

Abstract
There have been a variety of approaches taken to try to characterize and identify the genetic basis of adaptation in nature, spanning theoretical models, experimental evolution studies and direct tests of natural populations. Theoretical models can provide formalized and detailed hypotheses regarding evolutionary processes and patterns, from which experimental evolution studies can then provide important proofs of concepts and characterize what is biologically reasonable. Genetic and genomic data from natural populations then allow for the identification of the particular factors that have and continue to play an important role in shaping adaptive evolution in the natural world. Further to this, experimental evolution studies allow for tests of theories that may be difficult or impossible to test in natural populations for logistical and methodological reasons and can even generate new insights, suggesting further refinement of existing theories. However, as experimental evolution studies often take place in a very particular set of controlled conditions – that is simple environments, a small range of usually asexual species, relatively short timescales – the question remains as to how applicable these experimental results are to natural populations. In this review, we discuss important insights coming from experimental evolution, focusing on four key topics tied to the evolutionary genetics of adaptation, and within those topics, we discuss the extent to which the experimental work compliments and informs natural population studies. We finish by making suggestions for future work in particular a need for natural population genomic time series data, as well as the necessity for studies that combine both experimental evolution and natural population approaches.
Keywords: adaptive evolution, experimental evolution, genomic data, natural populations
Introduction
We have numerous examples of adaptations inferred from times series in the fossil record and from studies of extant natural populations adapting to changing environmental conditions. Elucidating the genetic basis of these adaptations, however, is still a formidable task. Natural selection acting on genetic variation is the only process that consistently produces biological adaptation. But the fraction of genetic variation enabling adaptation is potentially minute and thus hard to study in natural populations. This has frustrated progress in uncovering the genetic basis of adaptation for a wide range of organisms. However, it is only by looking broadly across taxa and environments that we can begin to identify the generalities vs. idiosyncrasies of adaptation.
Several research programmes have developed almost independently over the years to tackle the genetics of adaptation. Two types of approaches, ‘top‐down’ and ‘bottom‐up’, aim to explore the genetics of adaptation by examining natural populations (NP), while a third uses experimental evolution (EE) of highly controlled populations in the laboratory. Studies taking the ‘top‐down’ approach scrutinize the fitness consequences of phenotypic variation in natural populations, and work out the genetics underlying these phenotypes. This approach is the most direct as it directly examines the phenotypic variation and its fitness consequences in the naturally occurring environment (Hoekstra et al. 2006). Identification of the links between genes and fitness in these natural populations has allowed for the inference of key historical molecular adaptations, and in some cases even experimental validation of these inferences through the reconstruction and analysis of those ancestral proteins (e.g. Harms & Thornton 2014; McKeown et al. 2014). This approach, however, can be greatly limited by the number of individuals that can be phenotyped and assayed for fitness. Furthermore, even in the few situations where phenotypic variation for a set of ecological traits has been related convincingly to fitness differences, elucidating the genetic basis of such phenotypic variation is still a formidable task despite the boost in the ability to survey almost any nucleotide level variation in genomes (Lamichhaney et al. 2015).
A large number of studies are taking the ‘bottom‐up’ approach and bypass phenotypic variation altogether. They rely exclusively on genetic data, probing patterns of polymorphism and divergence at the nucleotide level in an attempt to identify regions/sites in the genome that are under selection (Vitti et al. 2013). All of these ‘bottom‐up’ methods invariably rely on characterizing a null distribution for a summary statistic describing some aspect of the data, for example the raw amount of polymorphism, proportion of rare vs. frequent alleles, or the mean amount of genetic differentiation. The null distributions are increasingly being specified by jointly fitting or prefitting a demographic model based on a fraction of the data that is deemed to be selectively neutral. Regions exhibiting discrepant values for these summary statistics are then candidates for selection. A considerable amount of method development has transformed the field, in particular with the introduction of computer intensive methods for fitting quite complex demographic scenarios to the data (Gutenkunst et al. 2009; Alves et al. 2012). Despite this leap in complexity, some claim that model misspecification – in particular when incorporating population subdivision – might be yielding spurious outliers that are not actually footprints of selection (Bierne et al. 2011, 2013; Fraisse et al. 2015). The use of an empirical null distribution obtained from a fraction of the genome that is deemed neutral may alleviate some of these issues (Lotterhos & Whitlock 2014). Crucially, these approaches rely on the assumption that most of the genome, or at least a sizable well‐identified class of sites in the genome, is truly neutral and therefore are only affected by demography (Li et al. 2012). However, there is mounting evidence that selection might be affecting, via linkage, a very substantial portion of the genome of species that are scrutinized to learn about the genetics of adaptation (Sella et al. 2009; Comeron 2014).
EE studies use an almost orthogonal approach relying on highly artificial environments created in the laboratory where repeated bouts of evolution from a common ancestral genotype or population can be tracked through time. Depending on the choice of organism, samples can be stored at regular time intervals and used for future in‐depth genetical or phenotyping studies including standardized fitness assays. The power of this approach is undeniable. Besides the added benefit of transforming evolutionary genetics into a prospectively experimental instead of mostly historical/retrospective enterprise, it has yielded a wealth of empirical information about the genetics of adaptation.
Experimental evolution can be used to characterize extensively the spectrum of mutations arising through their effects on fitness in well‐defined laboratory conditions. This is important because mutations are ultimately the raw stuff that enable adaptation. Other aspects of the dynamics of adaptation that have been studied intensely in the EE programme include the dynamics of selected mutations through time and independent selection of rare beneficial mutations vs. massive selective interference. Finally, EE has also begun to explore how environmental variation – both temporal and spatial – shapes selection and in turn the dynamics of adaptation.
Although the EE research programme has been traditionally focused on fitness as the ultimate phenotype, whole‐genome sequencing has been invading the field over the last 5 years (Barrick et al. 2009; Brockhurst et al. 2011; Dettman et al. 2012). The EE programme has been very successful on various fronts but one can wonder how widely applicable the findings of EE are and whether or not the observed patterns are too idiosyncratic of the laboratory environment to be of broad interest to a scientific community focused on understanding the genetics of adaptation in naturally occurring populations (for further discussion of these criticisms, see for example Buckling et al. 2009). There has also been a paucity of direct comparisons of patterns emerging from EE and NP studies, with some important exceptions in the study of antibiotic resistance of human pathogens (e.g. Wong et al. 2012; Ariey et al. 2014) and now recent work in yeast and bacteria (Maddamsetti et al. 2015; Metzger et al. 2015). As is clear from these emerging examples, and as we highlight further in this review, merging insights from the EE and NP research programmes can provide powerful and so far largely unexplored ways to borrow strength from both approaches.
Here the specific aim of our review is to revisit a number of outstanding questions on the genetics of adaptation and critically assess if the EE and NP centric approaches can complement each other. Our review is placed within the context of three interdependent poles of research in this field – Theory, Natural Populations, Experimental Evolution – with Theory and EE providing important proof of concept for theories testable in nature, and both EE and NP providing insight for development and ongoing refinement of the theory (Servedio et al. 2014). We focus on key topics for the evolutionary genetics of adaptation and critically assess what key pieces of knowledge have been gained using the EE approach. We ask to what extent these insights are specific to idiosyncratic laboratory conditions and how EE can help to complement other research strategies aimed at understanding the genetics of adaptation.
What kinds of mutations enable adaptation?
Mutations provide the variation that enables populations to evolve. Thus, knowledge of the properties of those mutations can help to guide our understanding of how evolution is expected to proceed, in particular the rate of adaptation, and how readily and repeatably populations adapt to novel environments. Specifically, we need to characterize the fitness effects of mutations that are available for selection to work with, as well as the types of mutations that tend to be selected and so rise to high frequencies in a population as it adapts. When a population is faced with a novel environment, its potential for adaptive evolution is provided by the already present standing genetic variation or by de novo genetic variation arising during that bout of adaptation. However, the relative importance of these two sources of variation in natural populations is still not clear. Another important part of understanding the drivers of adaptation is examining whether a substantial heterogeneity exists at the genome level between different types of mutations (loss‐ vs. gain‐of‐function, types of genes, etc.) that can also impact the dynamics of adaptation. We discuss these elements below.
The distribution of fitness effects of new mutations
A number of experimental studies have made strides towards characterizing the effect and size of new mutations affecting fitness. Mutation accumulation and directed mutagenesis experiments, now in combination with genome sequencing, have given us much insight into the distribution of effects of all arising mutations in a range of model organisms (Halligan & Keightley 2009), while experimental evolution studies have given us insight into the effects of mutations directly responsible for increases in fitness (recent review by Bataillon & Bailey 2014). In general, these experiments suggest that while the majority of mutations appear to have nearly neutral effects, it is mutations of intermediate effects that tend to drive adaptive evolution. These studies have also provided more quantitative tests for the distribution of fitness effects (DFEs) of novel mutations and in particular beneficial mutations (Fig. 1). Overall, experimental data tend to agree with the theoretical predictions – fitness effects of new mutations tend to be gamma distributed (although there are exceptions, e.g. Hietpas et al. 2011), and when the population in question is already quite well adapted, the effects of beneficial mutations follow a generalized Pareto distribution (e.g. Rokyta et al. 2008; Bataillon et al. 2011; although there are exceptions, e.g. Levy et al. 2015).
Figure 1.
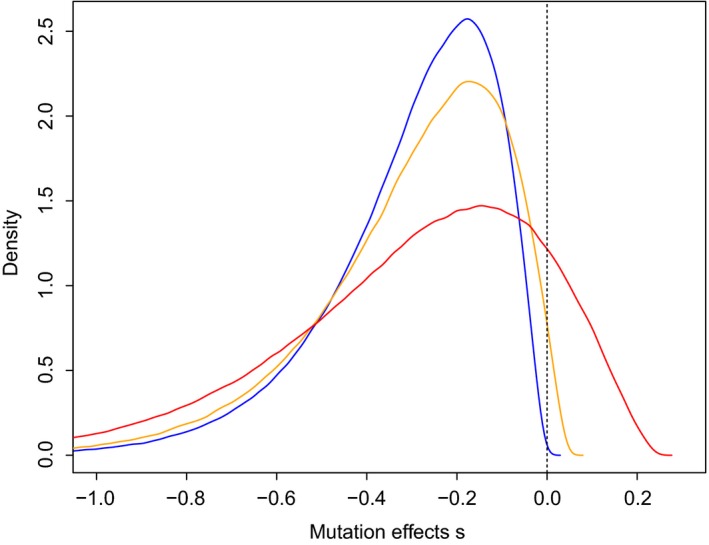
Distributions of fitness effects (DFEs) vary strongly with current level of adaptation. Three distinct DFEs are shown for populations with different levels of adaptation: (i) perfectly adapted (blue), (ii) almost perfectly adapted (orange) and (iii) poorly adapted (red). DFEs were obtained by simulations of 106 independent mutations affecting fitness according to Fisher's geometric model at three levels of initial adaptation. Similar variation in DFEs across level of adaptation has been found empirically in Escherichia coli (Hietpas et al. 2013; Perfeito et al. 2014). Further theoretical results on DFEs expected under Fisher's model can be seen in Martin & Lenormand (2006) and Tenaillon (2014).
In natural populations, polymorphism data possibly complemented by measures of divergence can be used to infer the DFE of mutations arising in gene coding regions (Keightley & Eyre‐Walker 2010) and more recently in functional but noncoding regions (Gronau et al. 2013; Racimo & Schraiber 2014). Inferences from these methods suggest that the DFE can vary widely (Kousathanas & Keightley 2013; Bataillon & Bailey 2014). However, direct comparisons between the insights obtained by EE and polymorphism/divergence surveys are quite problematic for a number of reasons. First there is almost zero taxonomic overlap between approaches although in a few taxa (such as yeast) both approaches could potentially be compared. The DFEs inferred from joint patterns of polymorphism and divergence are cast in terms of an effective selection coefficient – s multiplied by a scaling factor that is the effective population size. This potentially complicates comparisons; however, as effective size can be seen as a scaling factor, emphasis should be on the general shape of the inferred distributions. Last, while a laboratory environment‐specific selection coefficient is straightforward to compute in an EE study, the (scaled) selection coefficients inferred from polymorphism and divergence data are in essence long‐term averages encompassing a mosaic of environments (see below for more on both the crucial importance of having a time perspective and the importance of environmental heterogeneity).
De novo vs. standing variation
While spontaneous mutation is the ultimate source of all variation on which selection acts to drive adaptation, when a population is adapting to a new environment selection may either drive fixation of mutations already present in the population (standing variation) or, alternatively, act on mutations arising de novo during that bout of adaptation. Quantification of the relative importance of these two types of mutations for adaptive evolution across a range of scenarios is important for predicting the rate of adaptation in novel environments (Hermisson & Pennings 2005; Barrett & Schluter 2008).
In populations of organisms with long generation times that typically also evolve at small demographic population sizes, the input of new mutations per generation is expected to be modest, and thus, the total waiting time for useful variation to appear de novo might be exceedingly long. In contrast, rapidly reproducing organisms, especially species evolving with very high demographic population sizes, might benefit from a substantial mutational input of de novo variation over a short time span. Theory predicts that the relative importance of de novo vs. standing variation might also depend on the mating system and the history of populations (Orr & Betancourt 2001; Glémin & Ronfort 2013).
Determining the importance of these two sources of variation is difficult in evolution experiments for several reasons. First, the majority of experimental evolution studies use asexual microbes living in relatively simple environments, effectively eliminating the possibility for any standing variation to be maintained long enough to be important for evolution. Thus, in these cases de novo mutation is the only source of variation for selection to act on. This type of experimental set‐up might also be critically lacking another important source of variability available for selection in nature, as many bacteria also take up DNA fragments present in the environment, incorporating these fragments into their genomes.
While there have also been a number of experimental evolution studies using sexual organisms (e.g. Drosophila: Burke et al. 2010; Caenorhabditis elegans: Gray & Cutter 2014), these sexual organisms have generation times that are orders of magnitude longer than microbes, and so it is logistically difficult to maintain these experiments for long enough to see new mutations, much less observe the contributions of those mutations to adaptive evolution. Thus, most experimental set‐ups lack the ability to observe the dynamics of de novo and standing variation simultaneously. An exception is a recent experiment with sexual yeast (Saccharomyces cerevisiae), initiated with diverse outcrossed populations and cultured with periodic bouts of sex (Burke et al. 2014; also see Parts et al. 2011). This study identified regions under selection as those that followed consistent monotonic temporal patterns across replicate experimental populations. However, while de novo mutation did have the potential to play a role in this experimental, in actuality genetic variation at all the sites identified as being under selection in this experiment was the result of standing variation.
Patterns of variation in genomes are affected jointly by demographic history (this affects the genome as a whole) and selection (this varies by genomic location). Several theoretical models have explored the effect of selection on the diversity at neighbouring linked neutral sites. One important theoretical finding is that selective sweeps that originate from a single mutation (so‐called hard sweeps) leave a different imprint on patterns of linked neutral diversity compared to sweeps where selected alleles are regularly introduced by mutation or pre‐existing alleles segregating in a population (soft sweeps) (Hermisson & Pennings 2005). Therefore, one can, in principle, infer the relative importance of both types of sweeps from genomewide patterns of diversity.
In that context, various methods have been recently proposed to try and distinguish between selection footprints left specifically by either soft vs. hard sweeps (Peter et al. 2012). At this stage we only have a cursory understanding of the power of these methods. Crucially, it is not clear yet to what extent the power of detection of soft vs. hard sweeps is influenced by the demographic history of the population and the effect of a favourable mutation relative to the polygenic background of variation for fitness of a trait under selection (Chevin & Hospital 2008; Stephan 2015). Merely exhibiting instances of selection leaving detectable footprints at the molecular level was once a feat in itself (Wang et al. 1999), but we have now transitioned to a situation where we have a plethora of instances of natural selection footprints but still a very cursory idea of how biased the list of reported instances is (Rockman 2012). This potential bias of unknown magnitude casts doubt on the validity of a strategy relying exclusively on detection of footprints of selection to answer the question of the relative importance of de novo vs. standing variation in enabling adaptation to new environments.
Molecular characteristics of adaptive mutations
If the particular mutations that contribute to adaptation tend to have some general characteristics, identifying those characteristics could help us to better predict which genes or regions of the genome are most likely to contribute to adaptive evolution. If there are multiple potential genetic routes to the same phenotypic outcome, we expect evolution to tend to follow the most readily available genetic route. In EE, the adaptive routes taken very often involve loss‐of‐function mutations. Loss‐of‐function mutations are a very common class of mutations because many types of mutations (e.g. frameshifts, missense, gain‐of‐stop‐codon) at many locations along a gene can result in the loss‐of‐function of the protein that particular gene codes for. Loss‐of‐function of a protein can be beneficial if that protein is costly and unnecessary, for example those proteins involved in motility in a population evolving in a well‐mixed laboratory environment (e.g. Zhong et al. 2009; Maughan & Nicholson 2011; Bailey et al. 2015).
Loss‐of‐function mutations are also beneficial when they occur in genes coding for proteins that repress the expression of others that are themselves beneficial proteins. For example, an evolution experiment tracking Pseudomonas aeruginosa populations adapting to a quinolone antibiotic, ciprofloxacin (Wong et al. 2012), found that loss‐of‐function of efflux pump repressors was the most common mode of resistance evolution. Efflux pumps actively transport unwanted substrates such as antibiotics out of the cell. Interestingly, loss‐of‐function of efflux pump repressors is a known mechanism for quinolone resistance in clinical isolates of P. aeruginosa (Wong & Kassen 2011), and more broadly for multidrug resistance in clinical isolates of a range of Gram‐negative bacteria (Blair et al. 2014). However, while these loss‐of‐function mutations are seen in the clinical setting, the most common mode of resistance is, in fact, via mutations that specifically target drug enzymes (Wong & Kassen 2011). Although these target‐specific mutations are likely a much smaller class of mutation compared to loss‐of‐function, it may be that they result in more effective antibiotic resistance. Thus, while in the longer‐term clinical setting the drug enzyme‐specific mutations tend to win out, populations in Wong et al.'s experimental populations did not evolve long enough for these rare target‐specific mutations to arise. An alternate possibility is that the drug enzyme‐specific mutations confer less of a fitness cost in the absence of antibiotic compared to the efflux pump mutations, and so in the clinical setting where antibiotic treatment often starts and stops, the less costly mutations prevail.
Another interesting example of the importance of loss‐of‐function of repressors in experimental populations is the ‘wrinkly spreader’ (WS) phenotype that consistently evolves in populations of P. fluorescens grown in static microcosms (Rainey & Travisano 1998). In these experiments, the WS phenotype evolves via a number of genetic routes, most often via mutations conferring loss‐of‐function to negative regulators of operons responsible for synthesis of cellulose – the proteins responsible for the WS phenotype (McDonald et al. 2009). However, there are many other less‐common routes to the WS phenotype that have been characterized by Lind et al. (2015), who classify and quantify this variety showing that 98% of mutations confer WS phenotype via loss‐of‐function of a negative regulator, and the other 2% through promoter and intragenic activation, and gene fusions.
There are a few examples of loss‐of‐function mutations thought to be tied to adaptive evolution in natural populations. One of the most well characterized cases is loss‐of‐function mutations to the FRIGIDA gene that have arisen independently in several populations of Arabidopsis thaliana (Corre et al. 2002). Loss‐of‐function of the FRIGIDA gene is associated with an early‐flowering phenotype and this is beneficial under certain environmental conditions (but note also, there is a strong genotype‐by‐environment interaction; Stinchcombe et al. 2004). It has been suggested that these loss‐of‐function mutations are recent adaptations to the new environments A. thaliana now occupies due to its relatively recent postglacial expansion.
On the short evolutionary timescale – one that experimental evolution is well suited to explore – loss‐of‐function mutations are important drivers of adaptation. However, over longer timescales, other classes of mutations begin to play more important roles because, of course, loss‐of‐function mutations are not sufficient to explain the origin of new functions. Indeed, the Escherichia coli populations in Richard Lenski's long‐term evolution experiment have seen many putatively beneficial loss‐of‐function mutations; however, after ~30 000 generations of evolution, a key innovation arose in one of the populations – a mutation that usurps a promoter, ultimately allowing for the organism to utilize citrate, a substrate that was previously inaccessible to E. coli (Blount et al. 2012).
How prevalent are stable/selectively maintained polymorphisms?
In a simple homogeneous environment, genetic diversity can be maintained by balancing selection when rare alleles have a fitness advantage. Two kinds of selection that have this effect are: (i) heterozygote advantage/overdominance, and (ii) negative‐frequency‐dependent selection. Alternatively, in a heterogeneous environment, temporal and spatial variation in selection can lead to the maintenance of balanced polymorphisms through the evolution of locally adapted specialists. In this section we explore the prevalence of balanced polymorphisms in natural and experimental populations, focusing on the evidence for adaptive mechanisms maintaining genetic diversity in the absence of environmental variation. A discussion of evolution in variable environments is deferred to later.
Heterozygote advantage/overdominance
Genome scans for selection typically focus on the detection of selective sweeps or alleles that underly local adaptation but there is also increasing interest in being able to detect instances of balancing selection. However, very often the tests employed for detecting balancing selection ignore the underlying process causing balancing selection and focus exclusively on the expected deviation from selective neutrality in patterns of sequence polymorphism (Andrés et al. 2009; Vitti et al. 2013). Although there are a few cases where we understand very precisely why we expect to see balanced polymorphisms (e.g. SI alleles, mating types in fungi), it is currently unclear how much balanced selection we expect in nature, and in particular, how prevalent balanced selection driven by heterozygote advantage is in outcrossing organisms.
Recent theory – so far based mostly on Fisher's fitness landscape (Manna et al. 2011; Sellis et al. 2011) but also older work based on metabolic control theory – make predictions on the levels of dominance we expect for mutations affecting fitness. Both metabolic control theory and Fisher's fitness landscape models map the effects of mutations at the phenotypic level to fitness. In the case of Fisher's model, traits are arbitrary but their phenotypic variation causes fitness variation. In metabolic models, the traits are enzyme activity in a pathway and flux through the pathway is the proxy for fitness. In both instances mutations have additive effects on the phenotype and the nonlinear mapping between phenotype and fitness is what generates dominance (Keightley & Kacser 1987; Lunzer et al. 2010; Jiang et al. 2013). A few EE studies, notably large data sets in yeast, allow for testing this theory (Agrawal & Whitlock 2011; Manna et al. 2011; Sellis et al. 2011).
Given that one can make testable predictions regarding the dominance of mutations affecting fitness, one could envision going one step further and also using such models to answer – at least theoretically – questions such as the following: How often do we expect balancing selection due to an overdominant mutation? The modelling approach used to derive theoretical expectations for dominance can be used to work out how pervasive heterozygote advantage is expected to be in populations. This type of theoretical approach could also be systematized to explore the consequences of fluctuating environments for the genetics of adaptation. Some progress has been made on this question by studies that consider how mutations differentially affecting sexes (Connallon & Clark 2014; Kirkpatrick & Guerrero 2014) or more broadly, different environments (Martin & Lenormand 2015). A better theoretical understanding of the potential environmental conditions and frequency at which we expect to observe balancing selection created by overdominant mutations would be valuable for boosting studies in both EE and NP that are targeted at studying balancing selection.
Together this growing body of theory could be tested using an EE approach employing diploid organisms – for which dominance can be measured (Agrawal & Whitlock 2011; Manna et al. 2011). This body of theory also has the potential to guide genomewide scans for balancing selection in natural populations by providing a clearer picture of the processes underlying balancing selection. It can also be the basis for designing more sensitive methods targeting balancing selection footprints caused by specific types of processes or help focus studies of natural populations on the genomic contexts that are most likely to harbour such polymorphisms (Qiu et al. 2015).
Frequency‐dependent selection
Another mechanism that can drive and maintain stable genetic diversity in populations is negative‐frequency‐dependent selection. Here, the relative fitness of a genotype depends on its frequency in the population and can result in the stable maintenance of multiple genotypes. There are a number of interesting examples of this type of selection in EE, many of which arise from ‘cross‐feeding’ between genotypes – when one genotype lives off the by‐products of another genotype. In this scenario, negative‐frequency‐dependent selection arises because the by‐product consuming genotype does very well when rare as it is surrounded by by‐product producing individuals. However, as it becomes more frequent, the by‐product producing genotype becomes less frequent resulting in a decrease in the per capita by‐product supply and so a decrease in fitness. Rosenzweig et al. (1994) first showed how cross‐feeding allows polymorphisms to be maintained by negative‐frequency‐dependent selection in experimentally evolved populations of E. coli, and subsequently the long‐term dynamics and genetic details of this type of balanced polymorphism have been examined in populations from Lenski's long‐term E. coli evolution experiment (Rozen & Lenski 2000; Le Gac et al. 2012; Plucain et al. 2014) and others (e.g. Herron & Doebeli 2013). While the molecular mechanisms and genetic details of the particular interactions seen in these experimental systems may not be readily transferable to understanding stable polymorphisms in natural populations, these experiments have allowed for the exploration the conditions under which frequency‐dependent selection is expected to arise, and characterization of the evolutionary dynamics of these types of interactions.
Many EE studies observe phenotypic and genotypic diversity that, while not stably maintained in the long term, is maintained for substantial periods of time, sometimes long enough to make it difficult to distinguish from truly stable polymorphisms (e.g. Kao & Sherlock 2008; Lang et al. 2011; Maharjan et al. 2015). This diversity is generated by ‘clonal interference’ (two competing beneficial mutations: Gerrish & Lenski 1998) or ‘multiple mutation effects’ (multiple competing beneficial mutations: Desai & Fisher 2007). In a population experiencing clonal interference, clones from a lineage harbouring a beneficial mutation rise in frequency but before that mutation reaches fixation, another beneficial mutation arises in the ancestral background and this second beneficial mutation also begins to rise in frequency. While only a single genotype is expected to win out in the end, competition between those two clonal lineages slows the process of fixation and so diversity may be present in the population for an extended period of time. ‘Multiple mutation effects’ is simply a generalization of this scenario where many different beneficial mutations arise and compete for fixation simultaneously. Lang et al. (2011) distinguish between mechanisms driving diversity in ~600 S. cerevisiae populations, classifying the evolutionary dynamics as dominated by sweeps, clonal interference, multiple mutation and frequency‐dependent selection by examining the frequency trajectories of the evolving lineages. The authors estimate that in these lines the majority of the observed diversity was generated by clonal interference or multiple mutation effects. Although some populations do appear to have stable polymorphisms, possibly maintained through frequency‐dependent selection, these are much less common. In sexual populations, the potential for recombination to combine multiple beneficial mutations into a single individual makes clonal interference much less important. However, these experiments show the diversity of mechanisms that can generate diversity in populations and underline the importance of developing methods to distinguish between mechanisms generating and maintaining polymorphisms in natural populations.
How repeatable is evolution?
Despite the stochastic nature of evolution, there are many examples of similar traits evolving repeatedly in natural populations faced with similar environmental challenges (e.g. reduction of body armour in freshwater sticklebacks, Colosimo et al. 2005; colour variation in mice, Hoekstra et al. 2006). These similar phenotypic changes are often interpreted as evidence of strong selection (Losos 2011). Selection will strongly bias the spectrum of fixed mutations as a function of the fitness advantage conferred by a mutation. However, there are often many possible genetic routes by which the same phenotypic changes can evolve (e.g. Lind et al. 2015) and so repeated evolution at the gene or even nucleotide level is expected to occur much less frequently. The presence of repeated independent changes at the gene level suggest that in addition to selection, gene‐to‐gene or region‐to‐region heterogeneity in mutation rates may be an important driver of these patterns. In fact, theory suggests that under the assumptions of a ‘strong selection–weak mutation’ regime (beneficial mutations fix sequentially through a series of selective sweeps), heterogeneity in mutation and selection have the potential to contribute equally to the probability of repeated evolution (Chevin et al. 2010; Lenormand et al. 2015). On the other hand, if the mutation supply rate is large (i.e. Neμ ≫ 1), multiple beneficial mutations may arise simultaneously and compete for fixation. Under this regime, selection is expected to play the dominant role in driving the emerging patterns of repeated evolution (Nagel et al. 2012; Lenormand et al. 2015). However, the relative effects of selection and mutation rate heterogeneity on observed patterns of repeated evolution, and how these contributions might shift under different types of selection regimes is still not clear, as studies typically concentrate on one cause of heterogeneity (e.g. selection) at the expense of the other (e.g. mutation).
The EE programme provides an excellent framework for the systemic examination of repeated evolution. Repeated evolution has been observed experimentally across multiple levels of organization (from nucleotide to gene to phenotype, e.g. Tenaillon et al. 2012), across a range of taxa (e.g. viruses: Bull et al. 1997; bacteria: Tenaillon et al. 2012; yeast: Spor et al. 2014; fruit flies: Simões et al. 2008), and across different types of selection regimes (e.g. different numbers and types of resources: Bailey et al. 2015; in the presence vs. absence of an antibiotic: Wong et al. 2012). In these experiments, the selection environment is highly controlled and usually relatively simple, so perhaps it is not surprising that the same phenotypes tend to evolve repeatedly in replicate experimental populations adapting to the same environment. Indeed, in this kind of set‐up, divergent phenotypes are the interesting exceptions; possible evidence for unexpectedly rugged fitness landscapes (e.g. Melnyk & Kassen 2011).
More difficult to explain by selection alone, are repeat changes observed at the gene, and even nucleotide levels in these experiments. The frequency of repeated evolution at the gene level varies greatly across organisms and environments, and EE studies offer, and in some cases explicitly test, a number of potential mechanisms for these observed differences. For example, one factor shown to influence the frequency of repeated evolution is population size – large populations show a higher frequency of repeated evolution compared to small populations (Lang et al. 2013). A large population can harbour a large supply of contending mutations and so it is expected to follow a more deterministic, predictable evolutionary trajectory where only the most beneficial mutations eventually fix, compared to a small population whose evolutionary trajectory will tend to be more stochastic (Nagel et al. 2012). Theoretical models suggest a number of other factors with the potential to affect the degree of repeated evolution, such as the current level of adaptation in the population and level of pleiotropy of new mutations affecting fitness (Orr 2005; Chevin et al. 2010), and there is great potential for EE studies to explore these predictions. Furthermore, future statistical and mathematical modelling approaches should be aimed at quantifying the relative effects of selection and mutation on patterns of repeated evolution in these experimental data, and then evaluate the potential for environmental factors to shift these patterns.
Despite the potential for highly complex and variable selection regimes in nature, there are numerous examples of repeated evolution in natural populations. For very specific selection pressures, repeated independent evolutionary changes have even been observed down to the amino acid level (e.g. echolocation in mammals: Parker et al. 2013; cardenolide resistance in insects: Zhen et al. 2012). More generally, many natural populations with parallel phenotypic changes have been shown to have parallel changes at the gene level. A meta‐analysis by Conte et al. (2012) quantified the probability of gene reuse in natural populations that had evolved the same phenotypic adaptations. They saw that the mean probability of gene reuse ranged from about 0.1 to 0.8 depending of the relatedness of the populations under comparison. This is not dissimilar from the range of gene reuse seen in experimental populations evolved in the same selection environment. For example, using Conte et al.'s metric to calculate gene reuse across experimental bacterial populations from Bailey et al. (2015), mean probabilities of gene reuse ranging from 0.08 to 0.59, depending on selection environment. For replicate bacterial populations adapting to a quinolone antibiotic (Wong et al. 2012), Conte et al.'s gene reuse metric ranges from 0.33 to 0.36 depending on other attributes of the selection environment. Thus, with respect to gene reuse, EE and NP studies appear to be quite comparable.
One potentially important difference between parallel evolution in the laboratory and in natural populations is the origin of the mutations giving rise to those parallel changes. As touched upon earlier in this review, the EE programme is often strongly biased towards experimental set‐ups for which genetic variation is generated via de novo mutations and so knowledge gained about patterns of repeated evolution from those studies is specific to independently arising mutations. However, as is pointed out by Martin & Orgogozo (2013), similar phenotypes arising in independently evolving populations can result from not only the repeated generation of the same mutations in multiple populations, but also the repeated propagation of pre‐existing mutations. In other words, repeated evolution can occur in independent populations via the repeated fixation of mutations that existed as standing variation in the ancestral population (e.g. reduction of body amour in freshwater stickleback; Colosimo et al. 2005). A few EE studies are starting to explore the characteristics of this type of repeated evolution (Parts et al. 2011; Burke et al. 2014).
Another potential mode of repeated evolution, largely unexplored within the context of the EE programme, is lateral transfer of beneficial mutations via introgression or horizontal gene transfer (but see Perron et al. 2012 for an exception). These processes have been implicated in driving repeated evolution in natural populations across a range of taxa (e.g. introgression: Kim et al. 2008; Song et al. 2011; horizontal transfer: Ochman et al. 2000; Cobbs et al. 2013). While lateral gene transfer can be difficult to incorporate into EE studies, it is certainly an important area for future work in understanding patterns of parallel evolution in natural populations, and in particular for more applied questions regarding the repeated evolution of antibiotic resistance in bacteria (Jansen et al. 2013).
It's the environment, stupid
In nature, environments are variable, and thus, selection strength and direction are trivially expected to vary as well. In experiments, we often strive to reduce environmental variation, working with very simple environments in an attempt to isolate the effects of other factors such as population size, sex, etc. on patterns of evolution. This is a potentially disadvantageous mismatch between the EE and NP programmes. However, there are also a number of EE studies that specifically aim to examine the effects of environmental variation on the process of adaptation. While it is clear that selection fluctuates and shifts in natural populations, the potential impact of that variation on evolution is only beginning to be explored with genomic data. It is important to consider that selection can vary not only as a result of extrinsic processes (e.g. spatial and temporal variations in climate), but also as a result of intrinsic (often biotic) processes that have the potential to feedback as evolution proceeds (e.g. host–pathogen co‐evolution). These different types of variation have the potential to have important and potentially very different impacts on the evolutionary dynamics of adapting populations as we discuss below.
Are rates of beneficial mutation conditioned by current level of adaptation?
Inferring the DFE of new mutation remains experimentally challenging even with model organisms in well‐controlled environments. As stated above, one of the obvious short coming of the EE approach is that fitness of strains are measured in very artificial environments. But one important empirical and robust insight emerges from the wealth of experiments carried out so far. Intuitively mutations, because they are random perturbations, often tend to deteriorate fitness. In a very well adapted population the intuition is that virtually all mutations are deleterious and beneficial mutations vanishingly rare. That argument has also been repeatedly used since Gillespie (Martin & Lenormand 2008) to make prediction not about the whole DFE but the DFE for the fractions of mutations that are beneficial. But this theoretical purely heuristic argument cannot account for how different environments and different initial level of adaptation condition the availability of beneficial mutations. Theory based on an explicit fitness landscapes – and Fisher's geometric model (FGM) is emerging as a popular model (see Tenaillon 2014; Martin & Lenormand 2015) – has allowed for a more rigorous validation of these intuitions and, importantly the formation of precise predictions on the fraction of beneficial mutations available as a function of the initial level of adaptation of a population. The key prediction is that the fraction of favourable mutations available in a population for adapting in a given environment is strongly conditioned by the current level of adaptation in that population (Fig. 1).
Direct experimental evidence from single‐step mutations in genes (Barrick et al. 2010; Hietpas et al. 2011; Bank et al. 2014) shows very convincingly that the DFE is strongly determined by the current level of adaptation in the particular environment where it is measured. Similarly, quantitative analysis of the fitness trajectories of populations through time supports this view (Schoustra et al. 2009; Gifford et al. 2011; Perfeito et al. 2014; Good & Desai 2015). In this respect, EE provides a robust empirical result that is very hard to obtain without the laboratory‐controlled condition approach. It is also likely to be very influential for directing future theoretical and empirical work aimed at understanding how novel mutations can enable further adaptation over time in populations that are initially poorly adapted to their current environment.
How specialized are adaptations?
A more challenging question to tackle is how environment‐specific beneficial mutations are. In some cases, as a population adapts its fitness improvements are very specific to the environment in which it is selected, but in other cases evolutionary adaptations seem to confer more general fitness improvements (Bataillon et al. 2011; Bank et al. 2014). The pervasiveness of local adaptation and the specificity of the particular mutations underlying those adaptations have important consequences for the evolution and maintenance of diversity, as well as the resilience of populations faced with changing environments (Collins & de Meaux 2009).
There have been numerous studies that look for evidence of local adaptation using reciprocal transplant experiments – an experimental design where fitness is quantified and compared between populations grown in their native environment and those transplanted and grown in a non‐native environment (Kawecki & Ebert 2004). While the empirical evidence suggests that fitness trade‐offs are far from ubiquitous a meta‐analysis of reciprocal transplant experiments reported local adaptation was detected in 71% of the studies examined, and collectively these studies showed a general negative (albeit weak) correlation between a population's fitness in its native environment and its fitness in a foreign environment (Hereford 2009). Another meta‐analysis of reciprocal transplant studies focused solely on plants shows the same frequency of local adaptation (Leimu & Fischer 2008). EE studies measuring the level of specialization in populations evolving in divergent environments suggest that significant fitness trade‐offs readily evolve (in 20 of the 21 cases reviewed by Kassen 2014).
In recent years, effort has been made to try to identify alleles responsible for local adaptation and understand their fitness effects. By identifying the adaptively important genes, we can explore whether trade‐offs are a result of specific genetic architecture and constraints on the traits under selection, or alternatively, whether trade‐offs arise as a result of additional mutations that are neutral in the selection environment but deleterious elsewhere. Experimental evolution shows evidence for both mechanisms (decay of unused trait: e.g. Hall & Colegrave 2008; antagonistic pleiotropy: e.g. MacLean et al. 2004), sometimes even within the same system (e.g. Presloid et al. 2008). In natural populations, a number of studies that detected trade‐offs using reciprocal transplant experiments have also gone on to identify genes responsible for these adaptations (Savolainen et al. 2013).
Genomewide scans for the detection of loci underlying local adaptation primarily seek genomic regions or SNPs exhibiting higher than average genetic differentiation among potentially locally adapted populations, and/or look for associations between SNP frequency and environmental variables among a set of populations (Coop et al. 2010; Günther & Coop 2013). There has been a recent upsurge in the use of these methods (Savolainen et al. 2013), in particular methods for detecting association between SNPs and environmental covariates especially because it can enable the study of recent/ongoing adaptation to climatic variables in long lived organisms such as trees where a direct experimental approach is laborious (Grivet et al. 2011; Jaramillo‐Correa et al. 2015). This is an area of very active research for both method development and testing of the relative performance of these methods (De Mita et al. 2013; Lotterhos & Whitlock 2014). Problems with these approaches can be numerous: false positives due to demographic effects, and the specification of a robust null model for the amount of correlation expected for a neutral SNP with no effect at all on adaptation. Environment association detection has to overcome the very same caveats that plague association studies seeking SNPs that are causal of phenotypic variation – association can be spuriously created by population structure, although methods have been developed deal with this issue (e.g. Yu et al. 2006; Kang et al. 2008). Ideally, these detection methods should be seen as a first step and candidate SNPs should be validated using more experimental approaches.
Environmental variability: generalists, specialists and diversity
Environments vary in both space and time across many different scales, and so given the potential for local adaptation and specialization, how do we expect natural populations to evolve in such a variable world? The answer to this question depends on a number of things including the particular nature of that variability, genetic and physiological constraints on the traits under selection, and rates of gene flow. In a temporally varying environment, individuals in the population are forced to deal with the full range of variation. In this case, the theoretical expectation is that the genotype with the highest geometric mean fitness will prevail (Haldane 1937, pp. 338), and thus, a population of generalists will evolve. For a population in a spatially varying environment (where dispersal distances are low relative to the spatial scale of the variation), different lineages within a population may only be exposed to part of the full range of variation within a single generation of evolution. In this case, the theoretical expectation is for multiple locally adapted specialists to evolve and, if there are fitness trade‐offs in adapting to one part of the environment vs. another, this diversity of specialist types may be stably maintained. Of course this outcome also depends on genetic constraints which can limit the extent of adaptation to a particular part of the environment, and gene flow, which can aid in the adaptive evolution of a population by increasing the supply of genetic variation for selection to act on, but on the other hand can overwhelm a population with maladapted alleles.
EE studies have made significant progress towards understanding some of the key factors driving adaptation in range of variable, albeit still relatively simple, environments. A number of EE studies document adaptation to temporally (e.g. Bennett et al. 1992; Kassen & Bell 1998; Condon et al. 2014) and spatially varying environments (e.g. Silver & Mateles 1969; Joshi & Thompson 1997; Jasmin & Kassen 2007; Bailey & Kassen 2012). Many of these studies support the theoretical expectations outlined above, but others contradict these expectations and suggest additional potentially important factors driving adaptation in variable environments. For example in the case of Bailey & Kassen (2012), large differences in the evolutionary potential for improvement in different parts of a spatially variable environment resulted in the evolution of a single specialist (instead of multiple specialists or a single generalist), an outcome that was then maintained due to large evolved differences in the productivity of the component parts of that spatially variable environment.
EE studies have used trait measurements to quantify quantitative genetic variance (Beardmore 1961; Mackay 1981; Venail et al. 2011), and others, molecular markers (Powell 1971; Haley & Birley 1983) to try to characterize the genetics underlying adaptation in different types of variable environments. Recently, whole‐genome sequencing has been used to obtain more complete characterizations of the genetics underlying adaptation in experimental populations evolving in variable environments. For example, consistent with the theoretical expectations discussed above, Huang et al. (2014) found that genetic diversity at sites likely under selection was highest in Drosophila melanogaster populations evolved in spatially variable environments compared to those evolved in temporally variable and constant environments. These EE studies have provided an important intermediate link between theoretical models and natural populations, confirming or rejecting expectations but also suggesting other important factors with the potential to influence evolution in variable environments.
Adaptation feeds back on environment: biotic interactions and co‐evolution
Selection environments can also change and vary as a direct result of evolutionary changes in the adapting population, creating a feedback between the population and its environment. This is perhaps most apparent when the selection environment is itself an evolving population, for example in a host–parasite system, but occurs whenever an important attribute of the selection environment involves ecological interactions with another evolving population. In a co‐evolving selection environment, a number of the general predictions about adaptive walks change. For example, as the populations do not move closer to an evolutionary optimum with each adaptive step, the fitness effects of mutations are no longer expected to decrease and the rate of adaptation remains constant. Another potential outcome is intensified local adaptation as populations become specifically adapted to interactions with their particular co‐evolutionary partner population.
There are now a few EE studies that have tracked the genomic changes of co‐evolving populations, in particular bacteria–phage co‐evolution (Paterson et al. 2010; Meyer et al. 2012). These studies support some of the basic theoretical expectations for key differences between evolution in abiotically vs. biotically varying environments (e.g. higher rates of adaptation and genetic divergence in phage with co‐evolving host populations compared to phage with fixed host populations; Paterson et al. 2010) and help add to our understanding of processes that likely play an important part in the evolution of natural populations that often live in biologically diverse communities. Because of the potential for such a diverse range of biotic interactions, isolating the effects of these types of interactions in many natural populations is difficult. However for some specific types of interactions, in particular host–pathogen interactions, there has been some progress (Woolhouse et al. 2002; Gagneux 2012; Sironi et al. 2015).
While many EE studies have been aimed at examining the effects of different types of environmental variation on adaptive evolution, the extent of variation and the complexity of these laboratory environments have still been arguably very simple in comparison with the types of variation experienced by natural populations. Although performing experiments in simple environments has been an immensely profitable approach for isolating and identifying factors driving evolutionary adaptation, it is also extremely important to expand the EE programme to more complex laboratory environments in order to better understand how adaptation proceeds in the complex natural world (Collins 2010).
Conclusions
What have we gained?
The EE programme has provided direct insights on a number of evolutionary processes that can be inferred only indirectly via the study of natural populations. In particular, information about mutation rates and effects of mutations on fitness is exceedingly hard to obtain, yet these are central quantities for population genetic models of adaptation. The empirical estimates of these quantities that have been obtained so far are important milestones for guiding the development of improved theoretical models and methods for detecting selection footprints.
EE has established that DFEs are also highly environment‐specific in the sense that current level of adaptation strongly conditions the DFE. This is a major building block to achieving a more mature understanding of how adaptation proceeds. A more thorough understanding of the heterogeneity of mutation rates and processes throughout the genome – still at a nascent stage – will also be a key to formulating testable quantitative theories on the relative importance of mutation and selection in shaping the evolution of populations and to inform us on how repeatable we can expect the process of evolution to be. It is also important to keep in mind that patterns of evolution have the potential to vary widely across both selection environment and organism, and so it is critical that we continue to broaden the range of biological systems explored if we are to gain a more complete understanding of how adaptation proceeds. This speaks not only to the NP programme but also to EE where experimental designs would benefit greatly from a wider range of organisms and environment‐specific manipulations.
All methods for detecting regions under selection invariably have to make assumptions about both demographics and the relative importance of various types of mutations. In particular, one can hope that a more thorough understanding of the DFE and its variation will translate into better expectations for the relative amounts of positive and purifying selection and more sensitive tests for the occurrence of favourable mutations. In this context, the EE programme can also be a source of inspiration to re‐emphasize the power of having a temporal perspective in surveys of genomewide polymorphism in natural populations. Indeed, this is perhaps the most conspicuous advantage of the EE set‐up – the ability to evolve in parallel and assay/resequence at regular time intervals. In natural populations, there could be a big advantage to using more systematic designs involving temporal samples to detect segments of the genome responding to selection. This has been done to some extent in a few human pathogen systems (Poon et al. 2012). Recent and ongoing advances in sequencing technologies make this kind of repeated sampling design more and more logistically (and financially) feasible (Thépot et al. 2015) even in nonmodel organisms with no reference genome (Jones & Good 2015). Furthermore, a range of methods are being developed to distinguish selection and drift using these temporal designs (Goldringer & Bataillon 2004; Feder et al. 2014; Gompert 2015).
The way forward
There are a few ways in which future EE and NP studies should be focused to best support continued progress in our understanding of the genetics of adaptation. First, we need time series data from natural populations. This point has been touched upon already, but its importance warrants it a second mention. Adding this temporal dimension will give genome scan approaches a chance to become much more powerful. We need more explicit modelling of the interaction of various forms of selection and demography to generate clear expectations for footprints of selection instead of merely ‘rejecting’ selective neutrality, and time series data will make distinguishing these different footprints of selection more clear.
Second, there is a need to refocus EE studies towards experimental designs where evolution is tracked across a set of different environments where we manipulate the most salient features of environmental variation. We know indirectly, for example, that many complex, and potentially biotic, environmental factors are important for the survival of microbes in nature, as the vast majority of microbes have in fact proved impossible to culture using standard laboratory growth conditions (Vartoukian et al. 2010). Designing experiments aimed at looking across a range of environmental conditions will allow for a more comprehensive test of emerging quantitative theories about the DFE in multiple environments, revisit experimentally the importance of fine‐grained vs. coarse‐grained environmental variation, and test more extensively if adaptation in one environment generally comes with a sharp loss of adaptation in adjacent environments. A long‐term ambitious goal is to examine in the laboratory the extent to which adaptation can be seen as a sequential fine‐tuning of a population to a fixed environment or whether evolution can be best described as being moulded by extensive frequency‐dependent selection among interacting genotypes.
Finally, we need to continue to merge the NP and EE approaches more extensively, directly comparing data obtained from both types of studies. This kind of combined approach is clearly achievable for a range of model species (e.g. numerous bacteria species, Arabidopsis, C. elegans, Drosophila) and, in fact, is already starting to be carried out in yeast (Metzger et al. 2015) and bacteria (Maddamsetti et al. 2015). For example, Metzger et al. (2015) use a combination of EE and NP approaches to look for evidence of selection on mutations in a S. cerevisiae promoter region (TDH3). The authors compare the distribution of effects on gene expression of a random set of experimentally derived mutants (EE data) with the distribution of effects arising from naturally occurring polymorphisms (NP data), suggesting that a mismatch between these two distributions indicates that selection has shaped the phenotypes seen in the naturally occurring isolates. Using this combined approach, they are able to show that the promoter of interest has been affected more by selection for low levels of expression noise than by selection for a particular level of expression.
Another recent study also makes direct comparisons between EE and NP data, this time in E. coli (Maddamsetti et al. 2015). This particular study is notable not only because it is one of the first to directly compare evidence from both EE and NP data to draw inferences about the processes driving evolution, but also because it suggests some key differences between the processes that drive evolution in EE and NP. These differences arise both from a mismatch in the timescale of evolution observed in EE and NP data and from differences between EE and NP in the potential for horizontal gene transfer (HGT) and recombination. As mentioned earlier in this review, HGT and recombination have been shown to be important processes involved in the adaptive evolution of a number of natural populations. However, to date there have been very few EE studies that explore the potential role of these processes – an important direction for future EE work. It is clear that studies that directly combine and compare data from the two very different but complimentary approaches of the EE and NP programmes will be key to revealing the true relevance and impact of the EE programme on our understanding of the genetics of adaptation in nature.
S.B. and T.B. both designed and wrote the manuscript.
Acknowledgements
SB and TB acknowledge financial support from the European Research Council under the European Union's Seventh Framework Program (FP7/20072013, ERC Grant 311341).
References
- Agrawal AF, Whitlock MC (2011) Inferences about the distribution of dominance drawn from yeast gene knockout data. Genetics, 187, 553–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves I, Šrámková Hanulová A, Foll M, Excoffier L (2012) Genomic data reveal a complex making of humans. PLoS Genetics, 8, e1002837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrés AM, Hubisz MJ, Indap A et al (2009) Targets of balancing selection in the human genome. Molecular Biology and Evolution, 26, 2755–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariey F, Witkowski B, Amaratunga C et al (2014) A molecular marker of artemisinin‐resistant Plasmodium falciparum malaria. Nature, 505, 50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey SF, Kassen R (2012) Spatial structure of ecological opportunity drives adaptation in a bacterium. The American Naturalist, 180, 270–283. [DOI] [PubMed] [Google Scholar]
- Bailey SF, Rodrigue N, Kassen R (2015) The effect of selection environment on the probability of parallel evolution. Molecular Biology and Evolution, 32, 1436–1448. [DOI] [PubMed] [Google Scholar]
- Bank C, Hietpas RT, Wong A, Bolon DN, Jensen JD (2014) A Bayesian MCMC approach to assess the complete distribution of fitness effects of new mutations: uncovering the potential for adaptive walks in challenging environments. Genetics, 196, 841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett RDH, Schluter D (2008) Adaptation from standing genetic variation. Trends in Ecology & Evolution, 23, 38–44. [DOI] [PubMed] [Google Scholar]
- Barrick JE, Yu DS, Yoon SH et al (2009) Genome evolution and adaptation in a long‐term experiment with Escherichia coli . Nature, 461, 1243–1247. [DOI] [PubMed] [Google Scholar]
- Barrick JE, Kauth MR, Strelioff CC, Lenski RE (2010) Escherichia coli rpoB mutants have increased evolvability in proportion to their fitness defects. Molecular Biology and Evolution, 27, 1338–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataillon T, Bailey SF (2014) Effects of new mutations on fitness: insights from models and data. Annals of the New York Academy of Sciences, 1320, 76–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataillon T, Zhang T, Kassen R (2011) Cost of adaptation and fitness effects of beneficial mutations in Pseudomonas fluorescens . Genetics, 189, 939–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beardmore JA (1961) Diurnal temperature fluctuation and genetic variance in Drosophila populations. Nature, 189, 162–163. [DOI] [PubMed] [Google Scholar]
- Bennett AF, Lenski RE, Mittler JE (1992) Evolutionary adaptation to temperature. I. Fitness responses of Escherichia coli to changes in its thermal environment. Evolution, 46, 16–30. [DOI] [PubMed] [Google Scholar]
- Bierne N, Welch J, Loire E, Bonhomme F, David P (2011) The coupling hypothesis: why genome scans may fail to map local adaptation genes. Molecular Ecology, 20, 2044–2072. [DOI] [PubMed] [Google Scholar]
- Bierne N, Roze D, Welch JJ (2013) Pervasive selection or is it…? why are FST outliers sometimes so frequent? Molecular Ecology, 22, 2061–2064. [DOI] [PubMed] [Google Scholar]
- Blair JM, Richmond GE, Piddock LJ (2014) Multidrug efflux pumps in Gram‐negative bacteria and their role in antibiotic resistance. Future Microbiology, 9, 1165–1177. [DOI] [PubMed] [Google Scholar]
- Blount ZD, Barrick JE, Davidson CJ, Lenski RE (2012) Genomic analysis of a key innovation in an experimental Escherichia coli population. Nature, 489, 513–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhurst MA, Colegrave N, Rozen DE (2011) Next‐generation sequencing as a tool to study microbial evolution. Molecular Ecology, 20, 972–980. [DOI] [PubMed] [Google Scholar]
- Buckling A, Craig Maclean R, Brockhurst MA, Colegrave N (2009) The Beagle in a bottle. Nature, 457, 824–829. [DOI] [PubMed] [Google Scholar]
- Bull J, Badgett M, Wichman H et al (1997) Exceptional convergent evolution in a virus. Genetics, 147, 1497–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke MK, Dunham JP, Shahrestani P et al (2010) Genome‐wide analysis of a long‐term evolution experiment with Drosophila . Nature, 467, 587–590. [DOI] [PubMed] [Google Scholar]
- Burke MK, Liti G, Long AD (2014) Standing genetic variation drives repeatable experimental evolution in outcrossing populations of Saccharomyces cerevisiae . Molecular Biology and Evolution, 31, 3228–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevin L‐M, Hospital F (2008) Selective sweep at a quantitative trait locus in the presence of background genetic variation. Genetics, 180, 1645–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevin L‐M, Martin G, Lenormand T (2010) Fisher's model and the genomics of adaptation: restricted pleiotropy, heterogenous mutation, and parallel evolution. Evolution, 64, 3213–3231. [DOI] [PubMed] [Google Scholar]
- Cobbs C, Heath J, Stireman JO III, Abbot P (2013) Carotenoids in unexpected places: gall midges, lateral gene transfer, and carotenoid biosynthesis in animals. Molecular Phylogenetics and Evolution, 68, 221–228. [DOI] [PubMed] [Google Scholar]
- Collins S (2010) Many possible worlds: expanding the ecological scenarios in experimental evolution. Evolutionary Biology, 38, 3–14. [Google Scholar]
- Collins S, de Meaux J (2009) Adaptation to different rates of environmental change in Chlamydomonas. Evolution; International Journal of Organic Evolution, 63, 2952–2965. [DOI] [PubMed] [Google Scholar]
- Colosimo PF, Hosemann KE, Balabhadra S et al (2005) Widespread parallel evolution in sticklebacks by repeated fixation of ectodysplasin alleles. Science, 307, 1928–1933. [DOI] [PubMed] [Google Scholar]
- Comeron JM (2014) Background selection as baseline for nucleotide variation across the Drosophila genome. PLoS Genetics, 10, e1004434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condon C, Cooper BS, Yeaman S, Angilletta MJ (2014) Temporal variation favors the evolution of generalists in experimental populations of Drosophila melanogaster . Evolution, 68, 720–728. [DOI] [PubMed] [Google Scholar]
- Connallon T, Clark AG (2014) Balancing selection in species with separate sexes: insights from Fisher's geometric model. Genetics, 197, 991–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte GL, Arnegard ME, Peichel CL, Schluter D (2012) The probability of genetic parallelism and convergence in natural populations. Proceedings of the Royal Society of London B: Biological Sciences, 279, 5039–5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coop G, Witonsky D, Di Rienzo A, Pritchard JK (2010) Using environmental correlations to identify loci underlying local adaptation. Genetics, 185, 1411–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corre VL, Roux F, Reboud X (2002) DNA polymorphism at the FRIGIDA gene in Arabidopsis thaliana: extensive nonsynonymous variation Is consistent with local selection for flowering time. Molecular Biology and Evolution, 19, 1261–1271. [DOI] [PubMed] [Google Scholar]
- De Mita S, Thuillet A‐C, Gay L et al (2013) Detecting selection along environmental gradients: analysis of eight methods and their effectiveness for outbreeding and selfing populations. Molecular Ecology, 22, 1383–1399. [DOI] [PubMed] [Google Scholar]
- Desai MM, Fisher DS (2007) Beneficial mutation–selection balance and the effect of linkage on positive selection. Genetics, 176, 1759–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettman JR, Rodrigue N, Melnyk AH et al (2012) Evolutionary insight from whole‐genome sequencing of experimentally evolved microbes. Molecular Ecology, 21, 2058–2077. [DOI] [PubMed] [Google Scholar]
- Feder JL, Nosil P, Flaxman SM (2014) Assessing when chromosomal rearrangements affect the dynamics of speciation: implications from computer simulations. Frontiers in Genetics, 5, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraisse C, Belkhir K, Welch JJ, Bierne N (2015) Local inter‐species introgression is the main cause of extreme levels of intra‐specific differentiation in mussels. Molecular Ecology, 25, 269–286. [DOI] [PubMed] [Google Scholar]
- Gagneux S (2012) Host–pathogen coevolution in human tuberculosis. Philosophical Transactions of the Royal Society B: Biological Sciences, 367, 850–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrish PJ, Lenski RE (1998) The fate of competing beneficial mutations in an asexual population. Genetica, 102, 127–144. [PubMed] [Google Scholar]
- Gifford DR, Schoustra SE, Kassen R (2011) The length of adaptive walks is insensitive to starting fitness in Aspergillus nidulans . Evolution, 65, 3070–3078. [DOI] [PubMed] [Google Scholar]
- Glémin S, Ronfort J (2013) Adaptation and maladaptation in selfing and outcrossing species: new mutations versus standing variation. Evolution; International Journal of Organic Evolution, 67, 225–240. [DOI] [PubMed] [Google Scholar]
- Goldringer I, Bataillon T (2004) On the distribution of temporal variations in allele frequency: consequences for the estimation of effective population size and the detection of loci undergoing selection. Genetics, 168, 563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompert Z (2015) Bayesian inference of selection in a heterogeneous environment from genetic time‐series data. Molecular Ecology, 25, 121–134. [DOI] [PubMed] [Google Scholar]
- Good BH, Desai MM (2015) The impact of macroscopic epistasis on long‐term evolutionary dynamics. Genetics, 199, 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JC, Cutter AD (2014) Mainstreaming Caenorhabditis elegans in experimental evolution. Proceedings of the Royal Society of London B: Biological Sciences, 281, 20133055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivet D, Sebastiani F, Alía R et al (2011) Molecular footprints of local adaptation in two Mediterranean conifers. Molecular Biology and Evolution, 28, 101–116. [DOI] [PubMed] [Google Scholar]
- Gronau I, Arbiza L, Mohammed J, Siepel A (2013) Inference of natural selection from interspersed genomic elements based on polymorphism and divergence. Molecular Biology and Evolution, 30, 1159–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günther T, Coop G (2013) Robust identification of local adaptation from allele frequencies. Genetics, 195, 205–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD (2009) Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genetics, 5, e1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane JBS (1937) The effect of variation of fitness. The American Naturalist, 71, 337–349. [Google Scholar]
- Haley CS, Birley AJ (1983) The genetical response to natural selection by varied environments. II. Observations on replicate populations in spatially varied laboratory environments. Heredity, 51(Pt 3), 581–606. [DOI] [PubMed] [Google Scholar]
- Hall AR, Colegrave N (2008) Decay of unused characters by selection and drift. Journal of Evolutionary Biology, 21, 610–617. [DOI] [PubMed] [Google Scholar]
- Halligan DL, Keightley PD (2009) Spontaneous mutation accumulation studies in evolutionary genetics. Annual Review of Ecology, Evolution, and Systematics, 40, 151–172. [Google Scholar]
- Harms MJ, Thornton JW (2014) Historical contingency and its biophysical basis in glucocorticoid receptor evolution. Nature, 512, 203–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hereford J (2009) A quantitative survey of local adaptation and fitness trade‐offs. The American Naturalist, 173, 579–588. [DOI] [PubMed] [Google Scholar]
- Hermisson J, Pennings PS (2005) Soft sweeps molecular population genetics of adaptation from standing genetic variation. Genetics, 169, 2335–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herron MD, Doebeli M (2013) Parallel evolutionary dynamics of adaptive diversification in Escherichia coli . PLoS Biology, 11, e1001490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hietpas RT, Jensen JD, Bolon DN (2011) Experimental illumination of a fitness landscape. Proceedings of the National Academy of Sciences, 108, 7896–7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hietpas RT, Bank C, Jensen JD, Bolon DNA (2013) Shifting fitness landscapes in response to altered environments. Evolution, 67, 3512–3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra HE, Hirschmann RJ, Bundey RA, Insel PA, Crossland JP (2006) A single amino acid mutation contributes to adaptive beach mouse color pattern. Science, 313, 101–104. [DOI] [PubMed] [Google Scholar]
- Huang Y, Wright SI, Agrawal AF (2014) Genome‐wide patterns of genetic variation within and among alternative selective regimes. PLoS Genetics, 10, e1004527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen G, Barbosa C, Schulenburg H (2013) Experimental evolution as an efficient tool to dissect adaptive paths to antibiotic resistance. Drug Resistance Updates, 16, 96–107. [DOI] [PubMed] [Google Scholar]
- Jaramillo‐Correa J‐P, Rodríguez‐Quilón I, Grivet D et al (2015) Molecular proxies for climate maladaptation in a long‐lived tree (Pinus pinaster Aiton, Pinaceae). Genetics, 199, 793–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasmin J‐N, Kassen R (2007) Evolution of a single niche specialist in variable environments. Proceedings of the Royal Society of London B: Biological Sciences, 274, 2761–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Mishra P, Hietpas RT, Zeldovich KB, Bolon DNA (2013) Latent effects of Hsp90 mutants revealed at reduced expression levels. PLoS Genetics, 9, e1003600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MR, Good JM (2015) Targeted capture in evolutionary and ecological genomics. Molecular Ecology, 25, 185–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A, Thompson JN (1997) Adaptation and specialization in a two‐resource environment in Drosophila species. Evolution, 51, 846–855. [DOI] [PubMed] [Google Scholar]
- Kang HM, Zaitlen NA, Wade CM et al (2008) Efficient control of population structure in model organism association mapping. Genetics, 178, 1709–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao KC, Sherlock G (2008) Molecular characterization of clonal interference during adaptive evolution in asexual populations of Saccharomyces cerevisiae . Nature Genetics, 40, 1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassen R (2014) Experimental evolution and the nature of biodiversity. Roberts.
- Kassen R, Bell G (1998) Experimental evolution in Chlamydomonas. IV. Selection in environments that vary through time at different scales. Heredity, 80, 732–741. [Google Scholar]
- Kawecki TJ, Ebert D (2004) Conceptual issues in local adaptation. Ecology Letters, 7, 1225–1241. [Google Scholar]
- Keightley PD, Eyre‐Walker A (2010) What can we learn about the distribution of fitness effects of new mutations from DNA sequence data? Philosophical Transactions of the Royal Society of London B: Biological Sciences, 365, 1187–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley PD, Kacser H (1987) Dominance, pleiotropy and metabolic structure. Genetics, 117, 319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Cui M‐L, Cubas P et al (2008) Regulatory genes control a key morphological and ecological trait transferred between species. Science, 322, 1116–1119. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick M, Guerrero RF (2014) Signatures of sex‐antagonistic selection on recombining sex chromosomes. Genetics, 197, 531–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kousathanas A, Keightley PD (2013) A comparison of models to infer the distribution of fitness effects of new mutations. Genetics, 193, 1197–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamichhaney S, Berglund J, Almén MS et al (2015) Evolution of Darwin/'s finches and their beaks revealed by genome sequencing. Nature, 518, 371–375. [DOI] [PubMed] [Google Scholar]
- Lang GI, Botstein D, Desai MM (2011) Genetic variation and the fate of beneficial mutations in asexual populations. Genetics, 188, 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GI, Rice DP, Hickman MJ et al (2013) Pervasive genetic hitchhiking and clonal interference in forty evolving yeast populations. Nature, 500, 571–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gac M, Plucain J, Hindré T, Lenski RE, Schneider D (2012) Ecological and evolutionary dynamics of coexisting lineages during a long‐term experiment with Escherichia coli . Proceedings of the National Academy of Sciences, 109, 9487–9492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leimu R, Fischer M (2008) A meta‐analysis of local adaptation in plants. PLoS One, 3, e4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenormand T, Chevin L‐M, Bataillon T (2015) Parallel evolution: what does it (not) tell us and why is it (still) interesting? In: Chance in Evolution. Chicago University Press, Chicago, Illinois. [Google Scholar]
- Levy SF, Blundell JR, Venkataram S et al (2015) Quantitative evolutionary dynamics using high‐resolution lineage tracking. Nature, 519, 181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Li H, Jakobsson M et al (2012) Joint analysis of demography and selection in population genetics: where do we stand and where could we go? Molecular Ecology, 21, 28–44. [DOI] [PubMed] [Google Scholar]
- Lind PA, Farr AD, Rainey PB (2015) Experimental evolution reveals hidden diversity in evolutionary pathways. eLife, 4, e07074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losos JB (2011) Convergence, adaptation, and constraint. Evolution, 65, 1827–1840. [DOI] [PubMed] [Google Scholar]
- Lotterhos KE, Whitlock MC (2014) Evaluation of demographic history and neutral parameterization on the performance of FST outlier tests. Molecular Ecology, 23, 2178–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunzer M, Golding GB, Dean AM (2010) Pervasive cryptic epistasis in molecular evolution. PLoS Genetics, 6, e1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay TFC (1981) Genetic variation in varying environments. Genetics Research, 37, 79–93. [Google Scholar]
- MacLean RC, Bell G, Rainey PB (2004) The evolution of a pleiotropic fitness tradeoff in Pseudomonas fluorescens . Proceedings of the National Academy of Sciences of the United States of America, 101, 8072–8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddamsetti R, Hatcher PJ, Cruveiller S et al (2015) Synonymous genetic variation in natural isolates of Escherichia coli does not predict where synonymous substitutions occur in a long‐term experiment. Molecular Biology and Evolution, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharjan RP, Liu B, Feng L, Ferenci T, Wang L (2015) Simple phenotypic sweeps hide complex genetic changes in populations. Genome Biology and Evolution, 7, 531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna F, Martin G, Lenormand T (2011) Fitness landscapes: an alternative theory for the dominance of mutation. Genetics, 189, 923–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Lenormand T (2006) A general multivariate extension of Fisher's geometrical model and the distribution of mutation fitness effects across species. Evolution, 60, 893–907. [PubMed] [Google Scholar]
- Martin G, Lenormand T (2008) The distribution of beneficial and fixed mutation fitness effects close to an optimum. Genetics, 179, 907–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Lenormand T (2015) The fitness effect of mutations across environments: fisher's geometrical model with multiple optima. Evolution; International Journal of Organic Evolution, 69, 1433–1447. [DOI] [PubMed] [Google Scholar]
- Martin A, Orgogozo V (2013) The loci of repeated evolution: a catalog of genetic hotspots of phenotypic variation. Evolution, 67, 1235–1250. [DOI] [PubMed] [Google Scholar]
- Maughan H, Nicholson WL (2011) Increased fitness and alteration of metabolic pathways during Bacillus subtilis evolution in the laboratory. Applied and Environmental Microbiology, 77, 4105–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald MJ, Gehrig SM, Meintjes PL, Zhang X‐X, Rainey PB (2009) Adaptive divergence in experimental populations of Pseudomonas fluorescens. IV. Genetic constraints guide evolutionary trajectories in a parallel adaptive radiation. Genetics, 183, 1041–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeown AN, Bridgham JT, Anderson DW et al (2014) Evolution of DNA specificity in a transcription factor family produced a new gene regulatory module. Cell, 159, 58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnyk AH, Kassen R (2011) Adaptive landscapes in evolving populations of Pseudomonas fluorescens . Evolution, 65, 3048–3059. [DOI] [PubMed] [Google Scholar]
- Metzger BPH, Yuan DC, Gruber JD, Duveau F, Wittkopp PJ (2015) Selection on noise constrains variation in a eukaryotic promoter. Nature, 521, 344–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer JR, Dobias DT, Weitz JS et al (2012) Repeatability and contingency in the evolution of a key innovation in phage Lambda. Science, 335, 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel AC, Joyce P, Wichman HA, Miller CR (2012) Stickbreaking: a novel fitness landscape model that harbors epistasis and is consistent with commonly observed patterns of adaptive evolution. Genetics, 190, 655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Lawrence JG, Groisman EA (2000) Lateral gene transfer and the nature of bacterial innovation. Nature, 405, 299–304. [DOI] [PubMed] [Google Scholar]
- Orr HA (2005) The probability of parallel evolution. Evolution, 59, 216–220. [PubMed] [Google Scholar]
- Orr HA, Betancourt AJ (2001) Haldane's sieve and adaptation from the standing genetic variation. Genetics, 157, 875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J, Tsagkogeorga G, Cotton JA et al (2013) Genome‐wide signatures of convergent evolution in echolocating mammals. Nature, 502, 228–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parts L, Cubillos FA, Warringer J et al (2011) Revealing the genetic structure of a trait by sequencing a population under selection. Genome Research, 21, 1131–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson S, Vogwill T, Buckling A et al (2010) Antagonistic coevolution accelerates molecular evolution. Nature, 464, 275–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfeito L, Sousa A, Bataillon T, Gordo I (2014) Rates of fitness decline and rebound suggest pervasive epistasis. Evolution; International Journal of Organic Evolution, 68, 150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perron GG, Lee AEG, Wang Y, Huang WE, Barraclough TG (2012) Bacterial recombination promotes the evolution of multi‐drug‐resistance in functionally diverse populations. Proceedings of the Royal Society of London B: Biological Sciences, 279, 1477–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter BM, Huerta‐Sanchez E, Nielsen R (2012) Distinguishing between selective sweeps from standing variation and from a de novo mutation. PLoS Genetics, 8, e1003011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plucain J, Hindré T, Gac ML et al (2014) Epistasis and allele specificity in the emergence of a stable polymorphism in Escherichia coli . Science, 343, 1366–1369. [DOI] [PubMed] [Google Scholar]
- Poon AFY, Swenson LC, Bunnik EM et al (2012) Reconstructing the dynamics of HIV evolution within hosts from serial deep sequence data. PLoS Computational Biology, 8, e1002753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JR (1971) Genetic polymorphisms in varied environments. Science, 174, 1035–1036. [DOI] [PubMed] [Google Scholar]
- Presloid JB, Ebendick‐Corpus BE, Zárate S, Novella IS (2008) Antagonistic pleiotropy involving promoter sequences in a virus. Journal of molecular biology, 382, 342–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S, Bergero R, Guirao-Rico S et al. (2015) RAD‐mapping reveals an evolving, polymorphic and fuzzy boundary of a plant pseudoautosomal region. Molecular Ecology, 25, 414–430. [DOI] [PubMed] [Google Scholar]
- Racimo F, Schraiber JG (2014) Approximation to the distribution of fitness effects across functional categories in human segregating polymorphisms. PLoS Genetics, 10, e1004697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey PB, Travisano M (1998) Adaptive radiation in a heterogeneous environment. Nature, 394, 69–72. [DOI] [PubMed] [Google Scholar]
- Rockman MV (2012) The QTN program and the alleles that matter for evolution: all that's gold does not glitter. Evolution, 66, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokyta DR, Beisel CJ, Joyce P et al (2008) Beneficial fitness effects are not exponential for two viruses. Journal of Molecular Evolution, 67, 368–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig RF, Sharp RR, Treves DS, Adams J (1994) Microbial evolution in a simple unstructured environment: genetic differentiation in Escherichia coli . Genetics, 137, 903–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen DE, Lenski RE (2000) Long‐term experimental evolution in Escherichia coli. VIII. Dynamics of a balanced polymorphism. The American Naturalist, 155, 24–35. [DOI] [PubMed] [Google Scholar]
- Savolainen O, Lascoux M, Merilä J (2013) Ecological genomics of local adaptation. Nature Reviews Genetics, 14, 807–820. [DOI] [PubMed] [Google Scholar]
- Schoustra SE, Bataillon T, Gifford DR, Kassen R (2009) The properties of adaptive walks in evolving populations of fungus. PLoS Biology, 7, e1000250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sella G, Petrov DA, Przeworski M, Andolfatto P (2009) Pervasive natural selection in the Drosophila genome? PLoS Genetics, 5, e1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellis D, Callahan BJ, Petrov DA, Messer PW (2011) Heterozygote advantage as a natural consequence of adaptation in diploids. Proceedings of the National Academy of Sciences, 108, 20666–20671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servedio MR, Brandvain Y, Dhole S et al (2014) Not just a theory—The utility of mathematical models in evolutionary biology. PLoS Biology, 12, e1002017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver RS, Mateles RI (1969) Control of mixed‐substrate utilization in continuous cultures of Escherichia coli . Journal of Bacteriology, 97, 535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simões P, Santos J, Fragata I et al (2008) How repeatable Is adaptive evolution? The role of geographical origin and founder effects in laboratory adaptation. Evolution, 62, 1817–1829. [DOI] [PubMed] [Google Scholar]
- Sironi M, Cagliani R, Forni D, Clerici M (2015) Evolutionary insights into host‐pathogen interactions from mammalian sequence data. Nature Reviews Genetics, 16, 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Endepols S, Klemann N et al (2011) Adaptive introgression of anticoagulant rodent poison resistance by hybridization between old world mice. Current Biology, 21, 1296–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spor A, Kvitek DJ, Nidelet T et al (2014) Phenotypic and genotypic convergences are influenced by historical contingency and environment in yeast. Evolution, 68, 772–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan W (2015) Signatures of positive selection: from selective sweeps at individual loci to subtle allele frequency changes in polygenic adaptation. Molecular Ecology, 25, 79–88. [DOI] [PubMed] [Google Scholar]
- Stinchcombe JR, Weinig C, Ungerer M et al (2004) A latitudinal cline in flowering time in Arabidopsis thaliana modulated by the flowering time gene FRIGIDA. Proceedings of the National Academy of Sciences of the United States of America, 101, 4712–4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon O (2014) The utility of Fisher's geometric model in evolutionary genetics. Annual Review of Ecology, Evolution, and Systematics, 45, 179–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon O, Rodríguez‐Verdugo A, Gaut RL et al (2012) The molecular diversity of adaptive convergence. Science, 335, 457–461. [DOI] [PubMed] [Google Scholar]
- Thépot S, Restoux G, Goldringer I et al (2015) Efficiently tracking selection in a multiparental population: the case of earliness in wheat. Genetics, 199, 609–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartoukian SR, Palmer RM, Wade WG (2010) Strategies for culture of “unculturable” bacteria. FEMS Microbiology Letters, 309, 1–7. [DOI] [PubMed] [Google Scholar]
- Venail PA, Kaltz O, Olivieri I, Pommier T, Mouquet N (2011) Diversification in temporally heterogeneous environments: effect of the grain in experimental bacterial populations. Journal of Evolutionary Biology, 24, 2485–2495. [DOI] [PubMed] [Google Scholar]
- Vitti JJ, Grossman SR, Sabeti PC (2013) Detecting natural selection in genomic data. Annual Review of Genetics, 47, 97–120. [DOI] [PubMed] [Google Scholar]
- Wang R‐L, Stec A, Hey J, Lukens L, Doebley J (1999) The limits of selection during maize domestication. Nature, 398, 236–239. [DOI] [PubMed] [Google Scholar]
- Wong A, Kassen R (2011) Parallel evolution and local differentiation in quinolone resistance in Pseudomonas aeruginosa. Microbiology, 157, 937–944. [DOI] [PubMed] [Google Scholar]
- Wong A, Rodrigue N, Kassen R (2012) Genomics of adaptation during experimental evolution of the opportunistic pathogen Pseudomonas aeruginosa. PLoS Genetics, 8, e1002928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolhouse MEJ, Webster JP, Domingo E, Charlesworth B, Levin BR (2002) Biological and biomedical implications of the co‐evolution of pathogens and their hosts. Nature Genetics, 32, 569–577. [DOI] [PubMed] [Google Scholar]
- Yu J, Pressoir G, Briggs WH et al (2006) A unified mixed‐model method for association mapping that accounts for multiple levels of relatedness. Nature Genetics, 38, 203–208. [DOI] [PubMed] [Google Scholar]
- Zhen Y, Aardema ML, Medina EM, Schumer M, Andolfatto P (2012) Parallel molecular evolution in an herbivore community. Science, 337, 1634–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Miller SP, Dykhuizen DE, Dean AM (2009) Transcription, translation, and the evolution of specialists and generalists. Molecular Biology and Evolution, 26, 2661–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]