

Abstract
Phenotypic and genotypic characteristics of the cat can be obtained from single nucleotide polymorphisms (SNPs) analyses of fur. This study developed miniplexes using SNPs with high discriminating power for random‐bred domestic cats, focusing on individual and phenotypic identification. Seventy‐eight SNPs were investigated using a multiplex PCR followed by a fluorescently labeled single base extension (SBE) technique (SNaPshot®). The SNP miniplexes were evaluated for reliability, reproducibility, sensitivity, species specificity, detection limitations, and assignment accuracy. Six SNPplexes were developed containing 39 intergenic SNPs and 26 phenotypic SNPs, including a sex identification marker, ZFXY. The combined random match probability (cRMP) was 6.58 × 10−19 across all Western cat populations and the likelihood ratio was 1.52 × 1018. These SNPplexes can distinguish individual cats and their phenotypic traits, which could provide insight into crime reconstructions. A SNP database of 237 cats from 13 worldwide populations is now available for forensic applications.
Keywords: forensic science, forensic genetics, animal forensics, Felis silvestris catus, single base extension, single nucleotide polymorphism
Cats are a common inhabitant and fixture in human households. Approximately 86.4 million cats are owned within the United States with approximately 30% of households having cats. Each cat‐owning household has an average of 2.2 cats 1. In the USA, 85–90% of household cats are from random‐bred populations. Pedigreed fancy breeds, such as Persian, Siamese or Maine Coon, represent only 10–15% of the USA cat population 1, 2, 3. A by‐product of owning, interacting with, or being in a household with a cat is the transfer of shed fur to clothing or personal objects 4. Cat fur obtained from crime scenes has the potential to link perpetrators, accomplices, witnesses, and victims. Cat hairs can persist and be transferred without the actual presence of the cat. In a simulated crime scene of a burglary and assault, the Angora cat witness transferred ~311 hairs during the burglary and ~255 hairs during the assault 5. As cats are incessant groomers, cat fur can have nucleated cells, not only in the hair bulb, but also as epithelial cells on the hair shaft deposited during the grooming process 6, 7. Although an abundance of cat hair trace evidence can be left behind at crime scenes, these hairs are a relatively untapped resource.
Animal forensics is implementing the same technologies and tools used in human forensics, thereby encouraging the development of more efficient identification systems and databases for animals. Single nucleotide polymorphisms (SNPs) are a complementary resource to short tandem repeats (STRs) for individual identification 8, 9, 10, 11, and SNPs can provide the added value of phenotypic (externally visible characters – EVC) characterization of the contributor as well as biogeographical ancestry (BGA) 12. Although individual identification cannot be solely established, EVCs and BGAs can provide forensic inferences in helping solve missing person's cases or unidentified remains. For example, in the Madrid bombing attack investigation in 2004, an ancestry informative SNP assay led to the apprehension of a suspect whose STR profile was not in a DNA database 13.
Feline phenotypic DNA variants can also be exploited for physical trait identification purposes. Most cats can be defined by their phenotypic appearance using a very limited number of single‐gene genetic traits with known variants that affect coat color, length, and texture (see reviews, 14, 15, 16). Some phenotypes are breed specific; however, many define coat colors and fur lengths that are common to the variation of randomly bred domestics, the most common of pet cats. Cats are an excellent species to demonstrate the “proof‐of‐principle” that a panel of variants can accurately predict the phenotype of a contributor, particularly using a few cat hairs. In addition, BGAs have been developed for cat breeds and these SNPs can also biogeographically define populations or “races” of random‐bred cats 17, 18.
This study initiated the development of a SNP‐based assay using highly discriminative feline‐derived BGA and EVC markers, based upon the SNaPshot® technology (Applied Biosystems, Foster City, CA). The cat SNP panels were validated following SWGDAM revised guidelines 11, 19. If DNA sources are available, specifically cat hair, the cat's genotypic profile and its predicted phenotypes may support the apprehension of appropriate suspects involved with crimes.
Materials and Methods
DNA Sample Selection and DNA Purification
Archival cat DNA samples represented both random‐bred cats from the USA and a wide geographical distribution of 13 populations from the Americas and Europe 17, 18, 20 (Table 1). Samples included approximately 16 unrelated cats per population (n = 203) and cats with genetically defined phenotypes (n = 48) to determine phenotypic concordance with the EVC SNPs. Related cats (n = 72) were genotyped for parentage analysis of an Oriental Shorthair family. Two additional cats were used for sensitivity, inhibition, and precision studies.
Table 1.
Cats genotyped using SNPplexes and population statistics
| Population | No. | Study | No. Faileda | Average Ho | Informative loci (H)b |
|---|---|---|---|---|---|
| Brazil_Rio de Jenerio | 16 | Validation/Concordance | 4 | 0.3495 | 54 |
| Canada_Vancouver | 15 | Validation | 0 | 0.3551 | 53 |
| France_Lyon | 16 | Validation | 1 | 0.3234 | 55 |
| Italy_Rome | 15 | Validation/Concordance | 2 | 0.3369 | 55 |
| United Kingdom_East Sussex | 16 | Validation/Concordance | 0 | 0.3117 | 55 |
| US_California | 16 | Validation | 0 | 0.3287 | 54 |
| US_Florida | 15 | Validation | 0 | 0.3399 | 58 |
| US_Kansas | 15 | Validation | 0 | 0.3863 | 58 |
| US_Missouri | 16 | Validation/Concordance | 0 | 0.3402 | 57 |
| US_NY | 15 | Validation | 5 | 0.3410 | 52 |
| US_Ohio | 16 | Validation | 0 | 0.2787 | 45 |
| US_Pennsylvania | 16 | Validation | 0 | 0.3415 | 57 |
| US_Texas | 16 | Validation | 4 | 0.3443 | 54 |
| 13 locations | 203 | 16 | 0.3352 ± 0.024 | 54 ± 3.8 | |
| Mixed bred controls | 48 | Phenotype Concordance | 0 | ||
| Oriental Shorthairs | 72 | Pedigree Analysis | 0 | 0.3141 | 47 |
| US_California | 2 | Sensitivity/Inhibition/ Precision | 0 | ||
| 325 | 16 |
All DNA archival samples were previously quantified and were not retested prior to genotyping. For the two cats used in the sensitivity, reproducibility, precision, and inhibition studies, DNA was quantified using a Biophotometer UV spectrophotometer (Eppendorf, Hauppauge, New York). Diluted samples with low DNA (< 100 pg) were quantified using a real‐time feline‐specific quantitative PCR (qPCR) on an ABI 7300 Real‐Time PCR System (Applied Biosystems, Foster City, CA) 21. An internal positive control was used to standardize DNA quantities 21.
SNP Selection
A previous study used 148 intergenic SNPs to examine domestic cat population structuring 17. The genotypic data from this previous study were analyzed to select a subset of BGA SNPs for the forensic applications in this study. The statistical program FSTAT V.2.9.3 was used to determine G ST and heterozygosity data, based on Nei, Weir, and Cockerham estimators 22, 23, 24. The software program POPGENE V.1.32 25 was used to perform the pair‐wise linkage disequilibrium (D') tests based on Ohta's method 26, 27. Total variance of linkage disequilibrium was measured in di‐loci (DIT)², within population (DIS)² and between populations (DST)². A subset of the 148 SNPs (n = 49) were chosen based upon the following criteria: (i) high heterozygosity (>0.35), (ii) low G st (≤0.06), and (iii) low linkage disequilibrium (LD) across all random‐bred populations used in the previous study.
SNPs from 13 genes that are causal for 29 EVCs in cats, including sex and also blood type, were included to complement the BGA SNPs 14, 15, 16 (Table 2). Combining 49 BGA and 29 EVC SNPs, 78 SNPs were analyzed to develop the SNP panels. Excluding the familial cats, the same statistical analyses, and Shannon's information index (H') 28, 29, were re‐calculated on the selected SNPs for the cat populations analyzed in this study.
Table 2.
Phenotypic cat traits (eternally visible characters, EVCs) with known mutations in SNPplexes.a
| Trait (alleles)b | MOIc | Phenotype | Breeds | Gene | Mutation & Position | SNP |
|---|---|---|---|---|---|---|
| Agouti (A + , a) | AR | Banded to solid Charcoal | All cats Bengal | ASIP |
c.41G>C, c.122_123delCA c.142C>T |
ASIP‐2, ASIP‐1 ASIP‐3 |
| Brown (B + , b, b l ) | AR | Brown, light brown chocolate | All cats | TYRP1 d | b = c.8C>G, b = c.120C>G, bl = c.298C>T | TYRP1‐1, TYRP1‐2, TYRP1‐3 |
| Color (C + , C b , C s , c) | AR | Burmese color, Siamese color Full albino | All breeds | TYR | cb = c.715G>T, cs = c.940G>A, c = c.975delC |
TYR‐1
TYR‐3 TYR‐2 |
| Dilution (D + , d) | AR | Black to gray/blue, | All cats | MLPH | c.83delT | MLPH |
| Extension (E + , e) – Amber | AR | Brown/red color variant | Norwegian Forest Cat | MC1R | c.250G>A | MC1R |
| Gloves (G + , g) | AR | White feet | Birman | KIT | c.1035_1036delinsCA | KIT |
| Hairless (Hr + , hr) | AR | Atrichia | Sphynx | KRT71 | c.816 + 1G>A | KRT71‐A |
| Long fur (L + , l) | AR | Long fur | All cats | FGF5 | c.356_367insT, c.406C>T, c.474delT, c.475A>C |
FGF5‐4, FGF5‐3,
FGF5‐1, FGF5‐2 |
| Rexing (R + , r) | AR | Curly hair coat | Cornish Rex | PYP2R5 | c.250_253delTTTG | PYP2R5 |
| Rexing (Re + , re) | AR | Curly hair coat | Devon Rex | KRT71 | c.1108‐4_1184del, c.1184_1185insAGTTGGAG, c.1196insT | KRT71‐B |
| Rexing (Re S , Re + ) | AD | Curly hair coat | Selkirk Rex | KRT71 | c.445‐1G>C | KRT71‐C |
| Tabby (T M , t b ) | AR | Blotched/classic pattern | All cats | TAQPEP | C.2524G>A | TAQPEP |
| AB Blood Type (A + , b) | AR | Determines Type B | All cats | CMAH |
c.139G>A, c.199G>A c.353C>T |
CMAH‐3, CMAH‐2
CMAH‐1 |
| Polydactyl (Pd, pd + ) | AD | Extra appendage | All cats | SHH |
c.257G>C, c.479A>G c.481A>T |
SHH‐1, SHH‐2, SHH‐3 |
| Sex | AR | Sex Determination | All cats | ZFXY | Y – 163 bp, X = 166 bp | ZFX, ZFY |
“+” implies the wild‐type allele.
MOI implies mode of inheritance: AR is autosomal recessive, AD is autosomal dominant.
The variant b = −5IVS6, in TYRP1, is associated with brown coloration. The tested variants are in linkage disequilibrium with this casual variant.
PCR Primer Design
The sequences for each SNP locus were obtained from either GenBank or prior studies 17, 30 and primers for PCR amplification were designed using online software Primer3Plus 31 and NetPrimer (www.premierbiosoft.com/netprimer/index.html). Each primer pair was tested for potential primer dimers and secondary hairpin structures using the Auto Dimer software (www.cstl.nist.gov/strbase/AutoDimerHomepage/AutoDimerProgramHomepage.htm). All SNP and single base extension (SBE) primers (see below) were verified with the sequence databases at the National Center for Biotechnology Information (NCBI) via BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Seven PCR amplicons amplifying the genes ASIP, CMAH, FGF5, SHH, TYR, TYRP1, and ZFXY were designed to evaluate multiple EVC variants within each amplicon. Primers for PCR amplification were desalted (Integrated DNA Technology (IDT) Coralville, IA) (Table S1).
A singleton PCR and direct Sanger‐sequencing was performed to test all primer pairs for amplification of the correct product and to verify correct genotype calls for each locus (data not shown). To optimize the multiplex PCRs, a temperature gradient of 50–70°C was tested, MgCl2 concentration was varied from 2.0–8.0 mM, and the PCR primer concentrations were adjusted to balance product amplification at each locus. The final PCR multiplex reactions were conducted in a 15 μL reaction volume, containing 1 U Taq polymerase (Denville Scientific, South Plainfield, NJ), 800 mM of each dNTP (ThermoFisher Scientific, Waltham, MA), 2 μL of the PCR primers, premixed with 0.2–1.0 μM of each primer (IDT), 1× PCR Buffer with 0.5% BSA, 5.0 mM MgCl2, and 5 μM of betaine. The thermal cycling conditions were 94°C for 4 min, 35 cycles of 94°C for 30 sec, 63°C for 30 sec, 72°C for 10 sec, and final extension of 72°C for 10 min. All amplifications were performed using the GeneAmp PCR Thermal Cycler 9700 System (Applied Biosystems). To verify all loci amplified their expected amplicon size, amplification products were examined on a 4% agarose gel with 0.05% ethidium bromide for 120 min at 90 V. Unincorporated dNTPs and excess primers were removed by adding 2 μL of ExoSAP‐IT (Affymetrix, Santa Clara, CA) to the post‐PCR product, which was incubated at 37°C for 20 min followed by 15 min at 75°C for enzyme inactivation.
SBE (mini‐sequencing) Primer Design
SBE primers were designed using online software, BatchPrimer3 (v.1.0) 32. SNPplexes containing >10 SNP loci per panel were developed following criteria per the ABI SNaPshot® Multiplex Kit protocol (Applied Biosystems) 33 and the Primer Focus kit 34. The melting temperature of the primers ranged from 60 ± 3°C. The mobility ranges were from 24 to 90 bp (Table S2). Opposite allele combinations such as A/T and C/G were used for overlapping SNP loci when necessary. All SBE primers were HPLC purified (IDT).
Multiplex Mini‐sequencing (SNaPshot®) Conditions
Each SNP was verified by a singleton mini‐sequencing reaction (data not shown). Multiplex reactions were conducted in 5 μL reaction volume including 1.0 μL of the SNaPshot® Multiplex Ready Reaction Mix, 2.0 μL of the 1:10 diluted purified PCR product, and 2.0 μL of the SBE primers, premixed with 0.15–0.8 μM of each primer. The thermal cycling conditions were as follows: 96°C for 30 sec, 50°C for 5 sec, and 60°C for 30 sec, for 25 cycles. Due to high GC content of the PCR templates, the initial denaturation was extended from the original SNaPshot® protocol of 96°C from 10 to 30 sec. Reaction products were purified as described above.
Allele Detection and Analysis
The purified mini‐sequencing products (1 μL) were combined with 8.9 μL of Hi‐Di formamide (Applied Biosystems) and 0.1 μL of GeneScan‐120 LIZ (Applied Biosystems) for a final volume of 10 μL. Products were electrophorectically separated on a 48‐capillary ABI PRISM 3730 DNA Analyzer equipped with 36‐cm capillaries using POP‐7™ and 10 × 3730 Running Buffer (Applied Biosystems). The injection time was 10 sec and run time was 1200 sec at 15 kV. The spectral calibration and instrument protocols were analyzed using the ABI SNaPshot® Multiplex Kit protocol (Applied Biosytems) 33. Genotyping results were analyzed using GeneMapper (v.3.5) (Applied Biosytems) 35.
Peak height ratios were used to support allele calls as previously described 36. The instrument detection limits were set with a minimum peak threshold of 120 Relative Fluorescent Units (RFUs) (dR110 – blue), 60 RFUs (dR6 – green), 30 RFUs (dTamara – yellow, dROX – red, and LIZ – orange). The minimum peak height threshold was ≥300 RFU. The overall ratio between the signal strengths of the different fluorophores dR110 (blue), dR6G (green), TAMARA (yellow), and dROX (red) was 4:2:1:1, respectively. Using the SNaPshot® Primer Focus kit (Applied Biosystems), all peaks relating to the SNPs were placed into bins based upon their sizes corresponding to the internal size standard. GeneMapper software (v.4.1) 35 was used to determine quality of the genotypes.
Concordance
For a subset of cats, concordance was determined by comparing genotyping data from other assay technologies. The BGA and the EVC SNPs were compared to data generated from 43 to 48 cats, respectively, that were previously genotyped using the GoldenGate arrays (illumina, Inc., San Diego, CA) 17. A subset of cats were also genotyped for eight BGA and the EVC SNPs using the iPlex assay (Sequenom, San Diego, CA). In addition, some EVC SNPs were evaluated based on the cat's physical appearance and their expected genotypes.
Pedigree Analysis
To determine whether the SNPplexes demonstrated Mendelian inheritance and to determine their power to resolve relationships, the parent–offspring trios were evaluated in an Oriental Shorthair pedigree (n = 72, Fig. S1) using both 20 microsatellites from a previous study 37 and the six SNPplexes. The likelihood of relatedness between parent–offspring combinations was calculated by the software program COLONY 38 considering 15 known (mother, father, offspring) trios within the pedigree.
Sensitivity, Reproducibility, Precision, and Inhibition
The sensitivity study was performed in duplicate using a two‐fold serial dilution of a DNA standard from 7.2 to 0.014 ng with known genotypes. The assay sensitivity was determined by examining peak height ratios. Peak heights below the stochastic threshold and the occurrence of allelic drop‐out defined the sensitivity limits.
Reproducibility of the SNPplexes was assessed using a well‐characterized and independently genotyped reference sample that was amplified independently using the same protocol by two laboratory technicians. Instrument variability was assessed using two different capillary electrophoresis instruments of the same model described above. Discrepancies were noted to evaluate deviations in genotype migration.
To test precision, the reference was genotyped in triplicate and each SNP was assessed based upon its migration variability on the DNA Analyzer. The mean and standard deviation were calculated for each of the common alleles at each locus (data not shown).
The effects of an environmental inhibitor on the six SNPplexes were tested using 10 ng of template DNA combined with humic acid concentrations of 0.0002%, 0.0001%, 0.00005%, and 0.000025% by volume.
Species Specificity
Nondomestic cat samples were amplified using the same standard procedures established for the SNPplexes to determine the specificity of the assay. Archival DNA from human (Homo sapien), rhesus macaque (Macaca mulatta), dog (Canis lupus familiaris), squirrel (Sciurus carolinensis), deer (Odocoileus hemionus), fox (Vulpes vulpes), mouse (Mus musculus), coyote (Canis latrans), cow (Bos Taurus), goat (Capra aegagrus hircus ), horse (Equus ferus caballus), pig (Sus scrofa), sheep (Ovis aries), bear (Ursus americanus californiensis), bobcat (Lynx rufus), and snow leopard (Uncia uncia) were genotyped. Most DNA samples were isolated from EDTA anticoagulated whole blood or tissue. However, coyote, bear, and bobcat was isolated from scat. Squirrel DNA was isolated from hair samples. The DNA concentrations ranged 4.42–64 ng/μL.
Forensic Statistical Analysis
The random match probability, discrimination power (DP), and likelihood ratios were estimated to discern how informative the SNP miniplexes were for forensic analysis. The random match probability was based upon the combined probabilities of each locus p4 + 4p2q2 + q4 39.
Results
Cat Samples, SNPs, and Primers
For the different aspects of the panel validation, 325 cats were analyzed. Sixteen cats from the population study (n = 203) had <85% of SNP calls and were excluded from further analysis. Combining the 187 remaining cats from the population study and the control cats for the concordance and sensitivity studies, a reference database of 237 cats (Table 1) was developed for the six SNPplexes. The genotypic data for the cats are presented in Table S3.
Most intergenic, BGA SNPs had low D' values with 56 of the initial 148 SNPs showing high heterozygosity. However, seven loci were excluded from the 56 SNPs due to high Gst. The remaining most polymorphic BGA SNPs (n = 49) and all available EVC SNPs (n = 29) were selected for primer design. Population statistics for the final set of SNPs is described below and is presented in Table 3.
Table 3.
Population statistics for final cat SNPs
| Chr_Position | HO | GST a | MAFb | Hc | Chr_Position | HO | GST a | MAFb | Hc |
|---|---|---|---|---|---|---|---|---|---|
| A1_69424718 | 0.3533 | 0.048 | 0.4182 | 0.6797 | F1_565223 | 0.5189 | 0.055 | 0.4894 | 0.6929 |
| A1_175780586 | 0.354 | 0.031 | 0.413 | 0.6779 | F1_26100599 | 0.4613 | −0.001 | 0.5 | 0.6931 |
| A2_202225770 | 0.4341 | 0.023 | 0.3624 | 0.6548 | F1_82716202 | 0.4251 | 0.039 | 0.2995 | 0.6104 |
| A3_91058022 | 0.438 | 0.057 | 0.3457 | 0.6448 | F2_8427817 | 0.5118 | −0.007 | 0.4892 | 0.6929 |
| A3_12082294 | 0.4082 | 0.037 | 0.3378 | 0.6395 | F2_38395360 | 0.3707 | −0.009 | 0.4465 | 0.6874 |
| A3_159537633 | 0.501 | 0.009 | 0.4599 | 0.6899 | F2_46855978 | 0.6485 | 0.004 | 0.4787 | 0.6922 |
| A3_162208567 | 0.4984 | −0.006 | 0.2626 | 0.5757 | F2_78303221 | 0.4288 | 0.023 | 0.4301 | 0.6833 |
| B3_13666494 | 0.5763 | −0.02 | 0.3995 | 0.6728 | ASIP–1 | 0.4058 | 0.0404 | 0.2989 | 0.61 |
| B1_12214271 | 0.5077 | −0.01 | 0.4179 | 0.6796 | ASIP–2 | 0.0312 | 0.0293 | 0.0159 | 0.0815 |
| B1_88148379 | 0.4523 | 0.032 | 0.4396 | 0.6858 | ASIP–3 | 0.0777 | 0.0459 | 0.0397 | 0.1669 |
| B3_57141954 | 0.4089 | 0.027 | 0.4759 | 0.692 | CMAH–1 | 0.3281 | 0.1324 | 0.25 | 0.5623 |
| B4_21098349 | 0.351 | 0.035 | 0.4538 | 0.6889 | CMAH–2 | 0.2529 | 0.1198 | 0.1739 | 0.462 |
| B4_105706694 | 0.353 | 0.061 | 0.3783 | 0.6632 | CMAH–3 | 0.2265 | 0.1438 | 0.1559 | 0.4328 |
| B4_144693308 | 0.3778 | −0.009 | 0.4068 | 0.6757 | FGF5–2 | 0.3823 | 0.0812 | 0.2926 | 0.6044 |
| B4_147206961 | 0.4381 | 0.005 | 0.2754 | 0.5886 | FGF5–3 | 0.0799 | 0.1127 | 0.0481 | 0.193 |
| C1_52456776 | 0.4226 | 0.021 | 0.4021 | 0.5638 | FGF5–4 | 0.1006 | 0.1052 | 0.0612 | 0.2302 |
| C1_116355295 | 0.3627 | 0.06 | 0.2513 | 0.5752 | KIT | 0.0972 | 0.077 | 0.0565 | 0.2171 |
| C1_123164748 | 0.4279 | 0.06 | 0.262 | 0.6712 | KRT71–A | 0.068 | 0.0378 | 0.037 | 0.1584 |
| C1_215441574 | 0.4089 | 0.039 | 0.3957 | 0.6739 | KRT71–C | 0.0061 | 0.9601 | 0.0063 | 0.038 |
| C2_147124460 | 0.5125 | −0.003 | 0.3087 | 0.6181 | MC1R | 0.0773 | 0.1503 | 0.0508 | 0.2009 |
| C2_156491175 | 0.5157 | 0.032 | 0.3803 | 0.6642 | MLPH | 0.4537 | 0.0368 | 0.381 | 0.6645 |
| D1_10789012 | 0.3456 | 0.056 | 0.3218 | 0.6282 | PYP2R5 | 0.0258 | 0.0911 | 0.0114 | 0.0622 |
| D1_101321498 | 0.4192 | 0.019 | 0.4974 | 0.6931 | SHH_1 | 0 | 1 | 0 | 0.4937 |
| D1_125811329 | 0.4273 | 0.03 | 0.2686 | 0.5819 | SHH_3 | 0.0551 | 0.0346 | 0.0293 | 0.1321 |
| D2_1020904 | 0.5327 | −0.007 | 0.3351 | 0.6377 | Taqpep | 0.4499 | 0.1 | 0.4867 | 0.6928 |
| D2_74293444 | 0.3637 | −0.008 | 0.2888 | 0.601 | TYR–1 | 0.0226 | 0.071 | 0.0132 | 0.0704 |
| D3_1810839 | 0.451 | −0.026 | 0.3995 | 0.6728 | TYR–2 | 0 | 0 | 0 | 0 |
| E1_4114158 | 0.532 | −0.007 | 0.4787 | 0.6922 | TYR–3 | 0.25 | 0.0811 | 0.1631 | 0.4448 |
| E1_5453028 | 0.4432 | −0.003 | 0.4973 | 0.6931 | TYRP1–1 | 0.3556 | 0.2464 | 0.375 | 0.6616 |
| E2_7950477 | 0.4269 | 0.035 | 0.4385 | 0.6856 | TYRP1_2 | 0.1724 | 0.079 | 0.1005 | 0.3262 |
| E2_22632289 | 0.4077 | −0.028 | 0.3 | 0.6109 | TYRP1–3 | 0.1605 | 0.1537 | 0.1064 | 0.3389 |
| E2_35914023 | 0.4298 | −0.017 | 0.4654 | 0.6365 | ZFXY | 0.3805 | 0.018 | 0.2606 | 0.5737 |
All seventy PCR primer pairs designed to amplify the 78 BGA and EVC SNPs had a successful single, robust amplification product that was sequence verified as the expected locus (data not shown). Primer melting temperatures, lengths, concentrations, purine:pyrimidine ratios, and amplicons size are presented in Table S1. Six miniplexes that contain 10 to 14 SNPs per panel were developed (Table S2, Fig. 1). The 29 EVC markers were combined into two multiplexes with 14 loci each (Panels 3a and 3b). Most SNPs had efficient amplification as determined by RFU values in the multiplex mini‐sequencing reactions with the exception of SNP B4_40319102, which failed to amplify in the multiplex PCRs and was removed from the panel. SNP D4_41078218 was monomorphic and was eliminated. These two SNPs were not included in further analyses.
Figure 1.
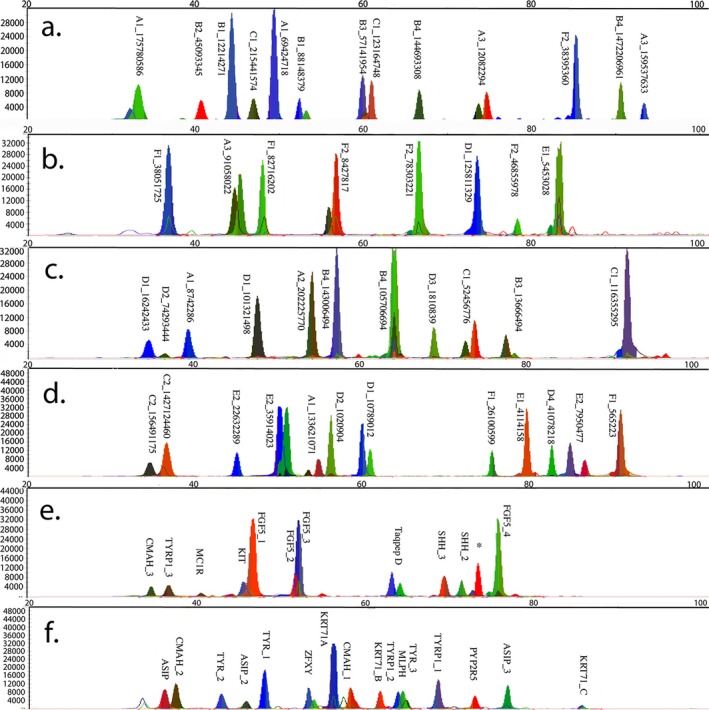
Electropherograms of six SNP miniplexes. Forty‐eight SNPs were successfully combined into six miniplexes. (a) represents Panel 1a, (b) represents Panel 1b, (c) represents Panel 2a, (d) represents Panel 2b, (e) represents the phenotypic Panel 3a, and (f) represents the phenotypic Panel 3b. Alleles are called as the following: A is green, T is red, G is blue, and C is black. SNP B4_40319102 did not amplify after multiplexing.
Allele Detection
The same principles of SNP allele assignments were used as previously described 40. Genotypes were considered homozygous if the peak height ratio was ≥5:1 and heterozygote if ≤3:1 after normalization. Peak ratios between 5:1 and 3:1 were considered inconclusive. However, loci A3_162208567, A3_159537633, and F1_82716202 required a peak height ratio of ≥6:1 to be considered a homozygote. These peak ratios were verified across the 385 cats amplified while considering the concordance with genotyping data for the same cats from other assays (See Concordance below). Six BGA SNPs had call rates <90% in the cat samples due to peak heights below the stochastic threshold (< 300 RFU) (Table 4), but only one BGA SNP was eliminated. Three loci (A1_69424718; B1_12214271; B1_881483379) improved in efficiency as well as concordance once primer concentrations were adjusted and therefore retained. SNP D1_18570323 did not improve with adjustments and allelic drop‐out was observed and was eliminated. Although two SNPs had poor call rates (D3_1810839 and E1_4114158), they were retained in the panel as they had balanced peak heights and later demonstrated proper Mendelian inheritance and had highly concordant genotypes across assays (Table 4). Three EVCs had call rates < 90%. SHH_1 and KRT71_B did not improve with adjustments and were eliminated. SNP CMAH_1 was retained for further analyses.
Table 4.
SNP call and concordance rates and cross‐species amplification
| Locus Leopard | Call Rate | Concordance | Coyote | Mouse | Human | Goat | Squirrel | Primate | Pig | Fox | Cattle | Dog | Deer | Bear | Horce | Sheep | Bobcat | Snow Leopard |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A1_69424718a, c | 85 | 78 | ||||||||||||||||
| A1_8742286a, b | 100 | 66 | ||||||||||||||||
| A1_133621071a, b | 99 | 50 | ||||||||||||||||
| A1_175780586 | 98 | 88 | x | x | x | x | x | |||||||||||
| A2_202225770 | 100 | 97 | x | x | x | x | ||||||||||||
| A3_12082294 | 100 | 94 | x | x | x | x | x | x | x | x | ||||||||
| A3_91058022 | 99 | 89 | x | x | x | x | x | |||||||||||
| A3_159537633 | 99 | 100 | x | x | x | |||||||||||||
| A3_162208567a | 94 | 89 | ||||||||||||||||
| B1_12214271 | 76 | 94 | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x |
| B1_88148379 | 79 | 96 | x | |||||||||||||||
| B2_45093345a, c | 100 | 63 | ||||||||||||||||
| B3_13666494 | 100 | 97 | x | x | x | x | x | x | x | x | ||||||||
| B3_57141954a | 99 | 97 | ||||||||||||||||
| B4_105706694 | 100 | 97 | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x |
| B4_143006494a, b | 90 | 56 | ||||||||||||||||
| B4_144693308 | 94 | 97 | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | |
| B4_147206961 | 99 | 92 | x | x | x | x | ||||||||||||
| B4_21098349 | 98 | 100 | x | x | ||||||||||||||
| C1_123164748 | 99 | 94 | x | |||||||||||||||
| C1_215441574 | 99 | 100 | x | |||||||||||||||
| C1_52456776 | 100 | 94 | x | x | x | x | ||||||||||||
| C1_116355295 | 100 | 94 | x | x | x | x | x | x | x | x | x | x | x | x | ||||
| C2_15641175 | 95 | 97 | x | |||||||||||||||
| C2_147124460 | 99 | 100 | x | x | x | x | x | |||||||||||
| C2_156491175 | 95 | 97 | x | |||||||||||||||
| D1_10789012 | 99 | 97 | x | x | x | x | x | x | x | x | ||||||||
| D1_16242433a, b | 99 | 59 | ||||||||||||||||
| D1_18570323a, b | 62 | 72 | ||||||||||||||||
| D1_101321498 | 100 | 100 | x | x | x | x | x | x | x | |||||||||
| D1_125811329 | 94 | 94 | x | x | x | x | x | x | x | x | ||||||||
| D2_1020904 | 100 | 97 | x | x | x | x | x | x | x | |||||||||
| D2_74293444 | 100 | 100 | x | x | ||||||||||||||
| D3_1810839b | 76 | 100 | x | x | x | x | x | |||||||||||
| E1_4114158b | 79 | 97 | x | x | x | x | x | x | x | x | x | x | x | x | x | |||
| E1_5453028 | 100 | 92 | ||||||||||||||||
| E2_7950477 | 100 | 97 | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x |
| E2_22632289 | 100 | 94 | ||||||||||||||||
| E2_35914023a, c | 99 | 53 | ||||||||||||||||
| F1_565223 | 98 | 97 | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x |
| F1_21799641a, b | 90 | 47 | ||||||||||||||||
| F1_26100599 | 94 | 100 | x | |||||||||||||||
| F1_38051725a, b | 99 | 72 | ||||||||||||||||
| F1_82716202 | 99 | 97 | x | x | x | x | x | x | x | x | ||||||||
| F2_8427817 | 98 | 100 | x | x | x | x | x | x | x | |||||||||
| F2_38395360b | 99 | 94 | x | |||||||||||||||
| F2_46855978 | 100 | 86 | x | x | x | x | x | x | x | x | x | |||||||
| F2_78303221 | 100 | 100 | x | |||||||||||||||
| ASIP | 100 | 100 | x | x | x | x | x | x | x | x | x | x | x | x | x | x | ||
| ASIP_2 | 100 | phen | x | x | x | x | x | x | x | |||||||||
| ASIP_3 | 100 | phen | x | x | x | x | x | x | x | x | x | |||||||
| CMAH_1 c | 85 | n/a | x | x | ||||||||||||||
| CMAH_2 a | 100 | — | ||||||||||||||||
| CMAH_3 | 100 | 100 | x | x | ||||||||||||||
| FGF5_1 a b | 99 | 0 | ||||||||||||||||
| FGF5_2 | 100 | 100 | x | x | x | x | ||||||||||||
| FGF5_3 | 99 | 100 | x | x | x | x | x | x | x | x | x | x | x | x | x | x | ||
| FGF5_4 | 100 | 100 | x | x | x | x | x | x | x | x | x | x | x | x | x | |||
| KIT | 99 | 100 | x | x | x | x | x | x | x | x | x | x | x | x | x | |||
| KRT71_A | 100 | 100 | x | x | x | x | ||||||||||||
| KRT71_B a b | 43 | — | ||||||||||||||||
| KRT71_C | 100 | n/a | x | |||||||||||||||
| MC1R | 99 | 100 | x | x | x | x | x | |||||||||||
| MLPH | 100 | 100 | x | x | x | x | ||||||||||||
| PYP2R5 | 93 | n/a | x | x | ||||||||||||||
| SHH_1 a b | 56 | — | ||||||||||||||||
| SHH_2 | 99 | n/a | x | x | ||||||||||||||
| SHH_3 | 100 | n/a | x | x | x | x | x | x | ||||||||||
| TAQPEP | 100 | phen | x | |||||||||||||||
| TYR_1 | 100 | 100 | x | x | x | |||||||||||||
| TYR_2 | 100 | 100 | x | x | ||||||||||||||
| TYR_3 | 99 | 100 | x | |||||||||||||||
| TYRP1_1 | 91 | 100 | x | |||||||||||||||
| TYRP1_2 | 100 | 100 | x | x | ||||||||||||||
| TYRP1_3 | 100 | 100 | x | |||||||||||||||
| ZFXY | 100 | 100 | x | x | x | x | x | x | x | x | x | x | x | x | x | |||
| % Call Rate | 17 | 21 | 26 | 26 | 26 | 26 | 26 | 29 | 32 | 33 | 33 | 36 | 36 | 37 | 63 | 73 |
SNPs not examined in the species specificity study.
SNPs eliminated from the panel.
SNPplex data are likely correct although discordant with GoldenGate assay data. SNP retained in the panels. Blanks are loci that failed to amplify during the species specificity study. SNPs with “n/a” could not be confirmed for concordance due to lack of sample representation. All cats were wild type for these mutations. SNPs with “phen” had correct genotypes inferred from phenotypes.
Concordance
Genotypes provided by an illumina GoldenGate and a Sequenom iPlex assay for BGA SNPs on 48 cats were compared to the SNPplex results. Genotypes were considered correct when concordant between two of the three assays. Thirty‐seven of 47 BGA SNPs were concordant for >85% of genotypes (Table 4). Seven of the ten SNPs with poor concordance were eliminated. Three SNPs improved with primer adjustments or the GoldenGate data were considered incorrect due to proper segregation of the SNPs in the pedigree (see below). Additionally, twelve cats were genotyped using both the iPlex and the Golden Gate assays and were cross‐examined with eight of the BGA SNPs overlapping the SNaPshot assay. Six of the SNPs were concordant when compared to both the iPlex and the Golden Gate assays. BGA SNPs A1_133621071 and A1_8742286 were concordant between the iPlex and the Golden Gate assays but discordant with the SNPplex, suggesting the SNPplex genotypes were incorrect.
Concordances of the EVC SNPs were determined by comparing genotypes from phenotypic control samples. Twenty SNPs had GoldenGate assay data and all had 100% concordancy. Five of six SNPs in the iPLex assay were concordant; however, one SNP was discordant (FGF5_1). Mutations ASIP_2 (non‐agouti), ASIP_3 (charcoal), and TAQPEP (Tabby) were verified by phenotypic verification based upon the known physical traits of the cats. SNPs KRT71_C (Selkirk Rex), PYPR25 (Cornish Rex), SHH_2, and SHH_3 (polydactyl) could not be phenotypically verified as samples from these breeds were not examined. All cats were wild type for these mutations, as would be expected from random population sampling. Although the cat blood‐type gene variants CMAH_1 and CMAH_2 are not associated with a cat's blood type, they were maintained within the miniplexes as informative polymorphisms. The CMAH_3 SNP is the only variant that is associated and concordant with the AB blood type in cats. Overall, three phenotypic SNPs were removed from the miniplexes based on poor amplification or discordancy. The final phenotypic SNPplexes contained 26 EVC SNPs.
Pedigree Analysis
Segregation analysis in a known pedigree was performed to confirm Mendelian inheritance of the BGA SNPs. The EVC SNPs were not informative in this pedigree. A1_133621071, F1_38051725, and F2_38395360 did not show proper segregation and were eliminated. One marker was not informative, D1_16242433. Five SNPs (A1_8742286, B2_45093345, B4_143006494, E2_35914023, and F1_21799641), segregated properly within the pedigree even though they were discordant with the overlapping GoldenGate genotypes. With further evaluation, SNPs B2_45093345 and E2_35914023 were retained within the SNPplexes as they demonstrated no peak height issues and segregated within the pedigree with no conflicting genotypes, suggesting the GoldenGate assay data may be inaccurate. Overall, the six SNPplexes contain 65 SNPs, including 39 BGAs and 26 EVCs.
Individuals from 15 known trios were examined with the remaining 38 informative BGA SNPs and 20 STRs (data not shown) to gauge ability to assign parentage. The COLONY program was used to assign the correct sires and dams to 15 offspring, from a pool of 10 dams and 8 sires (Fig. S1). The SNPplex panels identified 15 sires, but two were incorrect. The two incorrectly assigned sires were actually both paternal grandfathers. Additionally, two of the correctly selected sires were identified with very low probabilities of 0.04 and 0.14. Nevertheless, these cats were the best sire candidates. Twelve of 15 dams were assigned and all were correct. One of the 12 correctly selected dams had a low probability of 0.68. The SNPs were not sufficiently polymorphic to assign three dams. The STRs correctly assigned 12 sires and all dams. Two correctly assigned parents had low probabilities (< 0.75). Three sires could not be assigned by STRs, but these three sires were correctly assigned using the SNPs.
Sensitivity, Reproducibility, Precision, Inhibition
A complete profile of all SNPs was observed at 7.2 ng–112.5 pg of DNA (Fig. 2). At 56 pg of input DNA, SNPs D1_16242433 and D1_18570323 had complete drop‐out or low peak heights below the stochastic threshold. At 28 pg of input DNA, 10 loci had either drop‐out/drop‐in alleles. With 14 pg DNA, 21 loci demonstrated drop‐out/drop‐in alleles.
Figure 2.
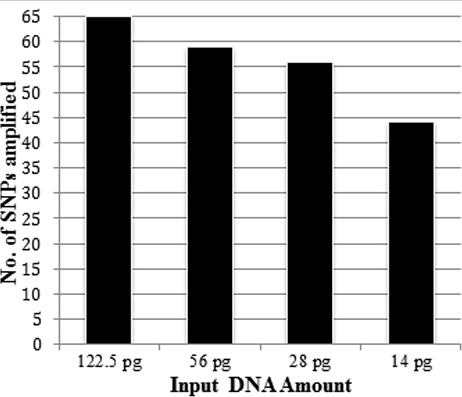
Sensitivity of the SNPs used in the six miniplexes. The SNPplexes produced complete profiles from 7.2 ng to 122.5 pg. Data for template DNA >122.5 pg not presented. Drop‐out of alleles was apparent at 56 pg and lower DNA concentrations.
To test the reproducibility of the SNP miniplex panels using a standard protocol, two different operators used the same protocol to genotype a control cat. Providing no additional instructions beyond the protocols, all genotype calls were identical between the two analysts (Fig. 3). Although some instrument variation was observed for peak heights, peak height ratios and genotypes were comparable. For one of the ABI 3730 DNA analyzer instruments, more background was observed in both the green and the red spectrums (Fig. 3 b–c).
Figure 3.
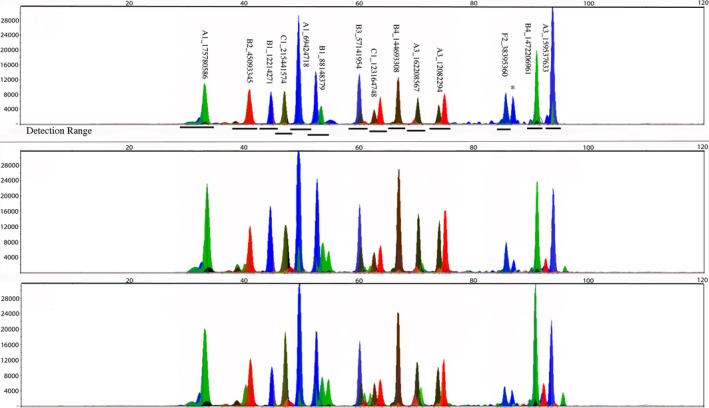
Electropherograms produced by the reproducibility study (Panel 1a). (a) Represents same operator and protocol using a different capillary electrophoresis ABI 3730 DNA analyzer instrument. (b) Represents normal conditions, (c) represents different operator same instrument. Alleles are called as the following: A is green, T is red, G is blue, and C is black.
Based upon three separate capillary electrophoresis injections using the same ABI PRISM 3730 DNA Analyzer, the precision of the mobility for each allele was evaluated. Migration differences of the originally selected SNPs ranged from 0 to 1.3343 bp. Some markers were above the suggested standard deviation of ±0.75 bp per protocol when developing bin ranges.
Possible inhibition issues related to common contaminants were tested on the six SNPplexes using different concentrations of humic acid (data not shown). All loci decreased in peak height with increased humic acid concentration. At 0.0002%, the panels showed the most inhibition. However, the majority of the loci at the highest acid concentration maintained a RFU >300, allowing the correct genotype to be called with the exception of E2_22632289 and SHH_2. Locus MLPH had complete allele drop‐out at 0.0001% humic acid concentration. All other loci did not have peak height imbalances and allelic drop‐in/drop‐out at any of the humic acid concentrations.
Species Specificity
To evaluate specificity and the possible amplification of contaminate DNA from common and related species, DNA from various mammals was assayed with the six SNPplexes (Table 4). Representatives of rodentia, carnivora, primate, and ungulata were included. On average, 19 of 64 (29%) SNPs amplified across species consistently. The bobcat (Lynx rufus) and snow leopard (Uncia uncia) had the most amplified SNPs, 54% and 65%, respectively. Besides the coyote, which was isolated from scat DNA, the rodents (mouse and squirrel) and primates (human and macaque) had the poorest amplification. Call rates were <31% for other species. Twenty‐two loci successfully amplified within the dog, with some polymorphisms noted. Seven BGA SNPs amplified relatively consistently across multiple species.
Statistical Analysis
SNPs were tested for Hardy–Weinberg expectations (HWE). HWEs were rejected (0.01< p <0.05) for three intergenic SNPs, F2_38395360, C1_123164748, and B1_12214271. Some phenotypic SNPs were not within HWE, which was anticipated as these variants are rare and distinctive to certain cat breeds. Across populations, 63 (98.41) SNPs were polymorphic. As expected, most of the EVC SNPs had the lowest amount of diversity, in particular the rare breed‐specific traits. For the BGA SNPs, the MAF ranged from 0.251 to 0.500 with an average of 0.371. The G ST ranged from 0.061 to ‐0.009 with an average of 0.015 (Table 3). All loci demonstrated low linkage disequilibrium. The average linkage disequilibrium of the individual compared to the total populations (D'IT) was ≤0.04866, and the average of linkage disequilibrium of the subpopulation compared to the total (D'ST) was ≤0.00109. Observed heterozygosity (HO) averaged 0.3352 ± 0.024, ranging from a high in the Kansas random‐bred population to the low in the Ohio populations, 0.3863 and 0.2787, respectively. Each population tested had a range of 45–58 polymorphic loci, with the population group from Ohio having the lowest amount of polymorphic loci (Table 1). The Ohio population was later confirmed to be a group of cats that had been obtained from a commercial breeding facility.
Forensic Statistical Analysis
All loci which had (i) high heterozygosity >0.35, (ii) minor allele frequency >0.25, and (iii) were polymorphic based upon H' were used to determine their efficacy as a forensic tool, excluding the rare phenotypic loci. Based upon the criteria, 45 SNPs were used to calculate random match probability (RMP), discrimination power (DP), and the likelihood ratio (LR) to infer the assay's power of discrimination. The combined random match probability (cRMP) was 6.58621 × 10‐19 across all Western populations and the likelihood ratio was 1.518 × 1018.
Discussion
As the genomes of several domestic animals, including cats 30, 41, 42, have been characterized and a wealth of SNPs identified, the broad application of SNP technology for autosomal loci in animal forensics is gaining momentum 43. SNPs for individual, phenotypic, and species identification, as well as, ancestry and parentage analysis are available in cats and can be assayed by various techniques 9, 44, 45, 46, 47. Hence, this study embarked upon developing an effective SNP identification panel that can be easily adapted into forensic laboratories for use with cat evidence. The SNaPshot® technology was selected as the required the capillary electrophoresis equipment is common in forensic laboratories.
Because a set of 148 SNPs had already been used to produce a commercially available ancestry assignment test for demarcating breeds and random‐bred cats 48, thereby establishing a database, this project selected a subset of the same 148 SNPs for use in forensic applications. Therefore, any data generated by this project add to the current feline SNP database. Several thousand individual cats have been genotyped on the illumina Infinium iSelect 63K cat DNA array; thus, an abundance of SNPs are available and can be mined to extend the currently validated panel (manuscripts in preparation 49, 50, 51, 52, 53). The SNPs in the cat SNPplex are assayed by the 63K array; thus, the array data could also be used to build the cat SNP database. As more cat genomes are sequenced, additional SNPs, DNA variants, and phenotypic SNPs will become readily accessible and not a limiting factor.
Initially, 49 intergenic BGA SNPs were combined into six panels, containing 11–14 SNPs. However, following the assay evaluation for call rate, concordance, and inheritance, 39 SNPs are suggested as an initial panel for cat identification. These loci proved to be polymorphic across diverse populations and no strong evidence of distortions from HWE or linkage disequilibrium was observed. Ten of the 49 SNPs were eliminated due to inaccurate genotypes most likely attributed to low peak height and poor amplification of the genomic region of interest caused by suboptimal affinities. Peak height ratios can be unbalanced due to the dye and or the nucleotide being assayed. Three of the 49 SNPs had genotyping conflicts within the pedigree analysis and were eliminated. However, these three SNPs had high call rates and one had high concordance with other data. Therefore, validation of this panel benefitted from using a pedigree to demonstrate Mendelian inheritance, thereby indicating a problem with a specific SNP. The SNaPshot® assay has been shown in previous studies to combine as many as 29 SNPs in a single reaction; thus, additional SNPs can be theoretically added to these panels to improve power. Additionally, primer, temperature, and salt concentration adjustments could further improve the balance of the allele amplification 36. These minor adjustments may improve some of the variation detected in the precision study.
Data that had been previously generated for the same loci using different technologies, such as illumina GoldenGate and Sequenom mass spectroscopy iPlex assay, were used to validate the SNP genotypes and to refine the peak height ratios. Several SNPs were not concordant between technologies, which implied a third assay or segregation analysis was required to determine the correct genotype. Typically, a minor allele frequency minimum threshold of <0.05 is used for genome‐wide association studies when analyzing array data due to the inherent error rate in these assay technologies. Similar discordancy rates between the SNPplex and the GoldenGate, Infinium, or mass spectroscopy technologies should be expected and replicate genotyping should be performed to confirm accuracy.
In addition to supporting SNP segregation, the 15 trios of cats that formed an extended pedigree also demonstrated the power of the panel for individual identification and kinship studies. The pedigree study indicated that the 20 STRs outperformed the 39 SNPs when determining parentage, which was not unexpected resulting from higher polymorphism of the STRs. To have the same power of paternity exclusion as seen in the 13 STR markers in humans, studies suggest 40–60 informative SNPs would be needed 10, 54, 55. The SNP miniplexes, however, proved to be beneficial when used concurrently with the STRs, specifically elucidating some maternal uncertainties in the highly inbred family evaluated. Increasing the SNP panels should improve the power to resolve potential parents or closely related cats, particularly those with have similar phenotypes.
The power of the cat SNP panel was evaluated by calculating the match probabilities and by performing a parentage analysis. The combined random match probability (cRMP) was 6.58 × 10‐19 across all Western populations of cats and the likelihood ratio was 1.52 × 1018. Ge et al. 56 examined the random match probabilities for humans from a set of commercially available STR kits. Although no population substructure correction was applied to the domestic cat populations, the cat SNPplex panels are comparable to the PowerPlex 16 with an RMP of 2.43 × 10−18, the Identifiler with an RMP of 5.93 × 10−18, and the 13 loci CODIS core with an RMP of 2.34 × 10−15. The New FBI core and Section A core have much better RMP on the magnitude of 10−25 to 30 56, 57.
Compared to other multiplex commercially available kits, the level of sensitivity of the SNP miniplexes is equivalent or better. With no allele drop‐in, or complete locus dropout across all samples tested, >56 pg of DNA should be proficient to create a complete SNP profile, more sensitive than the >100–250 pg needed for most STR typing systems 58, 59, 60. However, the DogFiler miniSTR panel has a reported higher sensitivity at ≥32 pg 61.
Species specificity was also sufficient for the cat SNP panel. A majority of SNPs were amplified in bobcat and snow leopard, which is expected as both species belong to the same family, Felidae. Other carnivores had some loci that amplified robustly and were also polymorphic. Human cross‐amplification was poor, 21%. Although the primers for the EVC SNPs may be in more conserved regions across species as compared to the intergenic BGA SNPs and may cross‐amplify, these loci would only be problematic in the case of cross‐species DNA contamination. With the wealth of SNP data across species, a cross‐species SNP panel that identified different species would be feasible.
In humans, the IrisPlex uses six highly informative markers to predict blue and brown eye color variants 62. A similar genetic test is able to predict eye color, as well as ancestry, based upon skin color variation using seven SNPs 9. As part of an individualization panel, the allele frequency of the EVC SNPs is not as important as presence or absence of the variant. Twenty‐six SNPs that confer a cat's phenotype were examined and combined into two panels. SNPs for polydactyl (SHH_1), curly fur of Devon Rex (KRT71_B), and one of four long‐hair phenotypes (FGF5_1) failed validation. These EVCs could be redesigned but they may have a lower priority as each one is less frequent in cat populations and is more specific to certain breeds or common to specific US random‐bred populations. Remaining in the panel are the DNA variants that identify several of the major coat colors of cats, including Agouti, Brown, Color, Dilute, and Extension. Agouti will determine whether a cat is solid or will display tabby markings. Brown indicates the tone of the color, from normal brown tabby to little chocolate or cinnamon tabbies. The Color locus is important as Siamese coloration is one of the most popular across several breeds and easily recognizable and preferred by many cat owners. Dilute is also a very common coloration, making a cat more of a bluish gray in coloration. The most common long‐hair variant for cats is present in the panel, as well as the variants that confer blotched/classic tabby pattern versus stripes or spots. Sex can be determined, as well as a few of the rare curly coats and white spotting phenotypes, such as Cornish and Selkirk Rex, and white only on the feet (a.k.a. gloves). Other important phenotypes that are missing to the panel include the Orange, Inhibitor, and Ticked loci (see review 63). The causal DNA variants have not yet been identified and their addition should improve the phenotypic discrimination power of the panel and better identify random‐bred cats. Other variants for dominant White and Spotting would also be beneficial for inclusion in the panel 64. Although morphological analyses of hairs should be predictive of a cat's phenotype, individual hairs can be different colors and a complete representation of the cats hair coat may not be available. As the SNP panel will be unlikely to find a matching cat in the database, the phenotypic traits could be identified first to provide a phenotypic prediction to what type of cat contributed the evidence.
Conclusion
Since the 1994 murder investigation of the “Snowball” case 65, there has been increased interest in developing efficient identification systems for animals. Tetranucleotide STRs panels have been developed for cats 66, 67, 68, 69, as well as a large dinucleotide STRs database that has been developed for worldwide efforts for determining parentage analysis and individual identification 18, 37. These same markers have been used to define eight major races of cats throughout the world and identify breeds 17, 18, 70. The cat control region mtDNA studies have expanded to include diverse worldwide populations including over 1000 cats 20, 71. This data has recently been used to support a homicide investigation involving cat hair as evidence in the State of Missouri (73). Both phenotypic and genotypic characteristics can be obtained from SNPs. These markers are also much more cost effective compared to STR typing systems due to their automated large‐screening capabilities. SNPs can be applicable to difficult forensic cases, specifically when handling degraded DNA samples.
This study has produced a dependable assay using 64 SNPs for individual identification with a combined match probability of 6.586 × 10−19 across all random‐bred Western populations. Six miniplexes were developed containing 39 intergenic SNPs and 26 phenotypic SNPs, including a sex identification marker, ZFXY, using the SNaPshot® platform. The panel should be increased with more BGA and EVC SNPs and all SNPplexes should be more robustly tested on poor quality and mixture samples, and specifically, at DNA quantities commonly isolated from a few cat hairs. The cat SNPplexes provide a novel tool for the analysis of frequently available, and underused, source of forensic evidence. The identification and phenotypic profiling panel should assist crime scene investigators with cat identification, potentially implicating individuals that have come into contact with the cat.
Supporting information
Figure. S1. Pedigree of the 15 parent‐offspring trios with known kinship for parentage validation.
Table S1. (a) PCR and primer information for cat SNPplexes.
Table S2. SBE primer information and concentrations.
Table S3. SNP genotypes of cat populations using the SNPplexes.
Acknowledgements
The authors appreciate commentary on the study by Drs. Hasan Alhaddad, Cecilia von Beroldingen, and Benjamin N. Sacks. We appreciate the Veterinary of Genetics Laboratory at the University of California – Davis for technical assistance, especially Julia Malvik and Tamala Gilliland. We appreciate the assistance with cat sample collection by Gordon A. Andrews, DVM, PhD; Betsy Arnold, DVM; Cats Protection, Sussex, UK; Norma Vollmer Labarthe, DVM, PhD; Julie K. Levy, DVM, PhD; Maria Longeri, DVM, PhD; William F. Swanson, DVM; Anne Thomas, PhD; and Barbara Zangerl, DVM, PhD.
Funding for this project was provided in part by the NIJ/FSF Forensic Science Research Grant Award # UCD2012G70, from the National Center for Research Resources R24 RR016094 and is currently supported by the Office of Research Infrastructure Programs/OD R24OD010928 and the University of California – Davis, Center for Companion Animal Health.
References
- 1. American Pet Products Manufacturers' Association . National Pet Owner's survey. Greenwich, CT: The Association, 2012. [Google Scholar]
- 2. American Veterinary Medical Association . US Pet Ownership and Demographics sourcebook. Schaumburg, IL: The Association, 2007. [Google Scholar]
- 3. Louwerens M, London CA, Pedersen NC, Lyons LA. Feline lymphoma in the post‐feline leukemia virus era. J Vet Intern Med 2005;19(3):329–35. [DOI] [PubMed] [Google Scholar]
- 4. Boehme A, Brooks E, McNaught I, Robertson J. The persistence of animal hairs in a forensic context. Aust J Forensic Sci 2009;41(2):99–112. [Google Scholar]
- 5. D'Andrea E, Fridez F, Coquoz R. Preliminary experiments on the transfer of animal hair during simulated criminal behavior. J Forensic Sci 1998;43(6):1257–8. [Google Scholar]
- 6. Peabody AJ, Oxborough RJ, Cage PE, Evett IW. The discrimination of cat and dog hairs. J Forensic Sci Soc 1983;23(2):121–9. [DOI] [PubMed] [Google Scholar]
- 7. Grahn RA, Huang TI‐T, Lyons LA. Impact of allogrooming in domestic cats (Felis silvestris catus) on mitochondrial DNA profiling of shed hairs. Open Forensic Sci J 2013;6:12–9. [Google Scholar]
- 8. Butler JM, Coble MD, Vallone PM. STRs vs. SNPs: thoughts on the future of forensic DNA testing. Forensic Sci Med Pathol 2007;3(3):200–5. [DOI] [PubMed] [Google Scholar]
- 9. Gettings KB, Lai R, Johnson JL, Peck MA, Hart JA, Gordish‐Dressman H, et al. A 50‐SNP assay for biogeographic ancestry and phenotype prediction in the U.S. population. Forensic Sci Int Genet 2014;8(1):101–8. [DOI] [PubMed] [Google Scholar]
- 10. Gill P. An assessment of the utility of single nucleotide polymorphisms (SNPs) for forensic purposes. Int J Leg Med 2001;114(4–5):4–5. [DOI] [PubMed] [Google Scholar]
- 11. Gill P, Werrett DJ, Budowle B, Guerrieri R. An assessment of whether SNPs will replace STRs in national DNA databases – joint considerations of the DNA working group of the European Network of Forensic Science Institutes (ENFSI) and the Scientific Working Group on DNA Analysis Methods (SWGDAM). Sci Justice 2004;44(1):51–3. [DOI] [PubMed] [Google Scholar]
- 12. Daniel R, Santos C, Phillips C, Fondevila M, van Oorschot RA, Carracedo A, et al. A SNaPshot of next generation sequencing for forensic SNP analysis. Forensic Sci Int Genet 2015;14:50–60. [DOI] [PubMed] [Google Scholar]
- 13. Phillips C, Prieto L, Fondevila M, Salas A, Gomez‐Tato A, Alvarez‐Dios J, et al. Ancestry analysis in the 11‐M Madrid bomb attack investigation. PLoS ONE 2009;4(8):e6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lyons LA. Genetic testing in domestic cats. Consultations on Feline Internal Medicine. St. Louis, MO: Saunders Elsevier, 2010. [Google Scholar]
- 15. Lyons LA. Feline genetics: clinical applications and genetic testing. Top Companion Anim Med 2010;25:203–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lyons LA. Genetic testing in domestic cats. Mol Cell Probes 2012;26:224–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kurushima J, Lipinski M, Gandolfi B, Froenicke L, Grahn J, Grahn R, et al. Variation of cats under domestication: genetic assignment of domestic cats to breeds and worldwide random‐bred populations. Anim Gene 2013;44(3):311–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lipinski MJ, Froenicke L, Baysac KC, Billings NC, Lyons LA, Leutenegger CM, et al. The ascent of cat breeds: genetic evaluations of breeds and worldwide random‐bred populations. Genomics 2008;91(1):12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Quality assurance standards for forensic DNA testing laboratories, Federal Bureau of Investigation, 2011 [updated 2011]; http://swgdam.org/SWGDAM_Validation_Guidelines_APPROVED_Dec_2012.pdf (accessed August 18, 2014).
- 20. Grahn RA, Kurushima JD, Billings NC, Grahn JC, Halverson JL, Hammer E, et al. Feline non‐repetitive mitochondrial DNA control region database for forensic evidence. Forensic Sci Int Genet 2011;5(1):33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lindquist CD, Evans JJ, Wictum EJ. Developmental validation of feline, bovine, equine, and cervid quantitative PCR assays. J Forensic Sci 2011;56(s1):S29–35. [DOI] [PubMed] [Google Scholar]
- 22. Goudet J. FSTAT (version 1.2): a computer program to calculate F‐statistics. J Hered 1995;86(6):485–486. [Google Scholar]
- 23. Nei M, Chakravarti A, Tateno Y. Mean and variance of FST in a finite number of incompletely isolated populations. Theor Popul Biol 1977;11(3):291–306. [DOI] [PubMed] [Google Scholar]
- 24. Weir BS, Cockerham CC. Estimating F‐statistics for the analysis of population structure. Evolution 1984;38(6):1358–70. [DOI] [PubMed] [Google Scholar]
- 25. Yeh FC, Yang R‐C, Boyle TBJ, Ye Z‐H, Mao JX. POPGENE, the user‐friendly shareware for population genetic analysis. Alberta, Canada: Molecular Biology and Biotechnology Centre, University of Alberta, 1997. [Google Scholar]
- 26. Ohta T. Linkage disequilibrium with the island model. Genetics 1982;101(1):139–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ohta T. Linkage disequilibrium due to random genetic drift in finite subdivided populations. Proc Natl Acad Sci USA 1982;79(6):1940–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shannon CE. The mathematical theory of communication. MD Comput 1963;14(4):306–317. [PubMed] [Google Scholar]
- 29. Lewontin RC. The apportionment of human diversity. New York, NY: Appleton‐Century‐Crofts, 1972. [Google Scholar]
- 30. Montague MJ, Li G, Gandolfi B, Khan R, Aken BL, Searle SMJ, et al. Comparative analysis of the domestic cat genome reveals genetic signatures underlying feline biology and domestication. Proc Natl Acad Sci USA 2014;111(48):17230–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JA. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 2007;35(Suppl. 2):W71–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. You FM, Huo N, Gu YQ, M‐c L, Ma Y, Hane D, et al. BatchPrimer3: a high throughput web application for PCR and sequencing primer design. BMC Bioinformatics 2008;9(1):253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. ABI PRISM® SNaPshot™Multiplex System . Life Technologies, 2014 [updated 2014]; http://tools.lifetechnologies.com/content/sfs/manuals/cms_041203.pdf (accessed August 18, 2014).
- 34. SNaPshot® Primer Focus® kit . Life Technologies, 2014 [updated 2014]; www.lifetechnologies.com/order/catalog/product/4329538? (accessed August 18, 2014).
- 35. GeneMapper® software version 4.0 and SNaPshot® kit analysis. Life Technologies, 2014 [updated 2014]; tools.lifetechnologies.com/content/sfs/manuals/cms_042037.pdf (accessed August 18, 2014).
- 36. Sanchez JJ, Phillips C, Børsting C, Balogh K, Bogus M, Fondevila M, et al. A multiplex assay with 52 single nucleotide polymorphisms for human identification. Electrophoresis 2006;27(9):1713–24. [DOI] [PubMed] [Google Scholar]
- 37. Lipinski MJ, Amigues Y, Blasi M, Broad TE, Cherbonnel C, Cho GJ, et al. An international parentage and identification panel for the domestic cat (Felis catus). Anim Genet 2007;38(4):371–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jones OR, Wang J. COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol Ecol Resour 2010;10(3):551–5. [DOI] [PubMed] [Google Scholar]
- 39. Jones DA. Blood samples: probability of discrimination. J Forensic Sci Soc 1972;12(2):355–9. [DOI] [PubMed] [Google Scholar]
- 40. Sanchez JJ, Borsting C, Balogh K, Berger B, Bogus M, Butler JM, et al. Forensic typing of autosomal SNPs with a 29 SNP‐multiplex–results of a collaborative EDNAP exercise. Forensic Sci Int Genet 2008;2(3):176–83. [DOI] [PubMed] [Google Scholar]
- 41. Pontius JU, Mullikin JC, Smith DR, Agencourt Sequencing T, Lindblad‐Toh K, Gnerre S, et al. Initial sequence and comparative analysis of the cat genome. Genome Res 2007;17(11):1675–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mullikin JC, Hansen NF, Shen L, Ebling H, Donahue WF, Tao W, et al. Light whole genome sequence for SNP discovery across domestic cat breeds. BMC Genom 2010;11:406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kakoi H, Kijima‐Suda I, Gawahara H, Kinoshita K, Tozaki T, Hirota K, et al. Individual identification of racehorses from urine samples using a 26‐plex single‐nucleotide polymorphism assay. J Forensic Sci 2013;58(1):21–8. [DOI] [PubMed] [Google Scholar]
- 44. Drobnic K, Børsting C, Rockenbauer E, Tomas C, Morling N. Typing of 49 autosomal SNPs by SNaPshot® in the Slovenian population. Forensic Sci Int Genet 2010;4(5):e125–7. [DOI] [PubMed] [Google Scholar]
- 45. Karniol B, Shirak A, Baruch E, Singrun C, Tal A, Cahana A, et al. Development of a 25‐plex SNP assay for traceability in cattle. Anim Genet 2009;40(3):353–6. [DOI] [PubMed] [Google Scholar]
- 46. Kidd KK, Pakstis AJ, Speed WC, Grigorenko EL, Kajuna SLB, Karoma NJ, et al. Developing a SNP panel for forensic identification of individuals. Forensic Sci Int 2006;164(1):20–32. [DOI] [PubMed] [Google Scholar]
- 47. Sanchez JJ, Borsting C, Hallenberg C, Buchard A, Hernandez A, Morling N. Multiplex PCR and minisequencing of SNPs: a model with 35 Y chromosome SNPs. Forensic Sci Int 2003;137(1):74–84. [DOI] [PubMed] [Google Scholar]
- 48. Kurushima JD. Genetic analysis of domestication patterns in the cat (Felis Catus): worldwide population structure, and human‐mediated breeding patterns both modern and ancient. Davis, CA: University of California – Davis, 2011. [Google Scholar]
- 49. Alhaddad H, Gandolfi B, Grahn RA, Rah HC, Peterson CB, Maggs DJ, et al. Genome‐wide association and linkage analyses localize a progressive retinal atrophy locus in Persian cats. Mamm Genome 2014;25(7–8):354–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gandolfi B, Alhaddad H, Affolter VK, Brockman J, Haggstrom J, Joslin SE, et al. To the root of the curl: a signature of a recent selective sweep identifies a mutation that defines the Cornish Rex cat breed. PLoS ONE 2013;8(6):e67105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gandolfi B, Alhaddad H, Joslin SE, Khan R, Filler S, Brem G, et al. A splice variant in KRT71 is associated with curly coat phenotype of Selkirk Rex cats. Sci Rep [Research Support, N.I.H., Extramural Research Support, Non‐U.S. Gov't] 2013;3:2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gandolfi B, Beresford LG, Myers JA, Pimentel M, Alhaddad H, Grahn JC, et al. The naked truth: Sphynx and Devon Rex cat breed mutations in KRT71 . Mamm Genome 2010;21(9–10):509–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gandolfi B, Gruffydd‐Jones TJ, Malik R, Cortes A, Jones BR, Helps CR, et al. First WNK4‐hypokalemia animal model identified by genome‐wide association in Burmese cats. PLoS ONE [Research Support, N.I.H., Extramural Research Support, Non‐U.S. Gov't] 2012;7(12):e53173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krawczak M. Informativity assessment for biallelic single nucleotide polymorphisms. Electrophoresis 1999;20(8):1676–81. [DOI] [PubMed] [Google Scholar]
- 55. Amorim A, Pereira L. Pros and cons in the use of SNPs in forensic kinship investigation: a comparative analysis with STRs. Forensic Sci Int 2005;150(1):17–21. [DOI] [PubMed] [Google Scholar]
- 56. Ge J, Eisenberg A, Budowle B. Developing criteria and data to determine best options for expanding the core CODIS loci. Investig Genet 2012;3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hares DR. Expanding the CODIS core loci in the United States. Forensic Sci Int Genet 2012;6(1):e52–4. [DOI] [PubMed] [Google Scholar]
- 58. Wallin JM, Buoncristiani MR, Lazaruk KD, Fildes N, Holt CL, Walsh PS. TWGDAM validation of the AmpFISTR blue PCR amplification kit for forensic casework analysis. J Forensic Sci 1998;43(4):854–70. [PubMed] [Google Scholar]
- 59. Mulero JJ, Chang CW, Lagace RE, Wang DY, Bas JL, McMahon TP, et al. Development and validation of the AmpFℓSTR® MiniFilerTM PCR Amplification Kit: a miniSTR multiplex for the analysis of degraded and/or PCR inhibited DNA. J Forensic Sci 2008;53(4):838–52. [DOI] [PubMed] [Google Scholar]
- 60. Lygo JE, Johnson PE, Holdaway DJ, Woodroffe S, Whitaker JP, Clayton TM, et al. The validation of short tandem repeat (STR) loci for use in forensic casework. Int J Leg Med 1994;107(2):77–89. [DOI] [PubMed] [Google Scholar]
- 61. Kun T, Lyons LA, Sacks BN, Ballard RE, Lindquist C, Wictum EJ. Developmental validation of Mini‐DogFiler for degraded canine DNA. Forensic Sci Int Genet 2013;7(1):151–8. [DOI] [PubMed] [Google Scholar]
- 62. Walsh S, Liu F, Ballantyne KN, van Oven M, Lao O, Kayser M. IrisPlex: a sensitive DNA tool for accurate prediction of blue and brown eye colour in the absence of ancestry information. Forensic Sci Int Genet 2011;5(3):170–80. [DOI] [PubMed] [Google Scholar]
- 63. Lyons LA. DNA mutations of the cat: the good, the bad and the ugly. J Feline Med Surg 2015;17(3):203–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. David VA, Menotti‐Raymond M, Wallace AC, Roelke M, Kehler J, Leighty R, et al. Endogenous retrovirus insertion in the KIT oncogene determines White and white Spotting in domestic cats. G3: Genes¦ Genomes¦ Genetics 2014;4(10):1881–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Menotti‐Raymond MA, David VA, O'Brien SJ. Pet cat hair implicates murder suspect. Nature 1997;386(6627):774. [DOI] [PubMed] [Google Scholar]
- 66. Butler JM, David VA, O'Brien SJ, Menotti‐Raymond M. The MeowPlex: a new DNA test using tetranucleotide STR markers for the domestic cat. Promega Corporation 2002;5(2):7–10. [Google Scholar]
- 67. Coomber N, David VA, O'Brien SJ, Menotti‐Raymond M. Validation of a short tandem repeat multiplex typing system for genetic individualization of domestic cat samples. Croat Med J 2007;48(4):547–55. [PMC free article] [PubMed] [Google Scholar]
- 68. Menotti‐Raymond M, David VA, Stephens JC, Lyons LA, O'Brien SJ. Genetic individualization of domestic cats using feline STR loci for forensic applications. J Forensic Sci 1997;42(6):1039–51. [PubMed] [Google Scholar]
- 69. Menotti‐Raymond MA, David VA, Wachter LL, Butler JM, O'Brien SJ. STR forensic typing system for genetic individualization of domestic cat (Felis catus) samples. J Forensic Sci 2005;50(5):1061–70. [PubMed] [Google Scholar]
- 70. Menotti‐Raymond M, David VA, O'Brien SJ, Johnson WE, Pflueger SM, Lindblad‐Toh K, et al. Patterns of molecular genetic variation among cat breeds. Genomics 2008;91(1):1–11. [DOI] [PubMed] [Google Scholar]
- 71. Tarditi CR, Grahn RA, Evans JJ, Kurushima JD, Lyons LA. Mitochondrial DNA sequencing of cat hair: an informative forensic tool. J Forensic Sci 2011;56(S1):S36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lyons LA, Grahn RA, Kun TJ, Netzel LR, Wictum EE, Halverson JL. Acceptance of domestic cat mitochondrial DNA in a criminal proceeding. Forensic Sci Int Genet 2014;13:61–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure. S1. Pedigree of the 15 parent‐offspring trios with known kinship for parentage validation.
Table S1. (a) PCR and primer information for cat SNPplexes.
Table S2. SBE primer information and concentrations.
Table S3. SNP genotypes of cat populations using the SNPplexes.