

Abstract Abstract
Congenital heart disease (CHD) is a frequent cause of pediatric pulmonary arterial hypertension (PAH), with diverse etiology and outcome. We aimed to describe phenotypic heterogeneity in pediatric PAH associated with CHD (PAH-CHD), assess the applicability of the Nice CHD classification, and explore whether this classification accurately reflects patient/disease characteristics and survival. All children with CHD from a contemporary cohort of consecutive pediatric PAH patients followed in three major referral centers (Denver, New York, the Netherlands) were characterized and classified on the basis of the latest proposed clinical classification for PAH-CHD (World Symposium on Pulmonary Hypertension, Nice, 2013). According to this classification, 24% of 134 children were classified into group 1, 14% into group 2, 19% into group 3, and 30% into group 4; 11% could not be classified. Types of CHD and hemodynamic profile differed between groups, with the highest right atrial pressure in group 4 (P < 0.040). Group 3 children had Down syndrome less frequently (P = 0.011) but other (un)defined syndromes most frequently (P = 0.063) and received most intense PAH-targeted therapy (P = 0.003). With 15 deaths and one lung transplant (12%; median follow-up: 4.3 years), survival differences could not be demonstrated between the groups in the Nice CHD classification. Pediatric PAH-CHD is a heterogeneous condition frequently associated with extracardiac, developmental factors that are believed to affect disease development. The Nice CHD classification identifies groups with specific patient/disease characteristics. However, a substantial proportion of children could not be classified. Group 3 forms a distinct disease entity. Its prognostic value could not be determined because of the low number of events. The Nice CHD classification supports clinical characterization of PAH-CHD; however, further refinement is needed to classify all children with PAH-CHD.
Keywords: pulmonary hypertension, congenital heart disease, pediatrics, Nice congenital heart disease classification, survival
Congenital heart disease (CHD) is the most common birth defect and comprises a broad spectrum of defects, varying from simple septal defects to complex cardiac malformations.1 Pulmonary arterial hypertension (PAH), a progressive disease of the pulmonary vasculature, is an important determinant of morbidity and mortality in CHD patients.
PAH associated with CHD (PAH-CHD) is a heterogeneous disease. Increased pulmonary blood flow, through a left-to-right shunt, has been recognized to trigger molecular and cellular changes in the pulmonary vasculature, leading to progressive PAH.2 The risk of developing PAH and its pace of progression highly depend on the anatomical location and size of the shunt defect.3-5 In patients with CHD and early-stage pulmonary vascular disease (PVD), shunt closure can reverse and resolve PAH.6,7 In time, PAH will pass a point of no return and become irreversible. Shunt closure is then contraindicated, as it is believed to worsen prognosis. PAH has also been reported in nonshunt CHD, such as aortic stenosis or coarctation.7-9
In some children, advanced PAH concurs with CHD that is considered not sufficient to explain the PAH.10 For example, pretricuspid shunts usually cause PAH from the third decade of life. Sometimes, severe PAH may be observed already in young children with such a shunt. In children with CHD, coexisting extracardiac factors may not only complicate clinical presentation but also increase susceptibility for developing PAH and thus play a modifying role.10 These include chromosomal or syndromal abnormalities but also developmental lung/airway anomalies and metabolic diseases.
Consequently, there is a need for adequate risk assessment and stratification in the individual child, which might be provided by a clinical classification for PAH-CHD. Such a classification would be most useful when it allows for identification of patient groups with specific disease characteristics, risks, or outcomes that allow tailored treatment approaches. Recently, an updated shunt-related classification was proposed for both adults and children at the Fifth World Symposium on Pulmonary Hypertension in Nice, France, in 2013 (the Nice CHD classification).11,12
Survival of both adults and children with PAH-CHD has been reported to be associated with the type of CHD.7,13,14 In adults, the value of the Nice CHD classification regarding differences in disease severity and survival between groups was recently reported.15 Although pediatric PAH-CHD has been evaluated with classifications based on hemodynamic relevance or anatomical location of the shunt defect, data regarding the Nice CHD classification in children are currently not available. We aimed to describe phenotypic heterogeneity in children with PAH-CHD, to assess the applicability of the Nice CHD classification, and to explore whether this classification accurately reflects patient/disease characteristics and survival.
Methods
Patients
For this study, all children with PAH and CHD were selected from a recently described9 contemporary cohort of consecutive pediatric PAH patients from the Children’s Hospital Colorado in Aurora, the Columbia University Medical Center in New York, and the Dutch National Referral Center for Pediatric Pulmonary Hypertension at the University Medical Center Groningen/Beatrix Children’s Hospital in the Netherlands. This cohort includes all children with PAH who visited these centers between 2000 and 2010 and had a diagnosis of PAH confirmed by a cardiac catheterization after 1997 at the age of ≥3 months and <18 years. PAH was defined as mean pulmonary artery pressure (mPAP) of ≥25 mmHg, mean pulmonary capillary wedge pressure of ≤15 mmHg, and indexed pulmonary vascular resistance (PVRi) of ≥3 Wood units⋅m2.
Children with other forms of pulmonary hypertension (PH), such as PH due to left heart disease, lung disease and/or hypoxia, or chronic thromboembolic disease, were not included in this cohort. In the event of mixed disease, meaning CHD and one of these conditions, it was at the discretion of the treating physician in the referral center, using the information from the cardiac catheterization, to define which was the most explanatory component for the PH: PAH-CHD or PH due to the coexistent condition. Only patients in the former group were included.
In this cohort, children with nonrepaired CHD were included only if the CHD was considered inoperable because of advanced PAH. In children with repaired CHD, diagnostic cardiac catheterization was performed more than 1 year after corrective surgery.
During cardiac catheterization, all three centers used the Fick principle to calculate cardiac output and pulmonary blood flow, using either measured or assumed oxygen consumption. In the latter case, oxygen consumption was assumed according to LaFarge and Miettinen16 (Aurora and New York) or Bergstra et al.17 (Netherlands). In patients with a nonrepaired patent arterial duct with right-to-left shunting, cardiac output calculations using Fick were not possible, and such patients are excluded from any analysis.
CHD
The anatomy, physiology, and repair status of the CHD were described. Children were classified according to the Nice CHD classification (Fig. 1).11,12
Figure 1.

Updated clinical classification for pulmonary arterial hypertension associated with congenital heart disease, Nice, France, 2013.
Cyanosis, required for group 1 of the Nice CHD classification, was defined as transcutaneous oxygen saturation of <90% at rest. If transcutaneous oxygen saturation was not available, systemic arterial oxygen saturation obtained during cardiac catheterization was used. Also, transcutaneous oxygen saturation of 90%–95% with systemic arterial oxygen saturation of <90% was considered cyanosis. Postductal saturations were used in children with patent arterial duct.
The distinction between CHD causal for the PAH and PAH with coincidental CHD was based on the expert opinion of the treating physician in the referral center and, for this study, was reviewed and agreed upon by two separate investigators (WMHZ and RMFB). In case of a residual shunt defect after corrective surgery, the patient was assigned to group 4, postoperative PAH-CHD, only if this shunt defect was considered not hemodynamically relevant. Otherwise, the patient was classified into group 1 or 2.
Assessments
Patient characteristics included age at PAH diagnosis, sex, and comorbidities. The moment of diagnostic cardiac catheterization, at either the referring or the referral center, was defined as baseline. Disease characteristics at baseline were collected and included both clinical and hemodynamic parameters. Ratios of mean pulmonary to systolic artery pressure (mPAP/mSAP) and pulmonary to systemic vascular resistance (PVR/SVR) were calculated.
During their disease course, children could receive PAH-specific therapies: calcium channel blocker (CCB) therapy or PAH-targeted therapy consisting of prostanoids, endothelin receptor antagonists, and/or phosphodiesterase 5 inhibitors. Treatment strategy was defined as previously described: CCB monotherapy if CCBs were the only PAH-specific drug used and otherwise the maximum number of PAH-targeted drugs used for at least 3 months or until end of follow-up (PAH-targeted mono-, dual, or triple therapy).9
Statistical analyses
Data are presented as mean ± SD, median (interquartile range), or number (percentage) as appropriate. One-way analysis of variance was used to compare normally distributed continuous variables. Nonnormally distributed continuous variables and ordinal variables were compared with the Kruskal-Wallis test. The χ2 and Fisher exact tests were used to compare categorical variables. Post hoc Bonferroni was used to correct for multiple comparisons, as appropriate.
Lung transplantation and death were defined as the primary end points. Children who did not die or undergo lung transplantation were censored at the last follow-up visit. Transplantation-free survival from diagnosis was depicted with Kaplan-Meier curves. To address potential survival bias due to the inclusion of prevalent patients, that is, patients receiving diagnoses between 1997 and 2000, data were left truncated. These patients entered the risk set at their truncation time, which was defined as the time between diagnosis and start of the study. Differences were explored with log-rank tests. A P value of <0.05 was considered significant.
Results
Patients
In total, 134 children had PAH and CHD (Table 1). Most of these children (77%) had simple pre- or posttricuspid shunt defects. Fourteen children (11%) had complex shunt defects (complete atrioventricular septal defect or nonrepaired functional univentricular heart). Seventeen children (13%) had miscellaneous CHD. Thirty-eight percent of the defects were repaired: 31% (n = 32) of the simple shunt defects and 62% (n = 8) of the complete atrioventricular septal defects. Most (70%) were repaired within 2 years after birth.
Table 1.
Congenital heart disease in the total cohort
| All (n =134) | Unrepaired (n = 83) | Repaired (n = 51) | |
|---|---|---|---|
| ASD±PAPVR | 28 (21) | 22 (79) | 6 (21) |
| VSD and/or PDA | 56 (42) | 37 (66) | 19 (34) |
| ASD+VSD and/or PDA | 19 (14) | 12 (63) | 7 (37) |
| Complete AVSD | 13 (10) | 5 (38) | 8 (62) |
| Nonrepaired functional univentricular heart | 1 (1) | 1 (100) | … |
| Miscellaneous | 17 (13) | 6 (35) | 11 (65) |
| Group 5 PH: MAPCAs | 2 | 1 | 1 |
| Not-classifiable PAH-CHD | 15 (11) | ||
| Aortic coarctation | 2 | 0 | 2 |
| Repaired TAPVR | 1 | 0 | 1 |
| Unilateral absence of PA | 5 | 4 | 1 |
| Scimitar syndrome | 1 | 1 | 0 |
| Neonatally repaired TGA±VSD | 6 | 0 | 6 |
Data are presented as no. (%) of patients; percentages are within column for “All” and within row for “Unrepaired” and “Repaired.” ASD: atrial septal defect; AVSD: atrioventricular septal defect; MAPCAs: major aortopulmonary collateral arteries; PA: pulmonary artery; PAH-CHD: pulmonary arterial hypertension associated with congenital heart disease; PAPVR: partial anomalous pulmonary venous return; PDA: patent arterial duct; PH: pulmonary hypertension; TAPVR: total anomalous pulmonary venous return; TGA: transposition of the great arteries; VSD: ventricular septal defect.
In 60 children (45%), comorbidities were described, most often Down syndrome (n = 30). Also, other syndromal abnormalities were reported: Noonan syndrome (n = 3), Di George syndrome (n = 1), Turner syndrome (n = 1), Robinow syndrome (n = 1), and undefined syndromes with dysmorphic features, psychomotor retardation, and/or developmental delay of unknown origin (3 children). Coexisting lung abnormalities were reported in 11 children (8%) and included (repaired) congenital diaphragmatic hernia, bronchopulmonary dysplasia, and chronic lung disease. Other described comorbidities were arteriovenous malformations in the lungs, hereditary telangiectasia, hyperthyroidism, glycogen storage disease 3A, ABCA3 gene mutation, ACTA2 gene mutation, Raynaud’s phenomenon, rheumatoid arthritis, VACTERL association, and Von Willebrand’s disease.
Nice CHD classification
Thirty-two children were classified as having Eisenmenger syndrome (ES; group 1), 19 as having PAH associated with left-to-right shunt (group 2), 26 as having PAH with coincidental CHD (group 3), and 40 as having postoperative PAH (group 4; Fig. 2). Of the 17 children (13%) with miscellaneous CHD, 2 children with major aortopulmonary collateral arteries could be classified as group 5 PH. The 15 remaining children (11%) could not be classified according to the Nice CHD classification.
Figure 2.
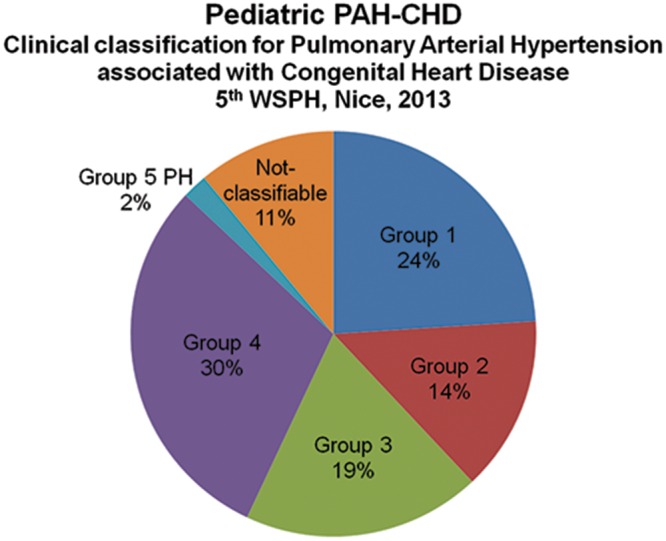
All children classified according to the Nice congenital heart disease classification. PAH-CHD: pulmonary arterial hypertension associated with congenital heart disease; WSPH: World Symposium on Pulmonary Hypertension; PH: pulmonary hypertension.
Within the four groups of the Nice CHD classification, age and sex were comparable (Table 2). In contrast, World Health Organization functional class (WHO FC) tended to differ between the four groups (P = 0.066): children with ES had most unfavorable WHO FC. Also, children with ES had hemodynamic profiles with the highest mPAP/mSAP and PVR/SVR. Cardiac index did not differ between the four groups (P = 0.270). Mean right atrial pressure (mRAP) differed between the four groups (P = 0.038) and was highest in postoperative PAH. In contrast to children in the other groups, children in group 3 most often had pretricuspid shunt defects (54% vs. 13%–21%, P = 0.001). Furthermore, while Down syndrome was less frequent in group 3 (P = 0.011), other (un)defined syndromes tended to be more frequent (19% vs. 0%–5%, P = 0.063).
Table 2.
Patient and disease characteristics at baseline stratified by the Nice CHD classification and not-classifiable PAH-CHD
| Group 1 | Group 2 | Group 3 | Group 4 | Not-classifiable PAH-CHD | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N | Value | N | Value | N | Value | N | Value | N | Value | P valuea | |
| Age at PAH diagnosis, years | 32 | 4.9 (1.5–11.9) | 19 | 6.8 (4.0–13.8) | 26 | 3.5 (1.1–9.4) | 40 | 5.4 (3.0–12.1) | 15 | 3.9 (2.5–9.0) | 0.268 |
| Female | 32 | 19 (59) | 19 | 16 (84) | 26 | 20 (77) | 40 | 29 (73) | 15 | 7 (47) | 0.240 |
| Down syndrome | 32 | 11 (34) | 19 | 4 (21) | 26 | 1 (4) | 40 | 14 (35) | 15 | 0 | 0.011*† |
| Cardiac defect | 32 | 19 | 26 | 40 | NA | 0.004*† | |||||
| ASD±PAPVR | 4 (13) | 4 (21) | 14 (54) | 6 (15) | |||||||
| VSD±PDA | 16 (50) | 12 (63) | 9 (35) | 19 (48) | |||||||
| ASD+VSD and/or PDA | 7 (22) | 2 (11) | 3 (12) | 7 (18) | |||||||
| Complete AVSD | 5 (16) | 0 | 0 | 8 (20) | |||||||
| Nonrepaired functional univentricular heart | 0 | 1 (5) | 0 | 0 | |||||||
| WHO FC | 29 | 16 | 21 | 34 | 11 | 0.066 | |||||
| I, II | 11 (38) | 10 (63) | 13 (62) | 24 (71) | 5 (46) | ||||||
| III, IV | 18 (62) | 6 (38) | 8 (38) | 10 (29) | 6 (55) | ||||||
| mPAP, mmHg | 32 | 60 ± 13 | 19 | 61 ± 20 | 26 | 58 ± 21 | 40 | 51 ± 17 | 15 | 49 ± 18 | 0.099 |
| mPCWP, mmHg | 32 | 9 ± 2 | 19 | 9 ± 2 | 26 | 8 ± 3 | 40 | 9 ± 3 | 15 | 10 ± 3 | 0.394 |
| mSAP, mmHg | 32 | 61 ± 15 | 19 | 67 ± 15 | 26 | 65 ± 12 | 39 | 70 ± 15 | 15 | 64 ± 14 | 0.087 |
| mRAP, mmHg | 31 | 6 ± 2 | 19 | 6 ± 3 | 25 | 6 ± 3 | 40 | 8 ± 3 | 13 | 7 ± 3 | 0.038‡ |
| PVRi, WU⋅m2 | 32 | 17.8 ± 10.5 | 19 | 18.5 ± 15.2 | 26 | 13.3 ± 9.7 | 40 | 13.2 ± 8.6 | 15 | 12.0 ± 7.0 | 0.119 |
| SVRi, WU⋅m2 | 16 | 16.8 ± 8.2 | 17 | 23.1 ± 14.0 | 20 | 19.5 ± 13.7 | 39 | 18.8 ± 8.5 | 13 | 18.7 ± 7.4 | 0.407 |
| CI, L/min/m2 | 17 | 4.1 ± 2.1 | 19 | 3.1 ± 1.2 | 25 | 3.8 ± 1.4 | 40 | 3.8 ± 1.4 | 15 | 3.4 ± 1.7 | 0.270 |
| Qpi, L/min/m2 | 32 | 4.1 ± 2.8 | 19 | 3.8 ± 1.7 | 26 | 4.5 ± 1.8 | 40 | 4.0 ± 2.0 | 15 | 3.6 ± 1.7 | 0.688 |
| mPAP/mSAP | 32 | 1.00 ± 0.19 | 19 | 0.92 ± 0.24 | 26 | 0.91 ± 0.37 | 39 | 0.75 ± 0.25 | 15 | 0.80 ± 0.31 | 0.002‡ |
| PVR/SVR | 16 | 1.36 ± 0.96 | 17 | 0.72 ± 0.25 | 20 | 0.71 ± 0.45 | 39 | 0.69 ± 0.33 | 13 | 0.69 ± 0.34 | <0.001*‡§ |
| Treatment strategy | 32 | 19 | 26 | 40 | 15 | 0.003*∥ | |||||
| No PAH-specific | 3 (9) | 3 (16) | 1 (4) | 3 (8) | 0 | ||||||
| CCB mono | 0 | 0 | 1 (4) | 4 (10) | 2 (13) | ||||||
| PAH-targeted mono | 13 (41) | 10 (53) | 5 (19) | 16 (40) | 6 (40) | ||||||
| PAH-targeted dual | 15 (47) | 5 (26) | 7 (27) | 11 (28) | 4 (27) | ||||||
| PAH-targeted triple | 1 (3) | 1 (5) | 12 (46) | 6 (15) | 3 (20) | ||||||
Data in “Value” columns are presented as no. (%), median (interquartile range), or mean ± SD. CCB: calcium channel blocker; CHD: congenital heart disease; CI: cardiac index; mPAP: mean pulmonary artery pressure; mPCWP: mean pulmonary capillary wedge pressure; mSAP: mean systemic artery pressure; mRAP: mean right atrial pressure; PAH: pulmonary arterial hypertension; PVRi: indexed pulmonary vascular resistance; PVR/SVR: ratio of pulmonary to systemic vascular resistance; Qpi: indexed pulmonary blood flow; SVRi: indexed systemic vascular resistance; WHO FC: World Health Organization functional class; WU: Wood units. For other abbreviations, see Table 1.
P value only for the four groups of the Nice CHD classification (excluding not-classifiable PAH-CHD). Symbols indicate P < 0.05 for indicated comparisons, from a post hoc test with Bonferroni correction.
Between group 1 and group 3.
Between group 3 and group 4.
Between group 1 and group 4.
Between group 1 and group 2.
Between group 2 and group 3.
In a separate secondary analysis, we compared patients with hemodynamically relevant shunt defects (groups 1 and 2 combined) with those without such defects (groups 3 and 4 and not-classifiable PAH-CHD; Table 3). Children with hemodynamically relevant shunt defects tended to have worse WHO FC (P = 0.076) than children without such defects. Furthermore, children with relevant shunt defects had unfavorable hemodynamics (higher mPAP, PVRi, mPAP/mSAP, and PVR/SVR; P < 0.020 for these parameters), whereas mRAP was higher in children without a hemodynamically relevant shunt defect (P = 0.031).
Table 3.
Patient and disease characteristics at baseline stratified by the hemodynamic relevance of the shunt defect
| Hemodynamically relevant shunt defect | No hemodynamically relevant shunt defect | ||||
|---|---|---|---|---|---|
| N | Value | N | Value | P value | |
| Age at PAH diagnosis, years | 51 | 6.1 (1.7–11.9) | 81 | 4.8 (2.5–10.1) | 0.865 |
| Female | 51 | 35 (69) | 81 | 56 (69) | 0.951 |
| WHO FC | 45 | 66 | 0.076 | ||
| I, II | 21 (47) | 42 (64) | |||
| III, IV | 24 (53) | 24 (36) | |||
| mPAP, mmHg | 51 | 60 ± 16 | 81 | 53 ± 19 | 0.019 |
| mPCWP, mmHg | 51 | 9 ± 2 | 81 | 9 ± 3 | 0.585 |
| mSAP, mmHg | 51 | 63 ± 15 | 80 | 67 ± 14 | 0.126 |
| mRAP, mmHg | 50 | 6 ± 3 | 78 | 7 ± 3 | 0.031 |
| PVRi, WU⋅m2 | 51 | 18.1 ± 12.3 | 81 | 13.0 ± 8.6 | 0.006 |
| SVRi, WU⋅m2 | 33 | 20.1 ± 11.8 | 72 | 19.0 ± 9.9 | 0.623 |
| CI, L/min/m2 | 36 | 3.6 ± 1.7 | 80 | 3.7 ± 1.5 | 0.628 |
| Qpi, L/min/m2 | 51 | 4.0 ± 2.4 | 81 | 4.1 ± 1.9 | 0.760 |
| mPAP/mSAP | 51 | 0.97 ± 0.21 | 80 | 0.81 ± 0.31 | 0.002 |
| PVR/SVR | 33 | 1.03 ± 0.75 | 72 | 0.70 ± 0.36 | 0.003 |
| Treatment strategy | 51 | 81 | 0.001 | ||
| No PAH specific | 6 (12) | 4 (5) | |||
| CCB mono | 0 | 7 (9) | |||
| PAH-targeted mono | 23 (45) | 27 (33) | |||
| PAH-targeted dual | 20 (39) | 22 (27) | |||
| PAH-targeted triple | 2 (4) | 21 (26) | |||
Therapy
In total, 117 children (87%) received PAH-targeted therapies during the study period: 51 monotherapy, 43 dual therapy, and 23 triple therapy. Ten children did not receive PAH-specific therapy. Seven children without hemodynamically relevant shunt defects received CCB monotherapy (1 in group 3, 4 in group 4, and 2 with not-classifiable PAH-CHD).
Treatment intensity differed significantly between the four groups of the Nice CHD classification (Table 2; P = 0.003). In particular, children in group 3 received PAH-targeted triple therapy more frequently than children in the other groups. Children without a hemodynamically relevant shunt defect received PAH-targeted triple therapy more frequently than those with a hemodynamically relevant shunt defect (Table 3; P = 0.001).
Not-classifiable PAH-CHD
Fifteen children (11%) could not be classified according to the Nice CHD classification. This was due to either the absence of a shunt defect (repaired aortic coarctation [n = 2] or corrected transposition of the great arteries with neonatal switch operation [n = 6]) or the presence of developmental anomalies of the pulmonary vasculature (including repaired total anomalous pulmonary venous return, absent pulmonary artery, or Scimitar syndrome [n = 7]). All children had normal pulmonary capillary wedge pressure (≤15 mmHg).
Transplantation-free survival
During a median follow-up of 4.3 years, 15 children died and 1 child underwent lung transplantation (Fig. 3). The cause of death was directly PAH related in 10 children and not directly PAH related in the remaining 5 children, although the condition causing death might have been poorly tolerated because of the presence of PAH.
Figure 3.

Transplantation-free survival of all children with pulmonary arterial hypertension associated with congenital heart disease. Survival at 1, 3, and 5 years was 96%, 91%, and 88%, respectively.
In group 1, 3 children died. Of interest, all three children were diagnosed with advanced PAH within 6 months after birth.
With the relatively low number of 16 events, an exploratory survival analysis could not demonstrate a significant survival difference between the four groups of the Nice CHD classification (Fig. 4A). Also, despite the fact that all three deceased children with not-classifiable PAH-CHD died within 3 years after diagnosis, no survival differences could be observed between this group and the four groups of the Nice CHD classification (P = 0.32). Finally, no survival differences between children with a hemodynamically relevant shunt defect (groups 1 and 2 combined) and those without such a shunt defect (groups 3 and 4 and not-classifiable PAH-CHD) could be demonstrated (Fig. 4B).
Figure 4.

Transplantation-free survival. A, Stratified by the Nice CHD classification. Survival at 1, 3, and 5 years was 96%, 92% and 82% for children in group 1, 100%, 100%, and 100% for children in group 2, 94%, 89%, and 89% for children in group 3, and 97%, 94%, and 89% for children in group 4, respectively (P = 0.45). Also shown is survival for children with not-classifiable PAH-CHD, with 1-, 3-, and 5-year survival of 90%, 70%, and 70%, respectively. B, Stratified by the hemodynamic relevance of the shunt defect. Survival at 1, 3, and 5 years was 98%, 95%, and 89% for children with a hemodynamically relevant shunt defect (groups 1+2) and 95%, 89%, and 86% for children without a hemodynamically relevant shunt defect (groups 3+4 and not-classifiable PAH-CHD), respectively (P = 0.30). CHD: congenital heart disease; PAH: pulmonary arterial hypertension.
Discussion
This study is the first to assess the applicability and value of the recently proposed Nice CHD classification in children with PAH-CHD. In our study, this classification identified clinically relevant differences in patient and disease characteristics between groups, especially regarding comorbidities, hemodynamic profile, and treatment intensity. However, a substantial proportion of children (11%) could not be classified, as their CHD did not fit in the predefined groups, which are based on congenital shunt defects. In this exploratory study with a relatively low event rate, no differences in transplantation-free survival between the four groups of the Nice CHD classification could be observed.
Nice CHD classification
ES, group 1, is the advanced stage of PAH associated with congenital shunt defects, with reversed or bidirectional shunting and the occurrence of cyanosis. This condition is distinguished from shunt defects with left-to-right shunting, group 2. However, this distinction is artificial, since a gradual progression of patients from the latter group to group 1 over time is to be expected (as we observed in 6 patients in this study), complicating group-specific outcome analyses. Furthermore, children in groups 1 and 2 had similar underlying CHD: predominantly posttricuspid shunt defects. Patients with ES had most advanced PAH, characterized by unfavorable pulmonary-to-systemic hemodynamics. However, cardiac index and mRAP seemed preserved in these children. This is likely to be associated with the hemodynamically relevant shunt defect that may function as “pop-off” for the right ventricle, preserving left ventricular filling and cardiac output. Despite this, children in group 1 were in higher WHO FCs, which might be explained by the accompanying cyanosis that aggravates during exercise.18
Objective criteria are lacking for “coincidental CHD,” in which the presence of a (small) shunt defect itself is not considered to account for the development of PAH. In our study, this was addressed by independent review of the treating physician’s assessment. Nevertheless, children in group 3 indeed seemed to form a distinct disease entity, with a preponderance of pretricuspid shunt defects and no complex CHD. Although comorbidities are frequent in all forms of pediatric PAH-CHD, as shown in this and other studies,10,19,20 children in group 3 showed a different pattern of extracardiac comorbidities, characterized by lower frequency of Down syndrome and greater frequency of other (un)defined syndromes. These features are in line with those found in children with idiopathic PAH, justifying previous reports that classified this condition as “idiopathic-like PAH.”10 It seems that, in children, extracardiac comorbidities, such as undefined syndromes or certain genetic traits, are associated with the development of PAH. Increased genetic susceptibility for a normally nonpathogenic “hit” may be the mechanism, where in these group 3 children the small heart defect and normally nonrelevant shunt may form this second hit and initiate or accelerate the pulmonary vascular remodeling of PAH.
Remarkably, in this contemporary cohort, group 4 (postoperative PAH) was the largest group. This is in line with earlier data from the Spanish Registry for Pulmonary Hypertension and the Tracking Outcomes and Practice in Pediatric Pulmonary Hypertension (TOPP) registry.20,21 Shunt closure in early stages of PVD can reverse and resolve PAH.6 Therefore, it is generally advised to close shunt defects early in life, usually within the first 1–2 years of life, depending on the defect, as was done in most of the children in this study. However, this study shows that early closure does not always prevent the development of progressive PAH. This is in line with epidemiological data on pediatric PAH showing that 1%–2% of children with PAH-CHD developed PAH despite early closure of their shunt defect.7 Also, in our study extracardiac factors were frequent in the group with postoperative PAH: one-third had Down syndrome, and 22% had other comorbidities. These factors may affect susceptibility for PVD and play a role in progression of PAH after shunt closure. Therefore, next to young age and hemodynamics, such factors should also be taken into account when determining eligibility for shunt closure.4,6,22
Therapy
Remarkably, although there is no evidence supporting the use of CCBs in PAH-CHD, 7 children without hemodynamically relevant shunt defects received CCB monotherapy in this study.
Most children received PAH-targeted therapies, confirming previous current-era reports.9,23,24 Interestingly, children in group 3 received more intense PAH-targeted therapy than children in other groups, although they did not have more advanced disease, according to WHO FC or hemodynamic profile. This suggests that these children were considered more similar to patients with idiopathic PAH and different from the other children with PAH-CHD.
In the original cohort, we have shown that children with PAH-CHD were treated less intensely than children with idiopathic/hereditary PAH.9,25 In the current study, about half of the children did not receive PAH-targeted dual or triple therapy. In light of the unfavorable prognosis for these children, with a 3-year mortality rate of approximately 10%, it should be questioned whether this less intense treatment is justified and whether more intense treatment might improve survival in these children. Further research is required to answer these questions.
Survival
Overall 5-year survival in this study was 88%, which was within the range of previously reported survival rates in pediatric PAH-CHD.7,20,24,26-29 In contrast to the recent report of Manes et al.15 describing outcomes in adult PAH-CHD, we could not demonstrate survival differences within the Nice CHD classification. The relatively low number of patients and end points and the relatively short follow-up time in this study, resulting in lack of power, hampered conclusions from the exploratory survival analysis in the current study.
In this study, ES was diagnosed in three infants within the first 6 months of life, and all died during follow-up. Rather than the classic disease evolution into ES, these infants probably had a failure of postnatal adaptation with remodeling of the pulmonary vascular bed. These observations are in line with the statement of the Pulmonary Vascular Research Institute (PVRI) task force that perinatal and in-utero factors affecting the pulmonary vascular bed are important in determining postnatal outcome.30 This maladaptation may contribute to a rapid progression of PAH, and such infants presenting with “accelerated PAH” have been shown to have a very poor prognosis.4,7,31 In fact, these children may represent a form of persistent pulmonary hypertension of the newborn with associated CHD or coinciding with CHD.
None of the children in group 2 died or underwent lung transplantation. This could very well be due to the artificial distinction between group 1 and group 2, as children in group 2 will first progress into group 1 before they die or undergo lung transplantation.
In this study, survival of children in group 3 seemed better than that reported for children with idiopathic PAH.27,32 Although not sufficient to explain the PAH, their shunt defect may contribute to preservation of cardiac output despite progression of PVD.15
While children and adults in group 4 have been reported to have worse survival than children with other CHD, this could not be demonstrated in this study.7,23 This may be due to patient selection and inclusion. Only children with PAH confirmed during cardiac catheterization at least 1 year after cardiac surgery were included in this study. Therefore, the sickest patients may not have been included, as they might have been too sick to undergo cardiac catheterization or died within the first year after cardiac surgery. Also, it may be that the worse survival of children with postoperative PAH occurs only after a longer period of follow-up.
It is important to notice that the inclusion criteria of this study were designed to study a contemporary cohort with predominantly incident patients but also allowed the inclusion of a small subset of prevalent patients (n = 18). Prevalent patients were seen in the referral centers between 2000 and 2010 but had diagnostic cardiac catheterization between 1997 and 2000. To avoid immortal-time bias, data of these patients were left truncated. For patients with PAH after repaired CHD, the inclusion criteria required a diagnostic cardiac catheterization performed more than 1 year after corrective surgery. Children who died within 1 year after surgery were thus not included in this cohort. This might have introduced a limited form of immortal-time bias.
Not-classifiable PAH-CHD
The Nice CHD classification is based on the presence or history of shunt defects. However, a significant number of children (13%) had CHD other than shunt defects. Only two of these children, with major aortopulmonary collateral arteries, could be classified as group 5 PH, according to the Nice classification, whereas the remaining children could not be classified. The heterogeneity of CHD associated with PH and the difficulties and limitations of simple classification have been repeatedly raised and are at odds with the advantages of stratification for risk or treatment effect that may be associated with such classification.33 The Panama classification, as proposed by the PVRI task force, aims to address this heterogeneity in pediatric PAH but awaits validation with respect to its value for risk stratification, prognostication, or treatment consequences.30 More research is needed to further characterize children with not-classifiable PAH-CHD.
Strengths and limitations
Bringing together the consecutive, contemporary cohorts of three major pediatric PAH referral centers has led to one of the largest contemporary cohorts in the field of pediatric PAH and provided the opportunity to assess the applicability of the Nice CHD classification in children. Nevertheless, this study was limited by the relatively low number of events, resulting in lack of power and thereby hampering definitive conclusions regarding survival. Also, its retrospective nature can be regarded a limitation. However, the standardized diagnostic and treatment strategies of three dedicated pediatric PH centers and a dedicated review at the level of individual patient data for this study allowed for a meticulous characterization of the patients and minimized differences in definitions or data inconsistencies. In our cohort, only children who had a diagnosis of PAH confirmed during cardiac catheterization were included, which strengthened disease definition but also introduced a selection bias by exclusion of children with PAH-CHD without cardiac catheterization. Furthermore, baseline was defined as the moment of diagnostic cardiac catheterization, which also strengthened disease definition but may have postponed time of diagnosis, affecting survival analyses.
Conclusions
Pediatric PAH-CHD is a heterogeneous condition frequently associated with extracardiac, syndromal, and developmental factors that are believed to affect PAH evolution and progression. The recently proposed Nice CHD classification identifies groups with specific patient and disease characteristics, also in children. However, the fact that a substantial proportion of children with PAH-CHD, 11%, could not be classified within this classification forms a serious limitation. Group 3, PAH with coincidental CHD, forms a distinct disease entity similar to idiopathic PAH. The prognostic value of the Nice CHD classification in children with PAH-CHD could not be determined by this study because of the relatively low number of events during a median follow-up of 4.3 years. The proposed Nice CHD classification supports clinical characterization of PAH-CHD; however, further refinement is needed in order to adequately classify all children with PAH-CHD.
Acknowledgments
We wish to thank Theresia Vissia-Kazemier, Kathleen Miller-Reed, and Beth Coleman for their contribution to this study.
Source of Support: This study was supported by the Sebald Foundation, the Frederick and Margaret L Weyerhaeuser Foundation, the Jayden de Luca Foundation, and grant UL TR001082 from the National Center for Advancing Translational Sciences/National Institutes of Health.
Conflict of Interest: EBR has received honoraria from Actelion, Gilead, and United Therapeutics for consultation on scientific advisory boards and is a consultant for Ikaria on a clinical trial. The University of Colorado has received consultancy fees for DDI from Actelion, Bayer, Gilead, Lilly, Pfizer, and United Therapeutics. The University Medical Center Groningen contracts with Actelion, Bayer, Glaxo-SmithKline, Lilly, Novartis, and Pfizer for consultant and steering committee activities of RMFB. All other authors declare no conflicts of interest.
References
- 1.Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol 2002;39(12):1890–1900. [DOI] [PubMed]
- 2.Berman EB, Barst RJ. Eisenmenger’s syndrome: current management. Prog Cardiovasc Dis 2002;45(2):129–138. [DOI] [PubMed]
- 3.Tulloh RM. Congenital heart disease in relation to pulmonary hypertension in paediatric practice. Paediatr Respir Rev 2005;6(3):174–180. [DOI] [PubMed]
- 4.van Albada ME, Berger RM. Pulmonary arterial hypertension in congenital cardiac disease—the need for refinement of the Evian-Venice classification. Cardiol Young 2008;18(1):10–17. [DOI] [PubMed]
- 5.Craig RJ, Selzer A. Natural history and prognosis of atrial septal defect. Circulation 1968;37(5):805–815. [DOI] [PubMed]
- 6.Rabinovitch M, Keane JF, Norwood WI, Castaneda AR, Reid L. Vascular structure in lung tissue obtained at biopsy correlated with pulmonary hemodynamic findings after repair of congenital heart defects. Circulation 1984;69(4):655–667. [DOI] [PubMed]
- 7.van Loon RL, Roofthooft MT, Hillege HL, ten Harkel AD, van Osch-Gevers M, Delhaas T, Kapusta L, et al. Pediatric pulmonary hypertension in the Netherlands: epidemiology and characterization during the period 1991 to 2005. Circulation 2011;124(16):1755–1764. [DOI] [PubMed]
- 8.Berger RM, Beghetti M, Humpl T, Raskob GE, Ivy DD, Jing ZC, Bonnet D, Schulze-Neick I, Barst RJ. Clinical features of paediatric pulmonary hypertension: a registry study. Lancet 2012;379(9815):537–546. [DOI] [PMC free article] [PubMed]
- 9.Zijlstra WM, Douwes JM, Rosenzweig EB, Schokker S, Krishnan U, Roofthooft MT, Miller-Reed K, Hillege HL, Ivy DD, Berger RM. Survival differences in pediatric pulmonary arterial hypertension: clues to a better understanding of outcome and optimal treatment strategies. J Am Coll Cardiol 2014;63(20):2159–2169. [DOI] [PubMed]
- 10.van Loon RL, Roofthooft MT, van Osch-Gevers M, Delhaas T, Strengers JL, Blom NA, Backx A, Berger RM. Clinical characterization of pediatric pulmonary hypertension: complex presentation and diagnosis. J Pediatr 2009;155(2):176–182.e1. [DOI] [PubMed]
- 11.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013;62(25 suppl):D34–D41. [DOI] [PubMed]
- 12.Ivy DD, Abman SH, Barst RJ, Berger RM, Bonnet D, Fleming TR, Haworth SG, et al. Pediatric pulmonary hypertension. J Am Coll Cardiol 2013;62(25 suppl):D117–D126. [DOI] [PubMed]
- 13.Daliento L, Somerville J, Presbitero P, Menti L, Brach-Prever S, Rizzoli G, Stone S. Eisenmenger syndrome: factors relating to deterioration and death. Eur Heart J 1998;19(12):1845–1855. [DOI] [PubMed]
- 14.Diller GP, Dimopoulos K, Broberg CS, Kaya MG, Naghotra US, Uebing A, Harries C, Goktekin O, Gibbs JS, Gatzoulis MA. Presentation, survival prospects, and predictors of death in Eisenmenger syndrome: a combined retrospective and case-control study. Eur Heart J 2006;27(14):1737–1742. [DOI] [PubMed]
- 15.Manes A, Palazzini M, Leci E, Bacchi Reggiani ML, Branzi A, Galiè N. Current era survival of patients with pulmonary arterial hypertension associated with congenital heart disease: a comparison between clinical subgroups. Eur Heart J 2014;35(11):716–724. [DOI] [PubMed]
- 16.LaFarge CG, Miettinen OS. The estimation of oxygen consumption. Cardiovasc Res 1970;4(1):23–30. [DOI] [PubMed]
- 17.Bergstra A, van Dijk RB, Hillege HL, Lie KI, Mook GA. Assumed oxygen consumption based on calculation from dye dilution cardiac output: an improved formula. Eur Heart J 1995;16(5):698–703. [DOI] [PubMed]
- 18.Diller GP, Dimopoulos K, Okonko D, Li W, Babu-Narayan SV, Broberg CS, Johansson B, et al. Exercise intolerance in adult congenital heart disease: comparative severity, correlates, and prognostic implication. Circulation 2005;112(6):828–835. [DOI] [PubMed]
- 19.Lévy M, Celermajer D, Szezepanski I, Boudjemline Y, Bonnet D. Do tertiary paediatric hospitals deal with the same spectrum of paediatric pulmonary hypertension as multicentre registries? Eur Respir J 2013;41(1):236–239. [DOI] [PubMed]
- 20.del Cerro Marín MJ, Sabaté Rotés A, Rodriguez Ogando A, Mendoza Soto A, Quero Jiménez M, Gavilán Camacho JL, Raposo Sonnenfeld I, Moya Bonora A, Albert Brotons D, Moreno Galdó A. Assessing pulmonary hypertensive vascular disease in childhood: data from the Spanish registry. Am J Respir Crit Care Med 2014;190(12):1421–1429. [DOI] [PubMed]
- 21.Beghetti M, Schulze-Neick I, Barst RJ, Kronmal D, Berger RMF, Humpl T. Clinical classification of congenital heart disease associated pulmonary hypertension. Does it work for pediatrics? analysis of the TOPP registry (tracking outcome and practice in pediatric pulmonary hypertension). Cardiol Young 2012;22(suppl S1):S3–S176. Abstract PW1-10.
- 22.Lopes AA, Barst RJ, Haworth SG, Rabinovitch M, Al Dabbagh M, del Cerro MJ, Ivy D, et al. Repair of congenital heart disease with associated pulmonary hypertension in children: what are the minimal investigative procedures? Consensus statement from the Congenital Heart Disease and Pediatric Task Forces, Pulmonary Vascular Research Institute (PVRI). Pulm Circ 2014;4(2):330–341. [DOI] [PMC free article] [PubMed]
- 23.Haworth SG, Hislop AA. Treatment and survival in children with pulmonary arterial hypertension: the UK Pulmonary Hypertension Service for Children 2001–2006. Heart 2009;95(4):312–317. [DOI] [PubMed]
- 24.Barst RJ, McGoon MD, Elliott CG, Foreman AJ, Miller DP, Ivy DD. Survival in childhood pulmonary arterial hypertension: insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Circulation 2012;125(1):113–122. [DOI] [PubMed]
- 25.Zijlstra WMH, Douwes JM, Rosenzweig EB, Schokker S, Krishnan U, Roofthooft MTR, Miller-Reed K, Hillege HL, Ivy DD, Berger RMF. Children with pulmonary arterial hypertension (PAH) associated with congenital heart disease are treated less intensively with PAH-targeted drugs compared to children with idiopathic/hereditary PAH. Eur Heart J 2013;34(suppl 1):40. Abstract P304.
- 26.Barst RJ, Ivy DD, Foreman AJ, McGoon MD, Rosenzweig EB. Four- and seven-year outcomes of patients with congenital heart disease-associated pulmonary arterial hypertension (from the REVEAL registry). Am J Cardiol 2014;113(1):147–155. [DOI] [PubMed]
- 27.van Loon RL, Roofthooft MT, Delhaas T, van Osch-Gevers M, ten Harkel AD, Strengers JL, Backx A, Hillege HL, Berger RM. Outcome of pediatric patients with pulmonary arterial hypertension in the era of new medical therapies. Am J Cardiol 2010;106(1):117–124. [DOI] [PubMed]
- 28.Douwes JM, van Loon RL, Hoendermis ES, Vonk-Noordegraaf A, Roofthooft MT, Talsma MD, Hillege HL, Berger RM. Acute pulmonary vasodilator response in paediatric and adult pulmonary arterial hypertension: occurrence and prognostic value when comparing three response criteria. Eur Heart J 2011;32(24):3137–3146. [DOI] [PubMed]
- 29.Ivy DD, Rosenzweig EB, Lemarié JC, Brand M, Rosenberg D, Barst RJ. Long-term outcomes in children with pulmonary arterial hypertension treated with bosentan in real-world clinical settings. Am J Cardiol 2010;106(9):1332–1338. [DOI] [PMC free article] [PubMed]
- 30.del Cerro MJ, Abman S, Diaz G, Freudenthal AH, Freudenthal F, Harikrishnan S, Haworth SG, et al. A consensus approach to the classification of pediatric pulmonary hypertensive vascular disease: report from the PVRI Pediatric Taskforce, Panama 2011. Pulm Circ 2011;1(2):286–298. [DOI] [PMC free article] [PubMed]
- 31.van Loon RL, Roofthooft MT, Berger RM. Pulmonary arterial hypertension in children with congenital heart disease. PVRI Rev 2009;1(4):203–207.
- 32.Moledina S, Hislop AA, Foster H, Schulze-Neick I, Haworth SG. Childhood idiopathic pulmonary arterial hypertension: a national cohort study. Heart 2010;96(17):1401–1406. [DOI] [PubMed]
- 33.Berger RM. Pulmonary hypertension: smaller kids, smaller steps. Lancet Respir Med 2014;2(5):348–350. [DOI] [PubMed]