MPP (methyl-piperidino-pyrazole) protects against hypoxia-induced pulmonary hypertension (PH) in female mice and reverses PH in mice overexpressing the serotonin transporter (SERT+ mice) via rescue of BMPR2 signaling.7,18
E2 synthesis and BMPR2 signaling
Two promoter single-nucleotide polymorphisms (SNPs) in the gene coding for aromatase that result in elevated E2 production have been associated with an increased risk of portopulmonary hypertension.19 The human pulmonary artery can synthesize E2; aromatase expression is present in the medial layer of human pulmonary arteries, and female hPASMCs have higher aromatase expression.7 The aromatase inhibitor anastrozole reversed pulmonary vascular remodeling, PH, and right ventricular hypertrophy observed in hypoxic mice and in the Sugen/hypoxic rat, but only in females in both models of PAH.7 The therapeutic effects correlated with decreases in plasma E2 levels. BMPR2 signaling is depressed in lungs from female hypoxic mice and Sugen/hypoxic rats, and anastrozole restores these levels to normal.7 This suggests that pulmonary arterial endogenous E2 in females can depress BMPR2 signaling and may contribute to the development of PAH. This is consistent with E2 suppressing BMPR2 expression via the ERα receptor.4 Compared to those of males, female non-PAH hPASMCs exhibit reduced messenger RNA (mRNA) and protein expression of BMPR2, Smad1, Id1, and Id3.20 Induction of phospho-Smad1/5/8 and Id protein by BMP4 is also reduced in female hPASMCs, and E2 decreased expression of Id genes in male hPASMCs. Under certain conditions, platelet-derived growth factor (PDGF), E2, BMP4, and serotonin induce proliferation only in non-PAH female hPASMCs, consistent with decreased BMPR2 signaling and elevated pERK (phosphorylated extracellular signal-regulated kinase) expression in female hPASMCs.20
E2 metabolism and PAH
Aberrant expression of CYP1B1 is reported in BMPR2 mutation–affected female hPAH patients21,22 and hPASMCs isolated from IPAH patients.23 A CYP1B1 SNP has also been identified that is in tight linkage disequilibrium with SNPs associated with PH, suggesting that these pathways may underpin sexual dimorphism in right ventricular failure.24 CYP1B1 plays a key role in the development of PAH; there is increased pulmonary artery CYP1B1 expression in PAH patients and in animal models of PH.25 CYP1B1 expression is also elevated in female SERT+ mice and demonstrating PH.25 There is a therapeutic effect of a CYP1B1 antagonist in four animal models of PAH: the hypoxic mouse, the Sugen/hypoxic mouse, the SERT+ mouse, and the dexfenfluramine-treated female mouse.25-27 In addition, a CYP1B1 antagonist markedly prolongs survival in the monocrotaline-treated pulmonary hypertensive male rat.27 This confirms that CYP1B1-induced metabolite accumulation likely contributes to the development of PAH. Further, the E2 metabolite 16α-OHE1 is increased in the hypoxic mouse, and 16α-OHE1-induced proliferation is greatest in hPASMCs from PAH patients.25 It has recently been shown that 16α-OHE1 causes proliferation of hPASMCs via the receptor ERα.27 When administered in vivo, 16α-OHE1 also caused PH in mice.25 This is consistent with a role for this metabolite in the development of PAH. Indeed, the 16α-OHE1∶2ME ratio was found to be high in hPAH patients, and 16α-OHE1 suppressed BMP signaling in mice.21,28
High E2 signaling drives penetrance
The finding of an elevated 16α-OHE1∶2ME ratio in patients, including male patients, raises the question of whether this is correlation or causation and, if it is causative, whether the issue is the high level of 16α-OHE1 or the low level of 2ME. In BMPR2-mutant mice, which like PAH patients will spontaneously develop PAH but with reduced penetrance, addition of 16α-OHE1 drives increased penetrance and increased severity of disease, including greater vascular pruning.28 Conversely, increased 2ME is not protective in this model and may slightly increase penetrance.28 This is because 16α-OHE1 is far more estrogenic than 2ME; 16α-OHE1 binds the E2 receptors covalently, whereas 2ME is a weak agonist.29 These data support the hypothesis that in PAH, increased estrogenicity is associated with increased disease penetrance and severity.
Increased E2 signaling drives metabolic defects
The female preponderance of disease has been a bit of a paradox in the past, because in classical rodent models of PAH, such as hypoxia and monocrotaline, E2 was usually published as protective: it is vasodilatory and anti-inflammatory, among other positive effects.30 However, these studies were all carried out in male rodents. The metabolite 16α-OHE1 is also anti-inflammatory in BMPR2-mutant mice;28 however, in addition to its effects in suppressing BMPR2 mentioned above, it appears to independently cause significant alterations in metabolism on many levels.
BMPR2-mutant cells treated with 16α-OHE1 have significantly decreased mobilization of the glucose transporter Glut4 in response to insulin, associated with reductions in Glut4 protein, reductions in lipid transporter CD36 protein, and reductions in the central metabolism regulator PPARγ (peroxisome proliferator–activated receptor gamma) protein, among others.31 This is associated with increased insulin resistance and decreased mitochondrial size in live animals.31 In multiple publications from groups in many different countries, alterations in metabolism have been causally linked to PAH not just in animal models but also in PAH patients.32,33 It is thus plausible that E2’s impact on metabolism is causal for increased penetrance.
Note that these metabolic shifts are probably linked to a proproliferative phenotype. These shifts result in outputs of the Krebs cycle changing from energy to synthetic intermediates, allowing the proliferation to continue. Thus, saying that E2 drives PAH through shifts in metabolism is not at odds with saying that E2 drives PAH through increasing proliferation. However, whether E2 drives proliferation directly in the context of PAH has not been tested; conversely, E2 does appear to directly drive alteration in metabolism, through regulation of miR29 (although possibly through other mechanisms as well).
E2 modulation of metabolism is at least partly through induction of miR29
MicroRNAs (miRs) are short, single-stranded, non–protein-coding gene products, typically 20–22 nucleotides long, that posttranscriptionally regulate the expression of target genes through interactions with specific mRNAs. E2-specific differential regulation of miRs in BMPR2-mutant mice are well matched by differential regulation of miRs in PAH patient lungs, as compared to control lungs or lungs from idiopathic pulmonary fibrosis patients.31In particular, on the basis of miR array data, although multiple miRs related to regulation of metabolism show altered expression in both BMPR2-mutant mice and patients, all three isoforms of the miR29 family are strongly upregulated in both.31
Blocking miR29 with an antagomiR results in rescue of PPARγ protein expression, significant reduction in penetrance of PAH in BMPR2-mutant mice, normalization of insulin resistance, and reduction in accumulation of the toxic lipid ceramide in lung tissues.31 Moreover, in both live mice and tissue culture, blocking miR29 results in normalization of average mitochondrial size, suggesting a return to more normal metabolic function.
Unfortunately, direct regulation of miR29 in patients to correct metabolic problems may have barriers. Although miR29 is strongly induced in PAH patients, it is strongly suppressed in idiopathic pulmonary fibrosis patients, and so correct titration of miR29 levels would likely be critical to its use as a therapeutic tool.
E2 regulation of the 5-hydroxytryptamine 1B receptor is via downregulation of miR96
The 5-hydroxytryptamine 1B (5HT1B) receptor mediates hPASMC proliferation, and miR96 regulates the gene for the 5HT1B receptor. Expression of miR96 is reduced in BMPR2R899X+/− PASMCs from female mice and hPASMCs from female patients with PAH.34 This is associated with increased 5HT1B receptor expression and serotonin-mediated proliferation, and this is driven by E2, which can reduce miR96 expression34 and increase 5HT1B receptor expression. Transfection of precursor miR96 into hPASMCs reduces 5HT1B receptor expression and inhibits serotonin-induced proliferation.34,35 Restoration of miR96 expression in pulmonary arteries in vivo via administration of a miR96 mimic reduces 5HT1B receptor expression and the development of hypoxia-induced PH in the mouse.34
MAPK signaling
The MAPK pathways consist of evolutionarily conserved kinases, including extracellular signal–regulated kinase (ERK1/2), p38 MAPK, and c-Jun NH2-terminal kinase (JNK). These 3-tiered MAPK modules (Fig. 2) receive and integrate extracellular or intracellular stimuli, phosphorylating downstream targets to regulate fundamental cellular processes, including proliferation, differentiation, metabolism, migration, survival, and apoptosis.36,37 Despite an increase in the number of publications describing dysregulated MAPK signaling in PAH, the role of altered MAPK activation in the development and/or progression of PAH is incompletely defined. A better understanding of these complex signaling cascades and their influence on pulmonary vascular remodeling in PAH is necessary to determine whether these pathways are promising therapeutic targets. This section focuses on the roles of Raf/ERK1/2 and p38 MAPK signaling in PAH.
Figure 2.
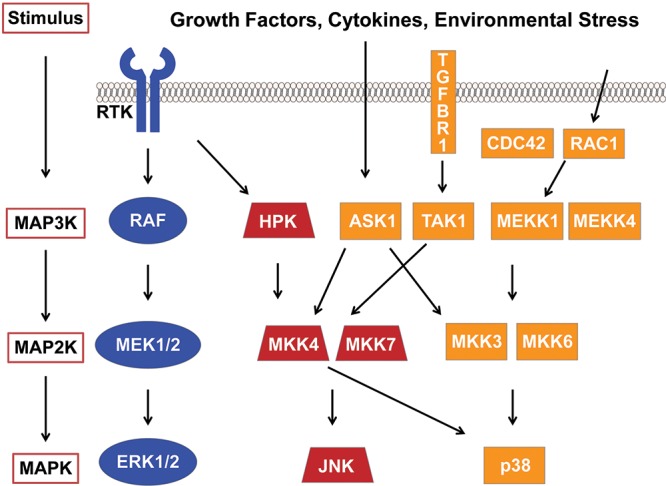
Mitogen-activated protein kinase (MAPK) signaling pathways. Simplified schematic depicting the activation of ERK1/2 (extracellular signal-regulated kinases), JNK (c-Jun NH2-terminal kinase), and p38 MAPK after growth factor–, cytokine-, and/or environmental stress–mediated phosphorylation of MAP3 kinases (MAP3K) followed by the phosphorylation and activation of MAP2 kinases (MAP2K). Growth factor–induced receptor tyrosine kinase (RTK) activation results in the subsequent activation of the Raf/MEK/ERK pathway. Growth factors can also mediate the activation of JNK through hematopoietic progenitor kinase (HPK), which phosphorylates MKK4 (mitogen-activated protein kinase kinase 4). Stress-dependent activation of ASK1 (apoptosis signal regulating kinase 1) also targets the MKK4/JNK pathway in addition to MKK3/p38 MAPK. TGFBR1 (transforming growth factor beta receptor 1)-induced activation of TAK1 (TGF-β-activated kinase 1) and ultraviolet radiation–induced RAC1 and/or CDC42 activation results in the activation of both JNK and p38 MAPK.
Dysregulated Raf/ERK signaling in PAH
ERK1/2 signaling is activated in response to growth factor and mitogen binding to their cognate RTKs, such as vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR), and PDGF receptor (PDGFR). Through parallels with cancer pathobiology, RTKs and their downstream signaling cascades have been linked to pulmonary vascular remodeling in PAH.38-40 More than a decade ago, Tuder et al.3 demonstrated endothelial expression of VEGF and VEGFR within plexogenic lesions in lung specimens from PAH patients. The implications of this finding, however, still remain unclear. Partovian et al.41 reported that overexpressing VEGF protected against hypoxia-induced PAH by improving endothelial function, and Taraseviciene-Stewart et al.42 demonstrated VEGFR blockade in combination with hypoxia-induced PAH in rats. One explanation for this unexpected finding is that pharmacological inhibition of VEGFRs results in upregulation of other RTKs. A detailed review by Voelkel and Gomez-Arroyo43 highlights the complexity of VEGF signaling in PAH. This paradoxical effect of activating RTKs by blocking others may also explain the differences between two PDGFR inhibitors, imatinib and dasatinib. Imatinib, an RTK inhibitor used to treat chronic myelogenous leukemia, reversed both monocrotaline-induced PH and PH induced by chronic hypoxia.38 Phase 244 and 345 trials of imatinib have yielded some evidence of efficacy in PAH but have also raised concerns about adverse effects because of a substantial incidence of subdural hematoma. In stark contrast, precapillary PH has developed in patients receiving dasatinib for imatinib-resistant chronic myelogenous leukemia.46 The profile of targets for dasatinib shows a range of RTKs broader than that for imatinib, including RTKs that regulate focal adhesions and the cytoskeleton.46 These studies highlight the significance of targeting RTKs in PAH, but further study of their effects on the downstream Ras/Raf/MEK signaling pathway may provide the necessary insight to develop safer therapies.
The Ras/Raf/MEK signaling cascade links upstream RTK to ERK1/2 activation47 and is implicated in carcinogenesis, with 30% of all cancers showing dysregulated MAPK signaling due to mutations in Ras and Raf.48 Aberrant ERK activation has been described in the pulmonary vasculature of patients with advanced PAH, implicating upstream activation of Ras/Raf/MEK in disease pathogenesis.49 Evidence that Ras/Raf/MEK activation plays an important role in pulmonary vascular responses to injury/stress comes from Raf-1 kinase inhibitor protein knockout mice that suffer from exaggerated hypoxia-induced PH as a result of the failure to inhibit MEK1/2 phosphorylation by Raf-1.50 The identification of Raf-1 gain-of-function mutations (p.Ser257Leu) leading to constitutive activation of the Raf/MEK/ERK1/2 pathway51 in 2 infants who developed severe PAH further implicates Raf/ERK dysregulation in PAH.52
Loss-of-function mutations in BMPR2 are the most common genetic cause of PAH, accounting for 70% of familial cases53 and 10%–40% of IPAH.54,55 Despite variable impact on canonical BMP/Smad signaling, PAH-associated BMPR2 mutations uniformly activate MAPK pathways.49,56 Importantly, BMPR2 expression is markedly reduced in end-stage PAH, even in patients not harboring mutations,57 underscoring the relevance of BMPR2-mediated regulation of MAPK signaling to the development of PAH. In BMPR2-silenced pulmonary artery endothelial cells (PAECs), we reported constitutive activation of Raf family members as well as ERK1/2. Two Raf inhibitors, sorafenib and AZ628, and a triple angiokinase inhibitor, nintedanib, decreased ERK1/2 activation and reversed the abnormal proliferation and hypermotility of BMPR2 deficiency,58 suggesting that inhibition of dysregulated Ras/Raf/ERK signaling may be useful in reversing vascular remodeling in PAH.
p38 MAPK contributes to the hyperproliferative and inflammatory PAH phenotype
Environmental stressors, as well as inflammatory cytokines, activate the p38 MAPK pathway through phosphorylation by MKK3 and MKK6. Independent of MAPK kinases, p38 MAPK is also activated through a direct interaction with TAB1 (TAK-1 binding protein)59 and subsequent phosphorylation by AMPK (AMP-activated protein kinase).60 Activation of p38 MAPK has been shown to have detrimental effects on endothelial function and is linked to many diseases, including cardiac hypertrophy61 and heart failure.62 In PAH, Rudarakanchana et al.56 demonstrated that the overexpression of BMPR2 mutants resulted in increased proliferation mediated by p38 MAPK activation. The increased growth of SMCs derived from PAH patients was reported by Wilson et al.63 to be caused by the activation of p38 MAPK and JNK. Serotonin, a potent mitogen implicated in the pathogenesis of PAH,64 activated p38 MAPK and increased the proliferation of pulmonary artery fibroblasts exposed to hypoxic conditions.65 Pharmacological inhibition of p38 MAPK in rat pulmonary artery fibroblasts exposed to chronic hypoxia66 or in the monocrotaline rat model of PH67 blocked hypoxia-induced proliferation.
In addition to its proproliferative effects, p38 MAPK activation also triggers inflammatory signaling cascades. Inflammation is increasingly recognized as playing a central role in PAH pathobiology.68-70 In IPAH, serum levels of the inflammatory mediator inerleukin-6 (IL-6) were significantly higher than those in controls,71 and mice deficient in IL-6 exposed to hypoxia showed decreased SMC migration and macrophage recruitment in the lungs compared to wild-type mice.72 Inhibition of p38 MAPK decreases IL-6 expression and reverses both chronic hypoxia– and monocrotaline-induced PAH, indicating that pulmonary vascular remodeling is due partly to p38 MAPK activation.67
Importantly, dysregulated MAPK signaling has been observed in various experimental models of PAH and in PASMCs from patients with BMPR2 mutations and is a prominent consequence of BMPR2 silencing in PAECs. Taken together, there is mounting evidence suggesting a central role for MAPK signaling in the proliferative and inflammatory PAH phenotype. A better understanding of these signaling pathways is essential for the design of safer and more effective therapies.
Wnt signaling pathways: key players in the preservation of pulmonary vascular homeostasis
Similar to the p38 signaling pathways, Wnt pathways are responsible for regulating baseline cellular activities involved in the preservation of homeostasis and in response to environmental stressors.73 Wnt signaling is the term used to describe a superfamily of evolutionarily conserved signaling pathways whose activity is indispensable for proper embryonic development and regeneration of damaged tissues in postnatal life. A common thread in Wnt signaling is the interaction of Wnt ligands with surface receptors that trigger unique signaling events with specific biological consequences related to the specific Wnt pathway involved. The best-characterized (i.e., “canonical”) of the Wnt signaling pathways is the Wnt/β-catenin (βC) pathway, known for targeting βC, a dynamic cytoplasmic protein that responds to ligand-receptor binding by relocating to the nucleus and modulating gene expression (Fig. 3A). In steady state, βC is bound to a cytoplasmic complex composed of axin, adenomatous poliposis coli (APC), and glycogen synthase kinase 3β (GSK3β), responsible for directing βC away from the nucleus and into proteasomal complexes.74 However, when Wnt ligands bind to a receptor complex composed of a member of the frizzled (FZD) family of receptors and the low-density lipoprotein receptor–related protein (LRP) 5/6, the cytoplasmic protein disheveled (Dvl) is activated, which then shuts down the βC degradation complex. Once released from the complex, βC translocates to the nucleus, where it targets the transcription of genes responsible for controlling cell fate, proliferation, and survival.75 Gain or loss of Wnt/βC activity is a feature of a wide range of clinical diseases, such as cancer, stroke, and heart failure; this has led to ongoing efforts to exploit opportunities to develop Wnt-based biomarkers and therapeutic strategies for many of these diseases.76,77
Figure 3.
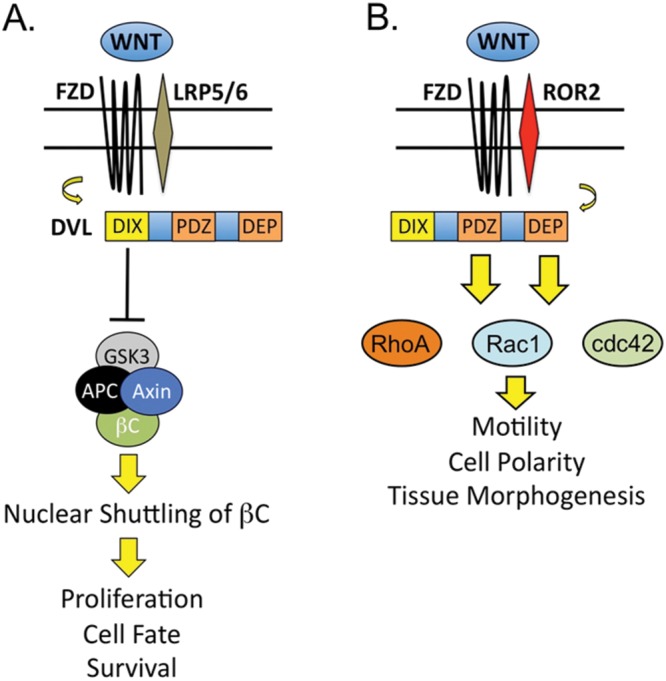
The Wnt signaling pathways regulate gene expression and cell movements. The Wnt/βC pathway (A) controls gene expression through DVL-dependent activation and nuclear shuttling of βC, while Wnt/PCP (B) modulates motility, polarity, and tissue morphogenesis thru DVL activation of RhoA/Rac1/cdc42. Specific domains within Dvl are responsible for differentia activation of βC (DIX) or PCP (PDZ/DEP). βC: β-catenin; DVL: protein disheveled; FZD: frizzled family of receptors; GSK3: glycogen synthase kinase 3; LRP: low-density lipoprotein receptor–related protein; PCP: planar cell polarity; ROR2: receptor tyrosine kinase-like orphan receptor 2.
Besides the Wnt/βC pathway, there are other Wnt pathways that have more specialized biological functions and whose activation is independent of βC accumulation. Among these “noncanonical” Wnt pathways, Wnt/PCP signaling is responsible for regulating cell movements during tissue morphogenesis. In mammals, activation of Wnt/PCP is necessary for neural-tube closure, and mutations that disrupt this event result in neural-tube defects, such as craniorachischisis and spina bifida.78 In contrast to Wnt/βC, recruitment of other coreceptors, such as receptor tyrosine kinase-like orphan receptor 2 (ROR2), is necessary to activate the pathway (Fig. 3B). Once again, the protein Dvl is activated, but in this case, it is responsible for activating the small GTPases Rac, Rho, and cdc42, which provoke cytoskeletal rearrangements that allow cells to navigate their immediate environment and synchronize their movements to those of other cells within a specific tissue plane.79,80 Like that of Wnt/βc, dysregulation of Wnt/PCP is a feature of various disorders, such as cancer, where it appears to contribute to progression and metastasis, thus making Wnt/PCP an attractive field for the development of novel therapeutics.
A study examining the expression profile of laser-microdissected plexiform lesions demonstrated significant upregulation of PCP pathway genes such as Wnt11, Rho, and Dvl.81 This was the first indication that Wnt signaling may play a role in PAH. A signaling crosstalk between the BMP and Wnt signaling pathways was shown to be partially responsible for the protective effects of BMP on the pulmonary endothelium. BMP2 triggers βC activation independent of Wnt ligand stimulation to upregulate genes such as VEGF-A, survivin, and cyclin D1 and induce proliferation and survival of PAECs. Downregulation of BMPR2 resulted in loss of βC activation, which significantly compromised the capacity of BMP2 to protect the cell against serum starvation and impair regeneration of PAECs lost to apoptosis. Concomitant to βC activation, BMPR2 also triggers PAEC motility in response to BMP2 by downstream activation of RhoA and Rac1, the signaling mediators of the Wnt/PCP pathway. Compromising activation of either βC or RhoA/Rac1 in PAECs, in an in-vivo matrigel plug angiogenesis model, prevented BMP2-induced angiogenesis, leading to a significant reduction of functional blood vessels.82 These observations suggest that concomitant activation of both Wnt signaling pathways is necessary for preservation of pulmonary vascular homeostasis and that loss of either Wnt pathway could contribute to PAH by limiting the extent of vascular regeneration in response to injury.
Lung pathology has shown that vascular remodeling is in great part caused by accumulation of PASMC-like cells in the neointima and media of small pulmonary arteries. BMP2 also activates both Wnt/βC and Wnt/PCP in PASMCs, but the order in which the pathways are activated differs from that observed in PAECs. As its final outcome, this chain of events can, in response to a vascular insult, mobilize PASMCs to reach the damage zone and aid in tissue repair while it prevents their accumulation by suppressing βC-dependent growth. The consequences of interfering with this protective signaling mechanism were shown in an in-vivo carotid stent model, where a mutant Dvl construct was found to result in severe neointima formation and medial thickening from PASMC accumulation.10 Consistent with this, PAH PASMCs (isolated from human PAH lungs obtained through the Pulmonary Hypertension Breakthrough Initiative) demonstrate increased baseline levels of active βC that correlated with their known proproliferative and apoptosis-resistant phenotype, and reduction of endogenous βC levels with small interfering RNA partially normalized growth and survival response. Intriguingly, application of Wnt5a (a ligand known to trigger Wnt/PCP signaling) was able to suppress βC levels in healthy PASMCs but not in PAH PASMCs.83 This suggests the existence of a crosstalk between Wnt/βC and Wnt/PCP that is responsible for appropriate PASMC response to injury regulation, which could be dysfunctional in PAH and contribute to the aberrant behavior of PASMCs in the disease.
More recently, a key role has been identified for Wnt/PCP in the assembly of the pulmonary microcirculation, orchestrating the establishment of endothelial-pericyte interactions during late angiogenesis. Pericytes are mural cells that provide structural support to capillaries and act as a source of cytokines that enhance endothelial viability and vessel maturation; absence of appropriate pericyte coverage can result in capillary fragility, endothelial cell apoptosis, and loss of microvessels.84-86 With matrigel-based coculture assays, it was shown that pericytes purified from explanted PAH lungs fail to reach healthy PAECs during tube assembly, resulting in tube networks smaller than healthy donor samples. In addition, Wnt/PCP genes frizzled 7 (Fzd7) and CDC42 were significantly downregulated in PAH pericytes and correlated with lower response to Wnt5a stimulation. Knockdown of both Fzd7 and CDC42 in healthy lung pericytes reproduced the abnormal PAH pericyte phenotype both in vitro and in an in-vivo matrigel plug assay, whereas restoring their expression in PAH pericytes resulted in increased association with endothelial cells and larger tube networks.87
Taken together, these studies suggest a model in which both Wnt/βC and Wnt/PCP participate in regeneration of pulmonary microvessels by promoting angiogenesis in response to injury (Fig. 4). The data suggest that gene mutations in BMPR2 and dysregulation of Wnt gene expression can contribute to impaired angiogenesis, and disruption could serve as future therapeutic targets for addressing defective angiogenesis and abnormal vascular remodeling in PAH.
Figure 4.

Proposed role of Wnt signaling in pulmonary vascular homeostasis. A, Wnt/βC and Wnt/PCP coordinate endothelial growth and movement during vascular repair in response to injury. B, Loss of Wnt signaling results in impaired angiogenesis and is permissive to PASMC-driven obliterative vasculopathy. βC: β-catenin; PASMC: pulmonary artery smooth muscle cell; PCP: planar cell polarity.
Summary
This review discusses novel signaling pathways in PAH, including estrogenic, MAPK, and Wnt signaling pathways. Female sex and sex hormones are risk factors for developing PAH. Endogenous E2 and E2 metabolites are pathogenic in the pulmonary circulation, and regulation may be controlled by miRAs. The metabolite 16α-OHE1 may regulate miR29, which modulates energy metabolism. E2 can decrease miR96 expression, leading to upregulation of the 5HT1B receptor, increasing serotonin-induced proliferation in female PASMCs. E2 can also inhibit BMPR2 expression and BMP signaling. Overall, in both humans and genetic mouse models of PAH, higher E2 signaling results in increased pulmonary vascular injury and higher PAH penetrance. These data suggest that inhibition of estrogenic signaling, either directly or through targeting intermediates, may be a viable therapeutic strategy in PAH.
Other viable therapeutic targets in PAH include MAPK and Wnt signaling pathways. Targeting these pathways may decrease proliferation, inflammation, and angiogenesis. The constitutive activation of Raf/ERK and p38 MAPK pathways results in increased proliferation of pulmonary cells and inflammation. Although inhibition of these pathways in animal models demonstrates some reversal of pulmonary hypertension, phase 2 and 3 trials of imatinib reported severe adverse side effects due to off-target effects. Dysregulation of Wnt pathways may contribute to pathogenesis in PAH, cancer, and heart failure. The Wnt pathway influences BMP2-induced angiogenesis, and loss of Wnt signaling results in severe pulmonary neointima formation, medial thickening, and impaired pulmonary vascular regeneration in response to injury. A clearer understanding of these intricate pathways will help in the development of safer and more effective therapies for PAH.
Acknowledgments
The opinions expressed in this article are the authors’ own and do not represent any position or policy of the National Institutes of Health, the Department of Health and Human Services, or the United States Government.
Source of Support: This research was supported in part by the Intramural Research Program of the National Institutes of Health Clinical Center.
Conflict of Interest: None declared.
References
- 1.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 2004;43(12 suppl):S13–S24. [DOI] [PubMed]
- 2.Cool CD, Stewart JS, Werahera P, Miller GJ, Williams RL, Voelkel NF, Tuder RM. Three-dimensional reconstruction of pulmonary arteries in plexiform pulmonary hypertension using cell-specific markers: evidence for a dynamic and heterogeneous process of pulmonary endothelial cell growth. Am J Pathol 1999;155(2):411–419. [DOI] [PMC free article] [PubMed]
- 3.Tuder RM, Chacon M, Alger L, Wang J, Taraseviciene-Stewart L, Kasahara Y, Cool CD, et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J Pathol 2001;195(3):367–374. [DOI] [PubMed]
- 4.Austin ED, Hamid R, Hemnes AR, Loyd JE, Blackwell T, Yu C, Phillips JA III, et al. BMPR2 expression is suppressed by signaling through the estrogen receptor. Biol Sex Differ 2012;3(1):6. doi:10.1186/2042-6410-3-6. [DOI] [PMC free article] [PubMed]
- 5.Lahm T, Crisostomo PR, Markel TA, Wang M, Wang Y, Weil B, Meldrum DR. Exogenous estrogen rapidly attenuates pulmonary artery vasoreactivity and acute hypoxic pulmonary vasoconstriction. Shock 2008;30(6):660–667. [DOI] [PubMed]
- 6.Tofovic SP. Estrogens and development of pulmonary hypertension: interaction of estradiol metabolism and pulmonary vascular disease. J Cardiovasc Pharmacol 2010;56(6):696–708. [DOI] [PMC free article] [PubMed]
- 7.Mair KM, Wright AF, Duggan N, Rowlands DJ, Hussey MJ, Roberts S, Fullerton J, et al. Sex-dependent influence of endogenous estrogen in pulmonary hypertension. Am J Respir Crit Care Med 2014;190(4):456–467. [DOI] [PMC free article] [PubMed]
- 8.McGoon MD, Benza RL, Escribano-Subias P, Jiang X, Miller DP, Peacock AJ, Pepke-Zaba J, et al. Pulmonary arterial hypertension: epidemiology and registries. J Am Coll Cardiol 2013;62(25 suppl):D51–D59. [DOI] [PubMed]
- 9.Hoeper MM, Huscher D, Ghofrani HA, Delcroix M, Distler O, Schweiger C, Grünig E, et al. Elderly patients diagnosed with idiopathic pulmonary arterial hypertension: results from the COMPERA registry. Int J Cardiol 2013;168(2):871–880. [DOI] [PubMed]
- 10.de Jesus Perez VA, Ali Z, Alastalo TP, Ikeno F, Sawada H, Lai YJ, Kleisli T, et al. BMP promotes motility and represses growth of smooth muscle cells by activation of tandem Wnt pathways. J Cell Biol 2011;192(1):171–188. [DOI] [PMC free article] [PubMed]
- 11.Shapiro S, Traiger GL, Turner M, McGoon MD, Wason P, Barst RJ. Sex differences in the diagnosis, treatment, and outcome of patients with pulmonary arterial hypertension enrolled in the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Chest 2012;141(2):363–373. [DOI] [PubMed]
- 12.Morse JH, Jones AC, Barst RJ, Hodge SE, Wilhelmsen KC, Nygaard TG. Familial primary pulmonary hypertension locus mapped to chromosome 2q31–q32. Chest 1998;114(1 suppl):57S–58S. [DOI] [PubMed]
- 13.Nichols WC, Koller DL, Slovis B, Foroud T, Terry VH, Arnold ND, Siemieniak DR, et al. Localization of the gene for familial primary pulmonary hypertension to chromosome 2q31–32. Nat Genet 1997;15(3):277–280. [DOI] [PubMed]
- 14.Loyd JE, Butler MG, Foroud TM, Conneally PM, Phillips JA III, Newman JH. Genetic anticipation and abnormal gender ratio at birth in familial primary pulmonary hypertension. Am J Respir Crit Care Med 1995;152(1):93–97. [DOI] [PMC free article] [PubMed]
- 15.Yang X, Long L, Southwood M, Rudarakanchana N, Upton PD, Jeffery TK, Atkinson C, Chen H, Trembath RC, Morrell NW. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ Res 2005;96(10):1053–1063. [DOI] [PubMed]
- 16.Ventetuolo CE, Baird GL, Barr RG, Bluemke DA, Fritz JS, Hill NS, Klinger JR, et al. Higher estradiol and lower dehydroepiandrosterone-sulfate levels are associated with pulmonary arterial hypertension in men. Am J Respir Crit Care Med 2016;193(10):1168–1175. [DOI] [PMC free article] [PubMed]
- 17.Simpson ER, Clyne C, Rubin G, Boon WC, Robertson K, Britt K, Speed C, Jones M. Aromatase—a brief overview. Annu Rev Physiol 2002;64:93–127. [DOI] [PubMed]
- 18.Wright AF, Ewart MA, Mair K, Nilsen M, Dempsie Y, Loughlin L, Maclean MR. Oestrogen receptor alpha in pulmonary hypertension. Cardiovasc Res 2015;106(2):206–216. [DOI] [PMC free article] [PubMed]
- 19.Roberts KE, Fallon MB, Krowka MJ, Brown RS, Trotter JF, Peter I, Tighiouart H, et al. Genetic risk factors for portopulmonary hypertension in patients with advanced liver disease. Am J Respir Crit Care Med 2009;179(9):835–842. [DOI] [PMC free article] [PubMed]
- 20.Mair KM, Yang XD, Long L, White K, Wallace E, Ewart MA, Docherty CK, Morrell NW, MacLean MR. Sex affects bone morphogenetic protein type II receptor signaling in pulmonary artery smooth muscle cells. Am J Respir Crit Care Med 2015;191(6):693–703. [DOI] [PMC free article] [PubMed]
- 21.Austin ED, Cogan JD, West JD, Hedges LK, Hamid R, Dawson EP, Wheeler LA, Parl FF, Loyd JE, Phillips JA III. Alterations in oestrogen metabolism: implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur Respir J 2009;34(5):1093–1099. [DOI] [PMC free article] [PubMed]
- 22.West J, Cogan J, Geraci M, Robinson L, Newman J, Phillips JA, Lane K, Meyrick B, Loyd J. Gene expression in BMPR2 mutation carriers with and without evidence of pulmonary arterial hypertension suggests pathways relevant to disease penetrance. BMC Med Genomics 2008;1:45. doi:10.1186/1755-8794-1-45. [DOI] [PMC free article] [PubMed]
- 23.White K, Loughlin L, Maqbool Z, Nilsen M, McClure J, Dempsie Y, Baker AH, MacLean MR. Serotonin transporter, sex, and hypoxia: microarray analysis in the pulmonary arteries of mice identifies genes with relevance to human PAH. Physiol Genomics 2011;43(8):417–437. [DOI] [PMC free article] [PubMed]
- 24.Ventetuolo CE, Mitra N, Wan F, Manichaikul A, Barr RG, Johnson C, Bluemke DA, et al. Oestradiol metabolism and androgen receptor genotypes are associated with right ventricular function. Eur Respir J 2016;47(2):553–563. [DOI] [PMC free article] [PubMed]
- 25.White K, Johansen AK, Nilsen M, Ciuclan L, Wallace E, Paton L, Campbell A, et al. Activity of the estrogen-metabolizing enzyme cytochrome P450 1B1 influences the development of pulmonary arterial hypertension. Circulation 2012;126(9):1087–1098. [DOI] [PubMed]
- 26.Dempsie Y, MacRitchie NA, White K, Morecroft I, Wright AF, Nilsen M, Loughlin L, Mair KM, MacLean MR. Dexfenfluramine and the oestrogen-metabolizing enzyme CYP1B1 in the development of pulmonary arterial hypertension. Cardiovasc Res 2013;99(1):24–34. [DOI] [PMC free article] [PubMed]
- 27.Johansen AK, Dean A, Morecroft I, Hood K, Nilsen M, Loughlin L, Anagnostopoulou A, Touyz RM, White K, MacLean MR. Interaction between bone morphogenetic protein receptor type 2 and estrogenic compounds in pulmonary arterial hypertension. Pulm Circ 2016;6(1):82–92. [DOI] [PMC free article] [PubMed]
- 28.Fessel JP, Chen X, Frump A, Gladson S, Blackwell T, Kang C, Johnson J, et al. Interaction between bone morphogenetic protein receptor type 2 and estrogenic compounds in pulmonary arterial hypertension. Pulm Circ 2013;3(3):564–577. [DOI] [PMC free article] [PubMed]
- 29.Swaneck GE, Fishman J. Covalent binding of the endogenous estrogen 16α-hydroxyestrone to estradiol receptor in human breast cancer cells: characterization and intranuclear localization. Proc Natl Acad Sci USA 1988;85(21):7831–7835. [DOI] [PMC free article] [PubMed]
- 30.Lahm T, Albrecht M, Fisher AJ, Selej M, Patel NG, Brown JA, Justice MJ, et al. 17β-estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am J Respir Crit Care Med 2012;185(9):965–980. [DOI] [PMC free article] [PubMed]
- 31.Chen X, Talati M, Fessel JP, Hemnes AR, Gladson S, French J, Shay S, et al. Estrogen metabolite 16α-hydroxyestrone exacerbates bone morphogenetic protein receptor type II-associated pulmonary arterial hypertension through microRNA-29-mediated modulation of cellular metabolism. Circulation 2016;133(1):82–97. [DOI] [PMC free article] [PubMed]
- 32.Fessel JP, West JD. Redox biology in pulmonary arterial hypertension (2013 Grover Conference Series). Pulm Circ 2015;5(4):599–609. [DOI] [PMC free article] [PubMed]
- 33.Ryan JJ, Archer SL. Emerging concepts in the molecular basis of pulmonary arterial hypertension: part I: metabolic plasticity and mitochondrial dynamics in the pulmonary circulation and right ventricle in pulmonary arterial hypertension. Circulation 2015;131(19):1691–1702. [DOI] [PMC free article] [PubMed]
- 34.Wallace E, Morrell NW, Yang XD, Long L, Stevens H, Nilsen M, Loughlin L, Mair KM, Baker AH, MacLean MR. A sex-specific microRNA-96/5-hydroxytryptamine 1B axis influences development of pulmonary hypertension. Am J Respir Crit Care Med 2015;191(12):1432–1442. [DOI] [PMC free article] [PubMed]
- 35.White K, Dempsie Y, Nilsen M, Wright AF, Loughlin L, MacLean MR. The serotonin transporter, gender, and 17β oestradiol in the development of pulmonary arterial hypertension. Cardiovasc Res 2011;90(2):373–382. [DOI] [PubMed]
- 36.Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev 2004;68(2):320–344. [DOI] [PMC free article] [PubMed]
- 37.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene 2007;26(22):3279–3290. [DOI] [PubMed]
- 38.Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest 2005;115(10):2811–2821. [DOI] [PMC free article] [PubMed]
- 39.Sakao S, Taraseviciene-Stewart L, Cool CD, Tada Y, Kasahara Y, Kurosu K, Tanabe N, et al. VEGF-R blockade causes endothelial cell apoptosis, expansion of surviving CD34+ precursor cells and transdifferentiation to smooth muscle-like and neuronal-like cells. FASEB J 2007;21(13):3640–3652. [DOI] [PubMed]
- 40.Izikki M, Guignabert C, Fadel E, Humbert M, Tu L, Zadigue P, Dartevelle P, et al. Endothelial-derived FGF2 contributes to the progression of pulmonary hypertension in humans and rodents. J Clin Invest 2009;119(3):512–523. [DOI] [PMC free article] [PubMed]
- 41.Partovian C, Adnot S, Raffestin B, Louzier V, Levame M, Mavier IM, Lemarchand P, Eddahibi S. Adenovirus-mediated lung vascular endothelial growth factor overexpression protects against hypoxic pulmonary hypertension in rats. Am J Respir Cell Mol Biol 2000;23(6):762–771. [DOI] [PubMed]
- 42.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, McMahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J 2001;15(2):427–438. [DOI] [PubMed]
- 43.Voelkel NF, Gomez-Arroyo J. The role of vascular endothelial growth factor in pulmonary arterial hypertension: the angiogenesis paradox. Am J Respir Cell Mol Biol 2014;51(4):474–484. [DOI] [PubMed]
- 44.Ghofrani HA, Morrell NW, Hoeper MM, Olschewski H, Peacock AJ, Barst RJ, Shapiro S, et al. Imatinib in pulmonary arterial hypertension patients with inadequate response to established therapy. Am J Respir Crit Care Med 2010;182(9):1171–1177. [DOI] [PMC free article] [PubMed]
- 45.Hoeper MM, Barst RJ, Bourge RC, Feldman J, Frost AE, Galiè N, Gómez-Sánchez MA, et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation 2013;127(10):1128–1138. [DOI] [PubMed]
- 46.Montani D, Bergot E, Günther S, Savale L, Bergeron A, Bourdin A, Bouvaist H, et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation 2012;125(17):2128–2137. [DOI] [PubMed]
- 47.McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene 2007;26(22):3113–3121. [DOI] [PubMed]
- 48.Gollob JA, Wilhelm S, Carter C, Kelley SL. Role of Raf kinase in cancer: therapeutic potential of targeting the Raf/MEK/ERK signal transduction pathway. Semin Oncol 2006;33(4):392–406. [DOI] [PubMed]
- 49.Lane KB, Blackwell TR, Runo J, Wheeler L, Phillips JA III, Loyd JE. Aberrant signal transduction in pulmonary hypertension. Chest 2005;128(6 suppl):564S–565S. [DOI] [PubMed]
- 50.Morecroft I, Doyle B, Nilsen M, Kolch W, Mair K, Maclean MR. Mice lacking the Raf-1 kinase inhibitor protein exhibit exaggerated hypoxia-induced pulmonary hypertension. Br J Pharmacol 2011;163(5):948–963. [DOI] [PMC free article] [PubMed] [Retracted]
- 51.Razzaque MA, Nishizawa T, Komoike Y, Yagi H, Furutani M, Amo R, Kamisago M, et al. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet 2007;39(8):1013–1017. [DOI] [PubMed]
- 52.Hopper RK, Feinstein JA, Manning MA, Benitz W, Hudgins L. Neonatal pulmonary arterial hypertension and Noonan syndrome: two fatal cases with a specific RAF1 mutation. Am J Med Genet A 2015;167(4):882–885. [DOI] [PubMed]
- 53.Machado RD, Aldred MA, James V, Harrison RE, Patel B, Schwalbe EC, Grünig E, et al. Mutations of the TGF-β type II receptor BMPR2 in pulmonary arterial hypertension. Hum Mutat 2006;27(2):121–132. [DOI] [PubMed]
- 54.Thomson JR, Machado RD, Pauciulo MW, Morgan NV, Humbert M, Elliott GC, Ward K, et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-β family. J Med Genet 2000;37(10):741–745. [DOI] [PMC free article] [PubMed]
- 55.Evans JD, Girerd B, Montani D, Wang XJ, Galiè N, Austin ED, Elliott G, et al. BMPR2 mutations and survival in pulmonary arterial hypertension: an individual participant data meta-analysis. Lancet Respir Med 2016;4(2):129–137. [DOI] [PMC free article] [PubMed]
- 56.Rudarakanchana N, Flanagan JA, Chen H, Upton PD, Machado R, Patel D, Trembath RC, Morrell NW. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet 2002;11(13):1517–1525. [DOI] [PubMed]
- 57.Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, Morrell NW. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 2002;105(14):1672–1678. [DOI] [PubMed]
- 58.Awad KS, Elinoff JM, Wang S, Gairhe S, Ferreyra GA, Cai R, Sun J, Solomon MA, Danner RL. Raf/ERK drives the proliferative and invasive phenotype of BMPR2-silenced pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol 2016;310(2):L187–L201. [DOI] [PMC free article] [PubMed]
- 59.Ge B, Gram H, Di Padova F, Huang B, New L, Ulevitch RJ, Luo Y, Han J. MAPKK-independent activation of p38α mediated by TAB1-dependent autophosphorylation of p38α. Science 2002;295(5558):1291–1294. [DOI] [PubMed]
- 60.Li J, Miller EJ, Ninomiya-Tsuji J, Russell RR III, Young LH. AMP-activated protein kinase activates p38 mitogen-activated protein kinase by increasing recruitment of p38 MAPK to TAB1 in the ischemic heart. Circ Res 2005;97(9):872–879. [DOI] [PubMed]
- 61.Behr TM, Nerurkar SS, Nelson AH, Coatney RW, Woods TN, Sulpizio A, Chandra S, et al. Hypertensive end-organ damage and premature mortality are p38 mitogen-activated protein kinase-dependent in a rat model of cardiac hypertrophy and dysfunction. Circulation 2001;104(11):1292–1298. [DOI] [PubMed]
- 62.Widder J, Behr T, Fraccarollo D, Hu K, Galuppo P, Tas P, Angermann CE, Ertl G, Bauersachs J. Vascular endothelial dysfunction and superoxide anion production in heart failure are p38 MAP kinase-dependent. Cardiovasc Res 2004;63(1):161–167. [DOI] [PubMed]
- 63.Wilson JL, Yu J, Taylor L, Polgar P. Hyperplastic growth of pulmonary artery smooth muscle cells from subjects with pulmonary arterial hypertension is activated through JNK and p38 MAPK. PloS ONE 2015;10:e0123662. doi:10.1371/journal.pone.0123662. [DOI] [PMC free article] [PubMed]
- 64.MacLean MR, Deuchar GA, Hicks MN, Morecroft I, Shen S, Sheward J, Colston J, et al. Overexpression of the 5-hydroxytryptamine transporter gene: effect on pulmonary hemodynamics and hypoxia-induced pulmonary hypertension. Circulation 2004;109(17):2150–2155. [DOI] [PubMed]
- 65.Welsh DJ, Harnett M, MacLean M, Peacock AJ. Proliferation and signaling in fibroblasts: role of 5-hydroxytryptamine2A receptor and transporter. Am J Respir Crit Care Med 2004;170(3):252–259. [DOI] [PubMed]
- 66.Welsh DJ, Peacock AJ, MacLean M, Harnett M. Chronic hypoxia induces constitutive p38 mitogen-activated protein kinase activity that correlates with enhanced cellular proliferation in fibroblasts from rat pulmonary but not systemic arteries. Am J Respir Crit Care Med 2001;164(2):282–289. [DOI] [PubMed]
- 67.Church AC, Martin DH, Wadsworth R, Bryson G, Fisher AJ, Welsh DJ, Peacock AJ. The reversal of pulmonary vascular remodeling through inhibition of p38 MAPK-alpha: a potential novel anti-inflammatory strategy in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2015;309(4):L333–L347. [DOI] [PMC free article] [PubMed]
- 68.Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, Jessup M, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;186(3):261–272. [DOI] [PMC free article] [PubMed]
- 69.Savai R, Pullamsetti SS, Kolbe J, Bieniek E, Voswinckel R, Fink L, Scheed A, et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;186(9):897–908. [DOI] [PubMed]
- 70.Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, Trembath RC, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010;122(9):920–927. [DOI] [PubMed]
- 71.Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, Duroux P, Galanaud P, Simonneau G, Emilie D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med 1995;151(5):1628–1631. [DOI] [PubMed]
- 72.Savale L, Tu L, Rideau D, Izziki M, Maitre B, Adnot S, Eddahibi S. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir Res 2009;10:6. doi:10.1186/1465-9921-10-6. [DOI] [PMC free article] [PubMed]
- 73.de Jesus Perez V, Yuan K, Alastalo TP, Spiekerkoetter E, Rabinovitch M. Targeting the Wnt signaling pathways in pulmonary arterial hypertension. Drug Discov Today 2014;19(8):1270–1276. [DOI] [PMC free article] [PubMed]
- 74.Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Regulation of intracellular β-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci USA 1995;92(7):3046–3050. [DOI] [PMC free article] [PubMed]
- 75.Papkoff J, Rubinfeld B, Schryver B, Polakis P. Wnt-1 regulates free pools of catenins and stabilizes APC-catenin complexes. Mol Cell Biol 1996;16(5):2128–2134. [DOI] [PMC free article] [PubMed]
- 76.Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell 2012;149(6):1192–1205. [DOI] [PubMed]
- 77.Leucht P, Minear S, Ten Berge D, Nusse R, Helms JA. Translating insights from development into regenerative medicine: the function of Wnts in bone biology. Semin Cell Dev Biol 2008;19(5):434–443. [DOI] [PubMed]
- 78.Ueno N, Greene ND. Planar cell polarity genes and neural tube closure. Birth Defects Res Pt C 2003;69(4):318–324. [DOI] [PubMed]
- 79.Shulman JM, Perrimon N, Axelrod JD. Frizzled signaling and the developmental control of cell polarity. Trends Genet 1998;14(11):452–458. [DOI] [PubMed]
- 80.Veeman MT, Axelrod JD, Moon RT. A second canon: functions and mechanisms of β-catenin-independent Wnt signaling. Dev Cell 2003;5(3):367–377. [DOI] [PubMed]
- 81.Laumanns IP, Fink L, Wilhelm J, Wolff JC, Mitnacht-Kraus R, Graef-Hoechst S, Stein MM, et al. The noncanonical WNT pathway is operative in idiopathic pulmonary arterial hypertension. Am J Respir Cell Mol Biol 2009;40(6):683–691. [DOI] [PubMed]
- 82.de Jesus Perez VA, Alastalo TP, Wu JC, Axelrod JD, Cooke JP, Amieva M, Rabinovitch M. Bone morphogenetic protein 2 induces pulmonary angiogenesis via Wnt-β-catenin and Wnt-RhoA-Rac1 pathways. J Cell Biol 2009;184(1):83–99. [DOI] [PMC free article] [PubMed]
- 83.Takahashi J, Orcholski M, Yuan K, de Jesus Perez V. PDGF-dependent β-catenin activation is associated with abnormal pulmonary artery smooth muscle cell proliferation in pulmonary arterial hypertension. FEBS Lett 2016;590(1):101–109. [DOI] [PMC free article] [PubMed]
- 84.Fuxe J, Tabruyn S, Colton K, Zaid H, Adams A, Baluk P, Lashnits E, et al. Pericyte requirement for anti-leak action of angiopoietin-1 and vascular remodeling in sustained inflammation. Am J Pathol 2011;178(6):2897–2909. [DOI] [PMC free article] [PubMed]
- 85.Patel MS, Taylor GP, Bharya S, Al-Sanna’a N, Adatia I, Chitayat D, Lewis ME, Human DG. Abnormal pericyte recruitment as a cause for pulmonary hypertension in Adams-Oliver syndrome. Am J Med Genet A 2004;129(3):294–299. [DOI] [PubMed]
- 86.Díaz-Flores L, Gutiérrez R, Madrid JF, Varela H, Valladares F, Acosta E, Martin-Vasallo P, Díaz-Flores L Jr. Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol Histopathol 2009;24(7):909–969. [DOI] [PubMed]
- 87.Yuan K, Orcholski ME, Panaroni C, Shuffle EM, Huang NF, Jiang X, Tian W, et al. Activation of the Wnt/planar cell polarity pathway is required for pericyte recruitment during pulmonary angiogenesis. Am J Pathol 2015;185(1):69–84. [DOI] [PMC free article] [PubMed]