

Abstract
Triple-transgenic mice (3xTgAD) overexpressing Swedish-mutated β-amyloid precursor protein (βAPPswe), P310L-Tau (TauP301L), and physiological levels of M146V-presenilin-1 (PS1M146V) display extracellular amyloid-β peptides (Aβ) deposits and Tau tangles. More disputed is the observation that these mice accumulate intraneuronal Aβ that has been linked to synaptic dysfunction and cognitive deficits. Here, we provide immunohistological, genetic, and pharmacological evidences for early, age-dependent, and hippocampus-specific accumulation of the β-secretase-derived βAPP fragment C99 that is observed from 3 months of age and enhanced by pharmacological blockade of γ-secretase. Notably, intracellular Aβ is only detectable several months later and appears, as is the case of C99, in enlarged cathepsin B-positive structures, while extracellular Aβ deposits are detected ∼12 months of age and beyond. Early C99 production occurs mainly in the CA1/subicular interchange area of the hippocampus corresponding to the first region exhibiting plaques and tangles in old mice. Furthermore, the comparison of 3xTgAD mice with double-transgenic mice bearing the βAPPswe and TauP301L mutations but expressing endogenous PS1 (2xTgAD) demonstrate that C99 accumulation is not accounted for by a loss of function triggered by PS1 mutation that would have prevented C99 secondary cleavage by γ-secretase. Together, our work identifies C99 as the earliest βAPP catabolite and main contributor to the intracellular βAPP-related immunoreactivity in 3xTgAD mice, suggesting its implication as an initiator of the neurodegenerative process and cognitive alterations taking place in this mouse model.
Introduction
Alzheimer's disease (AD) is an age-related neurodegenerative disorder characterized by the accumulation of extracellular senile plaques mainly composed of hydrophobic amyloid-β (Aβ) peptides (Glenner and Wong, 1984; Masters et al., 1985) and intracellular filamentous aggregates of the microtubule-associated protein Tau (neurofibrillary tangles) (Grundke-Iqbal et al., 1986). The amyloid cascade hypothesis predicts that the accumulation and aggregation of Aβ, and particularly small aggregated Aβ oligomers (Walsh and Teplow, 2012), triggers a pathological chain of events that ultimately produces the pathological and clinical symptoms of AD (Hardy and Higgins, 1992; Hardy and Selkoe, 2002). Aβ has also been reported to accumulate inside neurons, but the role of intracellular Aβ in AD pathogenesis is still debated (Gouras et al., 2005; LaFerla et al., 2007; Bayer and Wirths, 2010).
Aβ is derived from combined proteolytic cleavages of the amyloid precursor protein (βAPP). The action of β-secretase [β-site APP cleaving enzyme 1 (BACE1)] liberates a membrane-associated C-terminal fragment C99, which is subsequently cleaved by γ-secretase, thereby yielding Aβ peptides of various lengths (38–43 aa) (Checler, 1995). Among them, Aβ42 has a higher propensity to aggregate than Aβ40 and is considered to be the most toxic Aβ species (Jarrett et al., 1993).
One of the most widely used AD animal models is a triple-transgenic mouse (3xTgAD) that overexpresses two mutant proteins, namely βAPPswe and TauP301L, and harbors normal levels of PS1M146V (Oddo et al., 2003). This mouse develops extracellular amyloid deposits and neurofibrillary tangles relatively lately and in expected AD-affected brains areas such as the hippocampus, amygdala, and cortex. Moreover, these animals develop an early and age-dependent increase of intraneuronal Aβ-related immunoreactivity (Oddo et al., 2003; Hirata-Fukae et al., 2008; Mastrangelo and Bowers, 2008) that was reported to correspond to genuine intracellular Aβ (Oddo et al., 2003; Billings et al., 2005). Interestingly, the deficiencies in long-term potentiation and cognitive impairments better correlated with the appearance of this intraneuronal staining than with the presence of extracellular Aβ deposits (Billings et al., 2005). However, the molecular nature of Aβ-related immunoreactivity has been recently disputed. Thus, a recent paper claimed that full-length βAPP rather than Aβ, accumulates in the 3xTgAD mice (Winton et al., 2011). We demonstrate here that most of the early and age-dependent accumulation of intracellular Aβ-like immunoreactivity corresponds to another βAPP by-product, the β-secretase-derived fragment C99, which accumulates in a region-specific manner.
Materials and Methods
Animals.
3xTgAD (harboring PS1M146V, βAPPswe, and TauP301L transgenes) and nontransgenic (wild-type) mice (Oddo et al., 2003) were generated from breeding pairs provided by Dr. F. LaFerla (University of California, Irvine, CA). To produce double-transgenic animals [2xTgAD (PS1WT, βAPPswe, and TauP301L)], triple-transgenic mice (3xTgAD) were crossed first with the wild-type mice, and the F1 progeny was then intercrossed giving 25% of homozygous mice expressing PS1WT, βAPPswe, and TauP301L, as described previously (Oddo et al., 2008). All mice were kept on the original 129/C57BL6 background strain. Animals were housed with a 12 h light/dark cycle and were given ad libitum access to food and water. Animals were 2–24 months of age of either sex. All experimental procedures were in accordance with the European Communities Council Directive of 24 November 1986 (86/609/EEC) and local French legislation.
Immunohistochemical analyses.
Animals were deeply anesthetized with pentobarbital and perfused transcardially with cold PBS followed by 4% paraformaldehyde/PBS. Brains were postfixed another 24 h and then embedded in paraffin using standard protocols. Coronal sections (8 μm) were cut on a microtome and processed for immunohistochemistry using the following antibodies: 2H3 [residues 1–12 of human Aβ; 1:800; Dr. D. Schenk, Elan Pharmaceuticals, South San Francisco, CA (Lefranc-Jullien et al., 2006)], FCA18 [free residue Asp 1 common in human and mouse Aβ and C99 sequences (Barelli et al., 1997); 1:800], 82E1 [human Aβ residues 1–16 (Horikoshi et al., 2004); IBL; 1:100], 4G8 (residues 17–24 of human Aβ; Covance; 1:1000), α-Aβ42 (Aβ42-specific; BioSource/Invitrogen; 1:1000), 22C11 (βAPP N-terminal; Millipore; 1:1000), βAPPcter [recognizing human and mouse βAPP C-terminal; 1:1000 (Pardossi-Piquard et al., 2009)], and α-Cathepsin B (Millipore; 1:200). Sections were treated for 6 min with formic acid (90%), blocked for 1 h with BSA (5%)/Tween 20 (0.05%), and then incubated at 4°C overnight with primary antibodies diluted in BSA (2.5%)/Tween 20 (0.05%). After washes, sections were incubated with secondary antibodies [HRP-conjugated (1:1000; Jackson ImmunoResearch) or fluorescent Alexa Fluor antibodies, Alexa 488- and Alexa 594-conjugated (Invitrogen; 1:1000)] at room temperature during 1 h. Cathespin B labeling was visualized using the Vectastain ABC kit (Vector) and streptavidin-Alexa 594 (Invitrogen; 1:1000). Fluorescent slides were incubated for 5 min with DAPI (Roche; 1:20,000) and coverslipped. Slides with HRP-conjugated antibodies were incubated with DAB-impact (Vector), rinsed, and counterstained with cresyl violet. For DAB development, slides were analyzed using an optical light microscope (DMD108; Leica). Immunofluorescence was visualized using either an epifluorescence microscope (Axioplan2; Zeiss), or a confocal microscope (Fluoview10; Olympus) using excitation filters 340, 488, and 594.
Preparation of soluble and insoluble brain fractions.
Mice were killed by intraperitoneal injection of a lethal dose of pentobarbital. Dissected hippocampi, cortices, or hemi-forebrains of 3xTgAD, 2xTgAD, and wild-type mice were homogenized, respectively, in RIPA buffer (50 mm Tris, pH 7.4, containing 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.5% deoxycholate, 0.1% SDS, and protease inhibitor mixture). After lysis with a Dounce homogenizer and brief sonication, proteins were ultracentifuged (100,000 × g, 1 h, 4°C), and supernatants were recovered as the soluble fractions. Pellets containing insoluble material were mechanically dissociated in formic acid (70%) and ultracentrifuged (100,000 × g, 1 h, 4°C). Supernatants were kept as the insoluble fraction. Before any analysis, these fractions were neutralized to pH 7.5 with 1 m Tris-HCl, pH 10.8, containing 25 mm betaine (20 times dilution).
Immunoprecipitation.
After preclearing of the lysates with protein A-agarose (Sigma-Aldrich) (1 h, 4°C), C83, C99, and APP intracellular domain (AICD) were immunoprecipitated using the Br188 antibody [directed against the βAPP C-terminal (Lefranc-Jullien et al., 2006), provided by Dr. M. Goedert, Medical Research Center, Cambridge, UK] from the soluble fractions, while Aβ peptides were immunoprecipitated from both soluble and insoluble fractions using the human-specific 2H3 antibody. Immunoprecipitates were recovered by overnight incubation at 4°C with protein A-agarose. Beads were washed three times with the above RIPA buffer and once with PBS, and then analyzed by Western blotting.
In vitro γ-secretase activity.
Membrane fractions of dissected cortices and hippocampi of 3xTgAD and nontransgenic (WT) mice were prepared as described previously (Flammang et al., 2012). Briefly, for γ-secretase assay, membrane fractions were solubilized in CHAPSO (3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonate) (0.25%) andincubated overnight at 37 or 4°C with recombinant substrate preparations (C100-FLAG; 1% v/v) in absence or in the presence of the γ-secretase inhibitor difluoroketone-167 (DFK-167) (50 μm) (Sevalle et al., 2009). Aβ and AICD-FLAG were detected by Western blotting using anti-Aβ 2H3 (1:1000; provided by Dr. D. Schenk) and/or anti-FLAG (1:5000; Sigma-Aldrich) antibodies.
SDS/PAGE and Western blot analyses.
Soluble or membrane fractions (50 μg) or immunoprecipitated proteins were dissolved in SDS sample buffer, separated on Tris-Tricine gels (16% for Aβ, AICD, C83, C99) or Tris-glycine gels (8% for βAPP and actin). Proteins were transferred to nitrocellulose membranes and probed with the adequate antibody: polyclonal anti-βAPP (Br188; 1:1000), monoclonal anti-Aβ (2H3; 1:1000), and anti-actin (1:5000; Sigma-Aldrich). Immunological complexes were revealed with anti-rabbit or anti-mouse peroxidase antibodies, followed by electrochemiluminescence (Lumilight Roche).
Sandwich ELISA analysis.
Aβ40 and Aβ42 levels were measured in the soluble and insoluble fractions (see above) using sandwich ELISA kits detecting human Aβ40 and Aβ42, respectively (BioSource/Invitrogen). Using these kits, the minimal detectable amounts of human Aβ42 is <10 pg/ml and of human Aβ40 is <6 pg/ml.
Surface-enhanced laser desorption ionization–time of flight mass spectrometry.
PS20 ProteinChip reactive arrays (Bio-Rad Laboratories) were used for the surface-enhanced laser desorption ionization–time of flight (SELDI-TOF) experiments. 6E10 (directed toward residues 1–17 of human Aβ; 1 μg) was added to the chips and incubated for 2 h at room temperature. Excess antibodies were then removed, and the chips were incubated during 30 min in 20 μl of blocking buffer (1% BSA in Tris-HCl, 100 mm, pH 8) at room temperature. Then, a 20 μl drop of samples was added to each spot, and the chips were incubated overnight at 4°C. After washing, chips were incubated with α-cyano-4-hydroxycinnamic acid matrix solution [1 μl of 20% saturated solution in 50% (v/v) acetonitrile and 0.25% trifluoroacetic acid]. The chips were air-dried, read with the ProteinChip Reader Series 4000, and analyzed with the ProteinChip System, series 4000 software.
In vivo treatment with the γ-secretase inhibitor, ELND006.
To confirm inhibition of γ-secretase activity in 3xTgAD mice after treatment with a γ-secretase inhibitor, a group of 8-month-old animals were treated with a single oral dose of ELND006 [30 or 100 mg/kg; Elan Pharmaceuticals, South San Francisco, CA (Brigham et al., 2010; Schroeter et al., 2010)] or with vehicle alone (methylcellulose/polysorbate 80; Sigma-Aldrich) via oral gavage. Animals were killed 6 h after administration, and γ-secretase activity was measured (see above). A second group of animals was treated once daily for 30 d with ELND006 (30 mg/kg) or with vehicle alone via oral gavage. For six ELND006-treated and control mice, hippocampi were frozen on dry ice and stored at −80°C until ELISA analysis of Aβ levels. For three animals of each group, one-half of the brains were postfixed in 4% paraformaldehyde and embedded in paraffin for histological analysis, and the other half were used for membrane preparation and analyzed for βAPP, C83, and C99 levels by Western blot analysis.
β-Secretase activity assay.
Dissected hippocampi and cortices from 4- and 13-month-old nontransgenic (WT) and 3xTgAD mice were collected and lysed with Tris-HCl (10 mm), pH 7.5, and then homogenates were monitored for their BACE1 activity as described previously (Andrau et al., 2003). Briefly, samples (30 μg of proteins) were incubated in a final volume of acetate buffer (25 mm, pH 4.5, 100 μl) containing BACE1 substrate [(7-methoxycoumarin-4-yl)-acetyl-SEVNLDAEFRK(2,4-dinitrophenyl)-RRNH2;10 μm; R&D Systems] in the absence or presence of β-secretase inhibitor I (50 μm; PromoCell). BACE1 activity corresponds to the β-secretase inhibitor I-sensitive fluorescence recorded at 320 and 420 nm as excitation and emission wavelengths, respectively.
Statistical analysis.
Statistical analyses were performed with Prism software (GraphPad Software) by using the Mann–Whitney test for pairwise comparisons or the Tukey one-way ANOVA test for multiple comparisons.
Results
3xTgAD mice display early and age-dependent intraneuronal FCA18-related immunoreactivity in the subiculum of the hippocampus
The 2H3 antibody recognizes the first 12 residues of the Αβ domain and therefore theoretically labels full-length βAPP, C99, and Αβ (Fig. 1Α). In 13-month-old 3xTgAD mice, 2H3 (Fig. 1B, left panels) reveals the presence of large dense senile plaques (arrowhead) as well as an intracellular labeling (arrows) in the neurons surrounding the senile plaques in the caudal hippocampus at the area of the subiculum/CA1 interchange. A similar staining pattern is observed with 4G8, another antibody recognizing residues 17–24 of the human Aβ domain of βAPP (data not shown).
Figure 1.
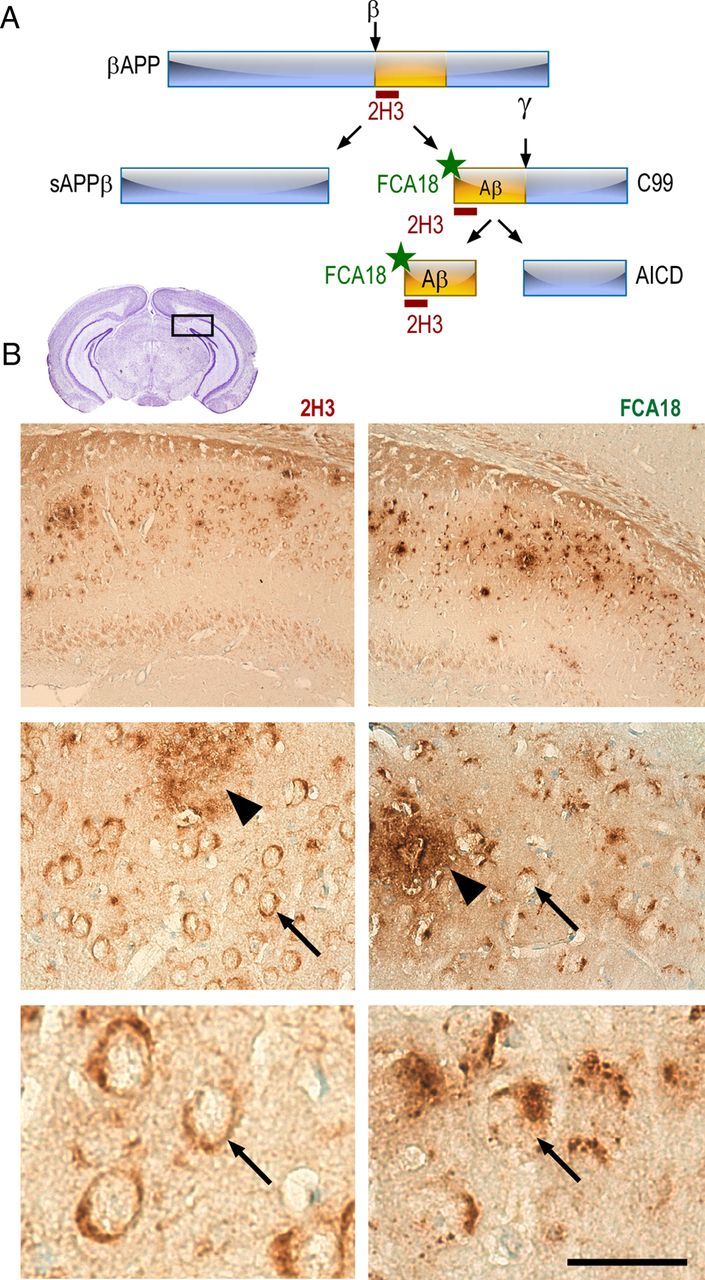
2H3 and FCA18 antibodies yield distinct intracellular staining in 3xTgAD mouse hippocampus. A, Schematic illustration of antibody epitopes and theoretical interaction with βAPP and its fragments. The 2H3 antibody directed against the first 12 aa of the N-terminal of Aβ recognizes βAPP, C99, and Aβ, but not AICD. The FCA18 antibody is directed against the free aspartyl N-terminal Aβ residue and recognizes C99 and Aβ but not βAPP or AICD. B, Immunohistochemical staining in the subiculum of the hippocampus (see inset) of 3xTgAD animals (13-month-old males) with 2H3 (left panels) or FCA18 (right panels). Immunoreactivity was revealed by horseradish peroxidase/DAB staining. Both antibodies yield a labeling that occurs mainly in the subiculum/CA1 interchange of the hippocampus (B, top panels). Higher magnification reveals both intracellular (arrows) and extracellular (arrowhead) staining (B, middle panels). Note that the intracellular staining obtained with 2H3 (B, bottom left panel) is evenly distributed within neurons soma, while FCA18 labeling appears clearly punctiform. Scale bar: (in B) Top panels, 250 μm; middle panels, 60 μm; bottom panels, 20 μm.
We earlier developed an antibody directed against the free aspartyl N-terminal residue of Aβ (FCA18) that therefore recognizes both the β-secretase cleaved fragment C99 and free Aβ peptides but neither full-length βAPP nor N-terminal truncated Aβ fragments (Barelli et al., 1997) (Fig. 1A). In the subiculum of 13-month-old 3xTgAD mice, FCA18 labels extracellular Aβ deposits (arrowhead) and also reveals a strong and punctiform intraneuronal labeling (arrows) clearly distinct from the more uniform labeling obtained with 2H3 (Fig. 1B, right panels).
To more specifically focus on βAPP catabolites and to avoid any cross labeling with βAPP itself, we examined the age-related progression of FCA18-associated intracellular staining in the hippocampus. We established that FCA18 staining appears in the subiculum as early as ∼3 months of age in 3xTgAD mice (Fig. 2A,B). A progressive age-related increase in the number of FCA18-positive neurons was observed until ∼16 months. In these old mice, FCA18 also stained extracellular deposits distributed randomly throughout the subiculum (Fig. 2A,B), while no labeling was observed in wild-type mice whatever their age (Fig. 2A). A similar immunolabeling was obtained when visualizing immunoreactivity by peroxidase/DAB development (data not shown). Interestingly, 82E1, another antibody recognizing C99 and Aβ, but not full-length βAPP (Horikoshi et al., 2004), revealed an immunostaining that perfectly matched the one observed with FCA18 whatever the age of the animals (Fig. 2B).
Figure 2.
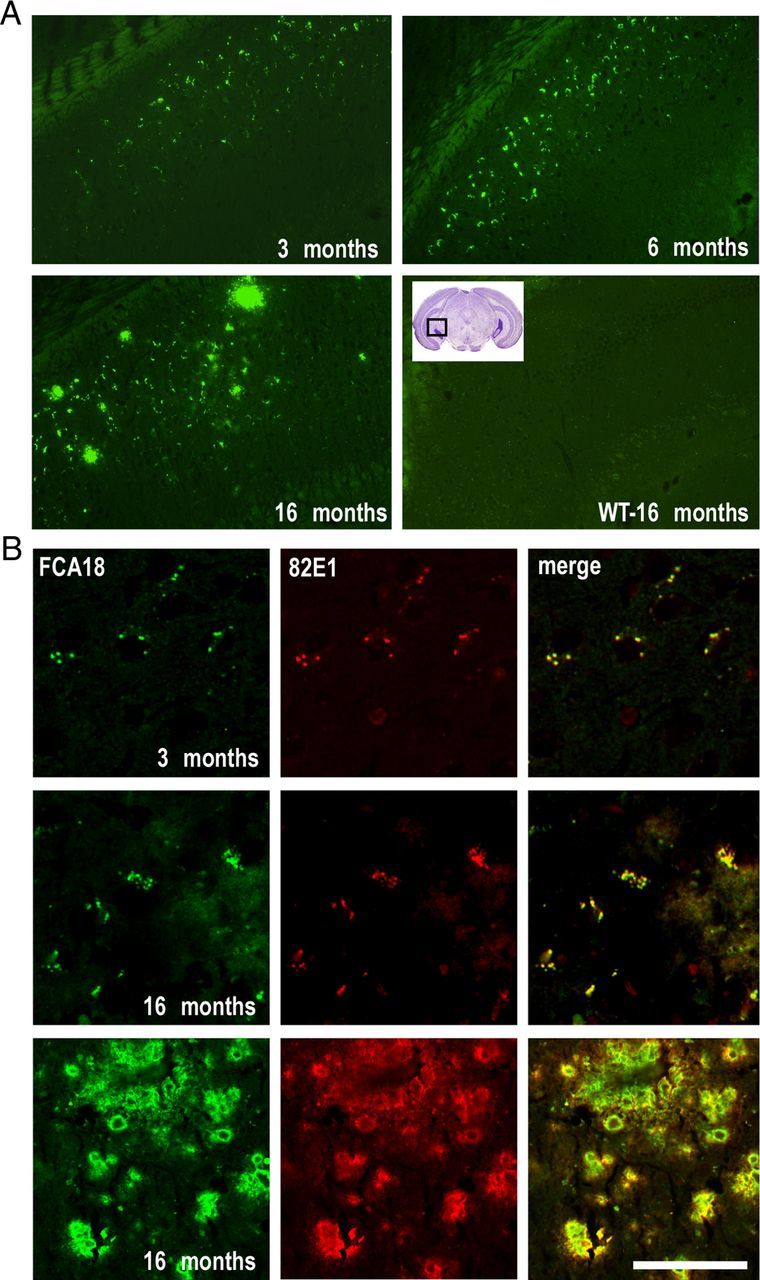
FCA18 reveals an early and age-dependent intracellular staining in the subiculum of 3xTgAD mice. A, FCA18-associated immunohistochemical staining of brain sections (level of the CA1/subiculum; see inset) of 3xTgAD (3-, 6-, and 16-month-old males) and WT (16-month-old males) mice. Epifluorescence images show FCA18-related intracellular punctiform staining as early as at 3 months. B, Confocal images obtained with FCA18 (green), 82E1 (red), and merged images from 3-month-old (top panels) and 16-month-old males (middle and bottom panels). Note the complete overlap between FCA18 and 82E1 in both young and old animals. Scale bar: (in B) A, 250 μm; B, top and middle panels, 50 μm; B, bottom panels, 100 μm.
To rule out any putative contribution of full-length βAPP to FCA18 and 82E1-associated immunoreactivities in young mice, we performed double-immunostaining experiments with FCA18 and 22C11 (a βAPP N-terminal specific antibody recognizing βAPP but not C99). 22C11 labeled a higher number of neurons than FCA18 in both the subiculum (Fig. 3A, top panels) and CA1 pyramidal layer (data not shown). Moreover, high-magnification images showed an evenly cytoplasmic/membrane-associated staining with 22C11 while the FCA18 labeling was more discrete and strongly punctuate (Fig. 3A, bottom panels). This dichotomy in this anatomical labels and the complete absence of overlap in merged images demonstrated that FCA18-linked immunoreactivity could not be accounted for an aspecific cross-reaction with βAPP. This was even reinforced by the comparison of the staining obtained with 2H3 and with a βAPPcter antibody in young 3xTgAD mice (Fig. 3B). Low-magnification analysis revealed an apparent overlap between the two labels (Fig. 3B, top right panel). However, high-magnification images delineated a 2H3-positive punctiform staining in a subset of neurons within the subiculum that did not colocalize with the βAPPcter antibody (Fig. 3B, bottom right panel). This punctiform labeling matched that observed with FCA18 (Fig. 3C). These data indicate that, as theoretically predicted, 2H3 antibody can label both βAPP, Aβ and C99, and, even if most of the 2H3 staining corresponds to full-length βAPP, the 2H3/FCA18 overlapping label indeed corresponds to either Aβ or C99, or to a mix of these βAPP-derived catabolites but not βAPP itself.
Figure 3.
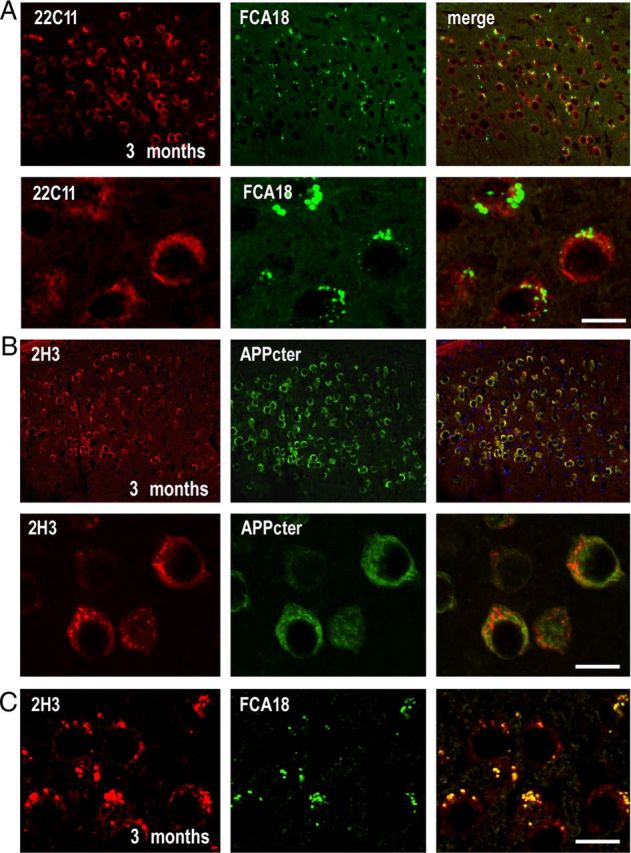
In 3-month-old animals, FCA18 reveals a punctuate labeling that does not correspond to full-length βAPP. A, Confocal images obtained with FCA18 (green), 22C11 (red), and merge images in the subiculum of 3-month-old males. Note the lack of overlap in the high-magnification images (bottom panels). B, Confocal images obtained with 2H3 (red), βAPP Cter (green), and merged images (right panels). Note that 2H3 reveals both punctiform and evenly distributed staining (see high magnification in B, left bottom panel), while only the latter staining is observed with the βAPPcter antibody. Accordingly, 2H3 reveals a subset of punctiform label that remains unlabeled by βAPPcter (see overlay in B, bottom right panel). C, Confocal images of double labeling with 2H3 (red) and FCA18 (green) in 3-month-old 3xTgAD mice. Note the perfect overlap between 2H3 and FCA18 staining in punctiform structures. Scale bar: (in C) A, B, Top panels, 50 μm; A, B, bottom panels, C, 10 μm.
FCA18-related intracellular label in young 3xTgAD mice hippocampi corresponds to C99 but not to Aβ peptides
FCA18 revealed an intracellular immunostaining in 3-month-old mice, but in these young animals, under our experimental conditions, we were unable to detect any staining when using specific C-terminal antibodies directed against Aβ40 (data not shown) or Aβ42 (Fig. 4A, top panel), whereas the neurons were indeed 22C11 positive. In older mice (16 months of age), the Aβ42-specific antibody stained a significant number of amyloid plaques and also revealed a low intracellular punctiform staining in the subiculum (Fig. 4A, bottom panels). The intracellular 22C11 staining was similar to the one obtained in young animals and was completely distinct from the Aβ staining (no overlap in the merged images; Fig. 4A, right panels). The Aβ-specific intracellular labeling was only detectable in animals of 6 months of age and beyond and was restricted to the neurons within the subiculum area. Thus, double-labeling experiments with 2H3 and the Aβ42-directed antibody in 8-month-old mice confirmed a punctiform staining localized to identical intracellular structures, although the labeling with Aβ42 remained low compared with that observed with 2H3 (Fig. 4B). Double labeling with the mouse monoclonal 82E1 and a rabbit polyclonal antibody specific to the lysosomal protein Cathepsin B (CatB) indicated that the intracellular sites accumulating C99 and Aβ correspond to lysosomes or lysosome-derived structures (Fig. 4C, top panels). Interestingly, CatB-positive structures were enlarged in 82E1-positive neurons (arrowhead) compared with those without 82E1 immunoreactivity (arrow), suggesting that these structures could correspond to autophagolysosomes (autophagic vacuoles fused with lysosomes) reflecting lysosomal dysfunction. Supporting the presence of autophagy, we were able to detect a small and age-dependent increase in autofluorescence also observed in absence of antibodies. This autofluorescence is observed only when increasing drastically the laser sensitivity and corresponds to lipofuscin granules reflecting lysosomal dysfunction (data not shown) (Nixon et al., 2000; Pasternak et al., 2004). Finally, as previously described, CatB was found in extracellular deposits (Cataldo and Nixon, 1990; Mueller-Steiner et al., 2006) (Fig. 4C, bottom panels).
Figure 4.
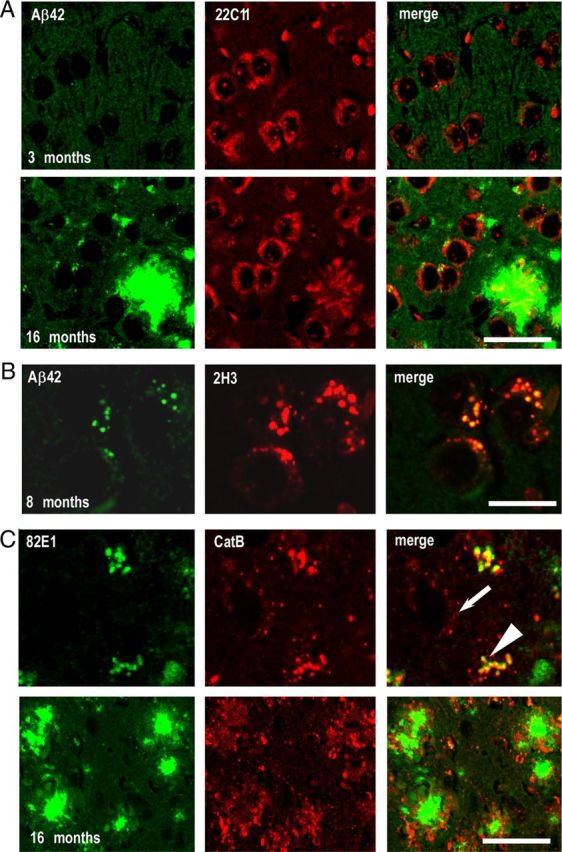
Genuine intraneuronal Aβ staining is absent in young mice and remains low at later ages. A, Confocal images of anti-Aβ42 (green) and 22C11 (red) staining in the subiculum of 3-month-old (top panels) and 16-month-old (bottom panels) 3xTgAD males. Note the absence of overlap in the merged images (right panels). B, Confocal microscopy images of double-staining experiments with Aβ42 (green) and 2H3 (red) in 8-month-old animals. The punctiform staining obtained with both antibodies is localized in identical intracellular structures (yellow color in merged images). C, Double immunostaining of 82E1 (green) and anti-cathepsin B (CatB) (red) in 16-month-old animals showed that the intracellular structures accumulating C99 and Aβ correspond to CatB-positive structures. Note the enlarged CatB structures in cells colabeled with 82E1 (arrowhead), compared with CatB-positive structures without 82E1 labeling (arrow). In these mice, CatB was also found in extracellular deposits (C, bottom panels). Scale bar: (in C) A, 25 μm; B, C, top panel, 10 μm; C, bottom panel, 50 μm.
We attempted to confirm the absence or very low levels of Aβ peptides in transgenic animals younger than 6 months of age by performing biochemical analysis of Aβ40 and Aβ42 levels in dissected hippocampi of 3xTgAD mice (Fig. 5). In 3-month-old mice, the levels of Aβ42 or Aβ40 in both soluble (RIPA-extracted) and insoluble (formic acid-extracted) fractions were below the limit of detection of the BioSource sandwich ELISA kit (see Materials and Methods) (Fig. 5A) and comparable with background levels measured in negative controls (10-month-old wild-type animals). Very low levels of Aβ40 and Aβ42 appeared detectable at 6 months of age in soluble and insoluble fractions, respectively. These peptides progressively increased with age and became particularly abundant in plaque-bearing animals (i.e., at 16 months of age and after). These findings were confirmed by SELDI-TOF mass spectrometry (Fig. 5B) that revealed the lack or very low levels of Aβ in 3- and 6-month-old animals, respectively, in both soluble and insoluble fractions. In agreement with ELISA data, SELDI-TOF analyses confirmed the occurrence of Aβ40 and Aβ42 in 10-month-old 3xTgAD mice but not in wild-type animals used as negative controls (with respect to human specificity of 6E10 antibodies; Fig. 5B). It should be noted that these analyses did not evidence other C-terminal truncated Aβ species, apart from a very faint detection of Aβ1–38 in 24-month-old 3xTgAD animals (Fig. 5B).
Figure 5.

ELISA and mass spectrometry analyses of Aβ species in 3xTgAD mice hippocampi. A, Aβ40 and Aβ42 levels were measured in soluble and insoluble fractions prepared from mice hippocampi 3xTgAD (3-, 6-, 10-, 16-, and 24-month-old males) and control wild-type (10-month-old males) mice by sandwich ELISA technique (see Materials and Methods). Bars corresponding to either Aβ40 or Aβ42 are expressed in nanograms per milligram of wet tissue. B, Aβ40 and Aβ42 levels were also analyzed at the indicated ages in soluble and insoluble fractions by SELDI-TOF mass spectrometry using the 6E10 antibody (recognizing the first 12 aa of the N-terminal of Aβ). Note that both Aβ40 and Aβ42 remained undetectable by both ELISA and SELDI-TOF in hippocampi of 3-month-old 3xTgAD animals.
The above set of biochemical and immunohistological data indicate that the FCA18-associated label in 3-month-old 3xTgAD mice cannot be accounted for Aβ species. As stated previously, FCA18 recognizes the free N-terminal aspartyl residue that occurs in both Aβ species but also in the β-secretase-derived catabolite C99. This fragment can undergo subsequent γ- or α-secretases cleavages yielding not only Aβ peptides but also C83 and AICD. In this context, we analyzed the levels of βAPP, C99, C83, Aβ, and AICD in whole-brain extracts of 2- to 18-month-old 3xTgAD and wild-type mice (Fig. 6A–C). As expected, the levels of βAPP measured by Western blot were higher in 3xTgAD mice (due to transgene expression) but remained unchanged with age (Fig. 6A). Using the more sensitive immunoprecipitation method, we observed an early (2-month-old) and age-dependent increase in the levels of C99, C83, and AICD, in 3xTgAD mice (Fig. 6B). Conversely, and in agreement with ELISA and mass spectrometry data, immunoprecipitable Αβ was only observed in 13-month-old mice brains and increased thereafter in an age-dependent manner (Fig. 6C). Since immunohistochemical experiments indicated an age-dependent increase within the subiculum/hippocampal regions, we isolated hippocampi of 6- and 18-month-old 3xTgAD and wild-type mice and analyzed βAPP levels by direct Western blotting and C99, C83, and AICD expression by combined immunoprecipitation/Western blot. In these conditions, C99, C83, and AICD levels were clearly increased with age in 3xTgAD mice, while βAPP levels remained unchanged (Fig. 6D--F). This transgene-associated enhancement of βAPP catabolites was particularly spectacular for C99 that was ∼16- and 25-fold higher than in wild-type mice at 6 and 18 months of age, respectively (Fig. 6E,F), while C83 and AICD levels increased by approximately fivefold to sixfold in 18-month-old 3xTgAD mice (Fig. 6E,F). In complete agreement with the biochemical data described above, immunohistochemical analysis comparing βAPP expression (using the 22C11 antibody) and C99/Aβ expression (using the FCA18 antibody) showed that the 22C11 staining was unchanged with age, whereas a clear age-dependent increase in intracellular and extracellular staining with FCA18 was observed (Fig. 6G).
Figure 6.
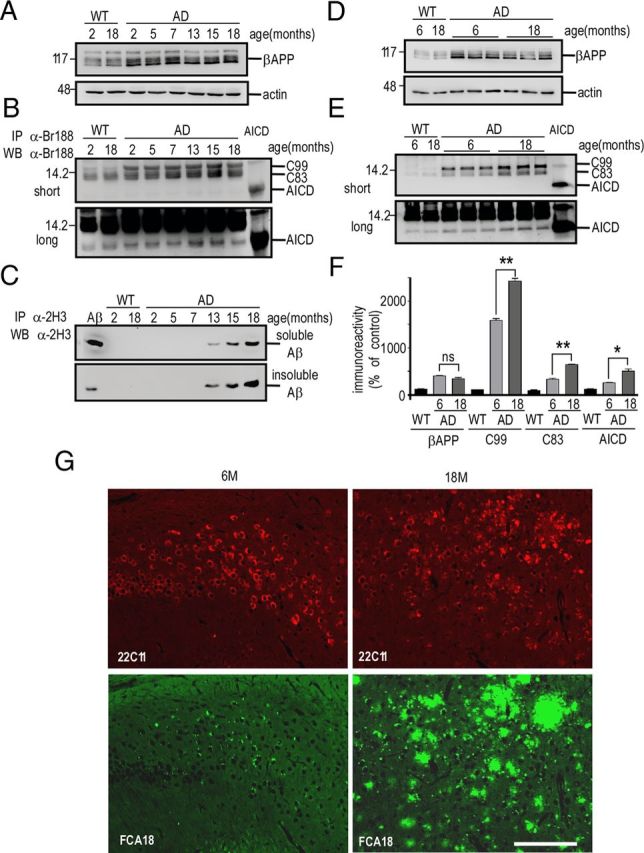
C99, C83, and AICD, but not βAPP, accumulate with aging in hippocampi of 3xTgAD mice. Soluble extracts of whole hemi-forebrains (A–C) or hippocampi (D–F) of wild-type (WT) or 3xTgAD (AD) females were prepared at the indicated ages and then analyzed by Western blot for βAPP [with Br188 in soluble fractions (A, D)], for C99, C83, and AICD [with Br188 in immunoprecipitates of soluble fractions (B, E)] and for Aβ [with 2H3 in immunoprecipitates of soluble and insoluble fractions (C)]. Actin was used as loading control, and synthetic Aβ and AICD (10 ng) were used as standards. The bars in F represent βAPP, C99, C83, and AICD immunoreactivities expressed as percentage of those measured in wild-type mice hippocampi (taken as 100) and are the means of three distinct animals. Error bars indicate SEM. *p < 0.05; **p < 0.001; ns, not statistically significant according to the Tukey one-way ANOVA test. G, Double immunostaining with 22C11 (red) and FCA18 (green) in the subiculum from 6- and 18 month-old 3xTgAD females. Note the high increase in FCA18 staining with age, while the 22C11 staining remained unchanged. Scale bar, 100 μm.
C99 accumulation is specific to the hippocampal region in the 3xTgAD mouse
Is the age-dependent modification of C99, C83, and AICD levels specific to the hippocampus? We performed biochemical analyses of βAPP and its catabolites in hippocampi and cortices of 13-month-old wild-type and 3xTgAD mice (Fig. 7A,B). Whereas the expression of βAPP in 3xTgAD mice was similar in these two structures, the levels of C99 were strongly increased in the hippocampi of 3xTgAD mice and only faintly modified in the cortices (Fig. 7A,B). Moreover, C83 and AICD levels were higher in 3xTgAD hippocampi than in wild-type mice while poorly detectable and not quantifiable in cortices (Fig. 7A,B). Immunohistochemical analyses corroborated the above data. FCA18 labeling was strong in the subiculum/hippocampus and spread out intracellularly and extracellularly, while a slight and restricted intracellular labeling was observed within cortical regions (Fig. 7C). ELISA and mass spectrometry analyses confirmed the occurrence of very low Aβ levels in cortices of transgenic animals whatever their age (data not shown).
Figure 7.

C99 strongly accumulates in hippocampi but not in cortices of 3xTgAD mice. A, Membrane fractions from cortices and hippocampi of wild-type (WT) and 3xTgAD (AD) females (13 months of age) were prepared as described in Materials and Methods and analyzed for βAPP, C99, C83, and AICD expression using the BR188 antibody and revealed upon short and long exposures. Actin was used as loading control. The bars in B are the means ± SEM of six animals and represent the quantification of βAPP, C99, C83, and AICD immunoreactivities expressed as respective expressions measured in wild-type mice (taken as 100). *p < 0.01; **p < 0.005; and ns, not significantly different (Mann–Whitney test). C, Coronal sections of hippocampus and cortex of 13-month-old 3xTgAD mice were immunostained with FCA18 to detect intracellular and extracellular labeling. Labeling was visualized using peroxidase/DAB development, as described in Materials and Methods. D, Dissected hippocampi and cortices from 4- and 13-month-old wild-type and 3xTgAD females were collected and monitored for their BACE1 activity in the absence (WT− and AD−) or presence (WT+ and AD+) of the BACE inhibitor I. Values of kinetic analyses of β-secretase activity corresponds to arbitrary fluorometric units recorded at 320 and 420 nm as excitation and emission wavelengths, respectively.
To establish whether the specific hippocampal accumulation of C99 could be explained by an enhanced production of C99 resulting from a higher β-secretase activity in this brain region, we measured BACE1 activity (Andrau et al., 2003) in hippocampi and cortices of both 4- and 13-month-old animals. A significant inhibitor-specific BACE1 activity was measured in both wild-type and 3xTgAD animals that remained similar in hippocampi and cortices of the two mice lines, whatever their age (Fig. 7D). This clearly indicates that region-specific and time-dependent increase of C99 in 3xTgAD mice was not due to abnormally high BACE1 activity.
In vivo treatment of 3xTgAD mice with a selective γ-secretase inhibitor, ELND006, increases the levels of intraneuronal FCA18-associated staining
We postulated that, if the above-described FCA18-associated intracellular labeling indeed corresponds to C99, γ-secretase inhibition should amplify this immunostaining, while conversely Aβ-related label should be wiped out or drastically decreased by γ-secretase blockade. We first established that a 6 h treatment of 8-month-old 3xTgAD animals with ELND006 (at both 30 and 100 mg/kg) fully reduced hippocampal γ-secretase-mediated productions of Aβ and AICD (Fig. 8A) measured in vitro, in a cell-free assay previously described (Flammang et al., 2012). We therefore examined the influence of a chronic treatment (30 d; 30 mg · kg−1 · d−1 of ELND006; 5- to 6-month-old mice) on Aβ levels in 3xTgAD mice brains. Although the Aβ levels are low at this age, ELISA measurements of Aβ40 and Aβ42 revealed a virtually complete reduction of both Aβ species in ELND006-treated 3xTgAD mice (Fig. 8B). Concomitantly, ELND006 increased the levels of the two γ-secretase substrates C99 and C83, while it did not influence βAPP expression (Fig. 8C,D). Immunohistochemical analysis of ELND006-treated animals also showed a dramatic enhancement of FCA18-associated intracellular punctiform staining (Fig. 8E, left panels), whereas 22C11 staining remained unchanged (Fig. 8E, right panels). ELND006 also enhanced the 2H3-associated labeling (Fig. 8F, middle panel) that overlaid that observed with FCA18 (Fig. 8F, right panel) and therefore confirmed that intracellular punctiform FCA18 and 2H3 labels correspond to the same βAPP-related fragment, the production of which is increased by γ-secretase inhibition and definitively identifies it as C99.
Figure 8.

In vivo treatment of 3xTgAD mice with a selective γ-secretase inhibitor, ELND006, reduces Aβ40 and Aβ42 levels and enhances C99 accumulation and FCA18-associated intracellular immunostaining. A, 3xTgAD mice were treated with a single oral dose of ELND006 of 30 mg/kg (D6-30) or 100 mg/kg (D6-100) (AD-D6) or with vehicle alone (AD-CT). Six hours after dosing, mice were killed and γ-secretase activity was measured in hippocampi using recombinant C100-Flag in a cell-free in vitro assay described in Materials and Methods. Aβ and AICD were monitored using a mix of two antibodies (2H3 and anti-Flag for Aβ and AICD detection, respectively) and revealed after short- and long-term gel exposures. Synthetic Aβ (10 ng) was used as standard. B–F, Mice (5- to 6-month-old females) were treated once daily for 30 d with ELND006 (30 mg/kg) or with vehicle. B, Aβ levels were quantified by ELISA in soluble and insoluble fractions prepared from hippocampi. The bars are the means of six distinct animals from each group (duplicate measurements). Error bars indicate SEM. C, D, Membrane fractions were analyzed for βAPP, C83, and C99 levels with BR188. Actin was used as loading control. The bars in D represent βAPP, C99, and C83 immunoreactivities expressed as percentage of their respective control levels recovered in wild-type mice (taken as 100) and are the means of three distinct animals. Error bars indicate SEM. E, Immunostaining with FCA18 (green) and 22C11 (red) in CA1/subiculum of vehicle- (AD-CT) or ELND006-treated (AD-D6) mice. Note the high increase in FCA18 staining in AD-D6-treated mice, while the 22C11 staining was unchanged. F, Confocal images of double labeling with 2H3 (red) and FCA18 (green) correspond to FCA18/2H3 double immunolabeling. Scale bars: E, 50 μm; F, 10 μm.
The accumulation of C99 is not due to a loss of γ-secretase function
The accumulation of C99 could result from a defect of its secondary processing by γ-secretase. In this context, it is noteworthy that 3xTgAD mice harbor the PS1M146V mutation that has been initially described as a loss-of-function mutant (Chen et al., 2002). Therefore, to determine whether a partial loss of γ-secretase function could be responsible for the C99 accumulation, we performed genetic selection to obtain double-transgenic mice (2xTgAD) harboring both βAPPswe and TauP301L mutant proteins but expressing endogenous wild-type PS1. In 13-month-old 3xTgAD mice, Aβ was readily immunoprecipitable, while it was almost undetectable in age-matched 2xTgAD mice [see the very faint label in 2xTgAD mice (2AD) only observed after long exposure in Fig. 9A]. ELISA analyses confirmed the barely detectable levels of Aβ40 and Aβ42 in insoluble fractions (Fig. 9B) as well as Aβ40 in soluble fraction (data not shown) of 2xTgAD mice brain samples. In 2xTgAD mice, expressions of βAPP, C99, C83, and AICD (Fig. 9C–E) were significantly higher than in wild-type mice (as expected from βAPP transgene expression), but remained identical to those recovered in 3xTgAD mice. It has to be noted that AICD levels were identical in 2xTgAD and 3xTgAD mice, indicating that the PS1M146V mutation triggers distinct effects on Aβ and AICD productions. Interestingly, punctiform intraneuronal FCA18-like immunoreactivity was similar in 3-month-old (Fig. 9F) and 13-month-old (Fig. 9G) 2xTgAD and 3xTgAD mice, while extracellular deposits were only detected in 3xTgAD mice (Fig. 9G). Thus, these deposits correlated with the presence of PS1M146V and were more likely associated with Aβ production (much higher in 3xTgAD mice; Fig. 9A,B) than with C99 (similar in 2xTgAD and 3xTgAD mice; Fig. 9D,E). Incidentally, this further confirms that FCA18-related intracellular staining in 3xTgAD mice corresponds to C99 and rules out the possibility that it could correspond to Aβ.
Figure 9.
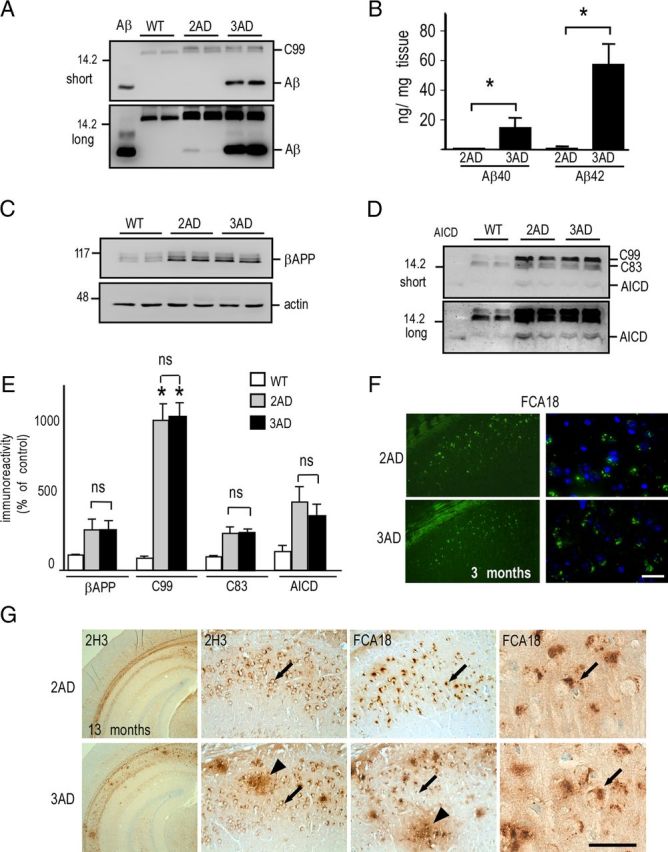
C99, C83, and AICD, but not Aβ, accumulate in 2xTgAD brain hippocampi. A, Aβ levels were analyzed in soluble fractions of hippocampi prepared from wild-type (WT), double-transgenic (2AD), and triple-transgenic (3AD) females (13 months of age) by immunoprecipitation/Western blot using the 2H3 antibody and revealed upon short and long exposures. Synthetic Aβ peptide (10 ng) was used as standard. B, ELISA sandwich analysis of Aβ40 and Aβ42 levels in insoluble fractions of hippocampi of 2AD and 3AD males (*p < 0.05, Mann–Whitney test). C–E, Membrane fractions of hippocampi from indicated mice (13 months of age of either sex) were analyzed for βAPP (C), C99, C83, and AICD (D) expressions by Western blot and revealed upon short and long exposures using the Br188 antibody. Actin was used as loading control, and synthetic AICD (10 ng) was used as standard. The bars in E correspond to βAPP, C99, C83, and AICD immunoreactivities expressed as percentage of respective expressions measured in wild-type mice (taken as 100) and are the means ± SEM of four animals. *p < 0.001; ns, not statistically significant according to the Tukey one-way ANOVA test. F, FCA18 labeling of 2xTgAD (2AD) and 3xTgAD (3AD) brain sections (subiculum; 3-month-old males) is revealed by fluorescence as described in Materials and Methods. Scale bar: Right panel, 80 μm; left panel, 20 μm. G, 2H3- and FCA18-associated immunostainings of indicated transgenes (13-month-old males; subiculum) using horseradish peroxidase/DAB development (see Materials and Methods). Both antibodies reveal both intracellular label (arrows) and extracellular plaques (arrowheads) in 3xTgAD mice, but no extracellular deposits were detected in 2xTgAD mice. Scale bar: Left panel, 500 μm; middle panels, 100 μm; right panel, 15 μm.
Finally, we used the enzymatic cell-free assay described above to measure γ-secretase activity in hippocampi and cortices of wild-type and 3xTgAD mice (Fig. 10). These data did not reveal a decrease in γ-secretase activity in 3xTgAD mice. Indeed, in 3xTgAD mice, Aβ in vitro production in hippocampi was slightly increased, rather than decreased when compared with wild-type mice (Fig. 10A,B), in accordance with the immunoprecipitation and ELISA data of 2xTgAD (PS1wt) versus 3xTgAD (PS1M146V) mice showing higher Aβ levels in 3xTgAD. Consistent with the small but significant increase in γ-secretase activity in 3xTgAD hippocampi, we observed a significant increase of the levels of PS1, Aph1, and Pen2, three protein partners of the γ-secretase complex, in hippocampi of 3xTgAD mice (data not shown). Together, these data show that a loss of function in γ-secretase activity is not responsible for the specific accumulation of C99 in the hippocampus.
Figure 10.

γ-Secretase activity and expression in wild-type (WT) and 3xTgAD mice. A, B, Membrane fractions of dissected cortices and hippocampi from WT and 3xTgAD (AD) females (13 months of age) were prepared as described in Materials and Methods. Then, γ-secretase cell-free assay using recombinant C100-Flag was performed for 16 h at 4 or 37°C, in presence or absence of DFK-167. C100, AICD, and Aβ were detected using a mix of anti-Flag and 2H3 antibodies, and synthetic Aβ peptide (10 ng) was used as standard (note that short and long exposure are shown). The bars in B correspond to AICD- and Aβ-like immunoreactivities expressed as percentage of control levels obtained in wild-type mice (taken as 100) and are the means ± SEM of six animals. *p < 0.05 according to the Mann–Whitney test.
Discussion
This paper unambiguously demonstrates an early, age-dependent, and region-selective intracellular accumulation of the β-secretase-derived βAPP fragment, C99, in the 3xTgAD mouse. This accumulation is particularly strong in the hippocampus and occurs firstly and mainly in the CA1/subiculum area. The conclusion that C99, rather than Aβ or full-length βAPP, accumulates in young 3xTgAD mice is based on multiple immunohistochemical, genetic, and pharmacological experimental approaches and is supported by four lines of independent data. First, two antibodies (FCA18 and 82E1) that recognize Aβ and C99, but not full-length βAPP, revealed an early (3-month-old mice) and age-dependent punctuate intraneuronal staining distinct from that observed with βAPP N- or C-terminal-directed antibodies. In addition, when using the immunoprecipitation method or direct Western blot using purified hippocampal membrane fractions, we observed an age-dependent increase in C99 levels and lack of changes in βAPP expression. Second, Aβ was undetectable in 3-month-old mice whatever the technique used (ELISA, immunoprecipitation, mass spectrometry, and immunohistochemical analysis using Aβ-specific antibodies) and became detectable only several months later, as previously observed by others (Hirata-Fukae et al., 2008). Third, the inhibitor (ELND006), which inhibits γ-secretase activity and thus Aβ production in 3xTgAD mice, triggered a strong augmentation of C99 levels assessed by biochemical approach and a concomitant increased intraneuronal FCA18- and 82E1-related immunohistochemical staining in young animals. Fourth, the comparison of 3xTgAD mice with age-matched 2xTgAD mice (expressing endogenous wild-type PS1) indicates that both mouse strains exhibit similar C99 expression and FCA18-associated intracellular staining, while 2xTgAD mice displayed barely detectable Aβ levels and do not show extracellular deposits. Overall, our study undoubtedly establishes that the punctiform intraneuronal label observed in situ in young 3xTgAD mouse hippocampi corresponds to C99, which remains the main intracellular accumulating catabolite in these mice.
Studies on the identification of intraneuronal label in 3xTgAD mice have led to contrasted conclusions. Thus, a recent study claimed that intracellular labeling in 3xTgAD mice corresponded to full-length βAPP but not Aβ (Winton et al., 2011), which was confirmed in another paper reporting mainly βAPP and only minor intraneuronal Aβ in hippocampi of 3xTgAD mice (Wirths et al., 2012). These reports were in strong contradiction with Dr. F. LaFerla's group, who originally argued that the intracellular immunoreactive material accumulating in 3xTgAD mice brain corresponded to Aβ (Oddo et al., 2003, 2008; Billings et al., 2005). Our study showing an early accumulation of C99 agrees to some extent with both reports. Thus, in agreement with Dr. F. LaFerla's group, we found an age-dependent increase in the levels of Aβ using ELISA and mass spectrometry approaches but only after 6 months of age in these mice. Therefore, clearly the earliest intracellular lesions are not due to genuine Aβ. If one sticks to this conclusion, then one can agree with Dr. V. Lee's group that the intracellular early label does not correspond to Aβ (Winton et al., 2011). However, our data do not support the conclusion of Dr. V. Lee's group that the age-dependent increase in intracellular immunostaining corresponds to full-length βAPP. Instead, we demonstrate that C99 constitutes the earliest intracellular βAPP-related immunoreactivity in 3xTgAD mice and that the levels of this catabolite increases with age, while βAPP levels remains unaffected.
How does C99 selectively accumulate at early stages in 3xTgAD mice hippocampus? The prime hypothesis could be an enhanced formation of C99. 3xTgAD mice harbor the βAPP Swedish mutation that has been shown to favor β-secretase-mediated cleavage of βAPP (Citron et al., 1992). However, although we found higher levels of C99 in these mice when compared with their wild-type congeners, we did not detect obvious quantitative differences in BACE1 [the main cerebral β-secretase (Vassar et al., 2009)] activity between wild-type and transgenic animals or between hippocampi or cortices at the two ages tested, 4 and 13 months of age. Therefore, BACE1 activity and region distribution could not explain the region-specific accumulation of C99.
Another possibility for explaining the accumulation of C99, based on a simple precursor–product relationship, could be a partial loss of γ-secretase function due to the PS1M146V mutation that would have impaired C99 secondary processing. However, two lines of data rule out this hypothesis. First, the C99 accumulation was identical in mice expressing endogenous wild-type PS1 or the PS1M146V mutated protein. Second, functional analysis of γ-secretase activity showed no evidence for a decrease of this activity in 3xTgAD hippocampi when compared with wild-type mice. Moreover, the treatment of the 3xTgAD mice with the potent and selective γ-secretase inhibitor ELND006 (Brigham et al., 2010) led to a rapid and massive increase in C99 levels, indicating a functional γ-secretase. Since C99 is produced in a brain area where γ-secretase is functional, this means that, even at young ages, mice likely produce Aβ, but that this fragment is extremely labile and barely detectable in brain. Therefore, the above data suggest that neither an enhanced BACE1-mediated production nor a defective γ-secretase-mediated secondary processing could account for C99 accumulation.
Our work evidenced an accumulation of C99, and later Aβ, in cathepsin B-positive granules corresponding to lysosomes or lysosome-derived structures. Lysosomes constitute important sites for βAPP proteolytic cleavages. They display the substrate βAPP, the protein machinery (β- and γ-secretases) and the environmental conditions (acidic pH) necessary and optimal for C99 and Aβ production (Golde et al., 1992; Haass et al., 1992; Pasternak et al., 2004). Furthermore, lysosomes that are enriched in lysosome-specific proteases and hydrolases (cathepsins) are also important in the degradation of βAPP catabolites. In this regard, it is important to note that amyloidogenic APP fragments have been seen to accumulate in lysosomes after treatment with alkalization drugs or protease inhibitors, as well as in Presenilin-1 knock-out cells lacking γ-secretase activity (Golde et al., 1992; Chen et al., 2000; Vingtdeux et al., 2007a,b; Asai et al., 2011).
It is well known that one of the earliest manifestations of Alzheimer's disease is an endosomal–lysosomal dysfunction with associated proliferation of lysosomes, upregulation of expression of lysosomal hydrolases, and presence of enlarged endosomal structures (Cataldo et al., 2000; Pasternak et al., 2004). Moreover, defective lysosomal proteolysis can be accompanied by a failure of autophagic degradation (Nixon et al., 2000; Nixon, 2006; Nixon and Yang, 2011) and autophagic vacuoles can themselves be sites for βAPP proteolysis since these structures are enriched in both β- and γ-secretase activities (Yu et al., 2004; Cai et al., 2012). Our findings showing the accumulation of C99 in enlarged cathepsin B-positive structures enriched in autofluorescent pigments would be in agreement with the presence of C99 in autophagolysosomal structures and therefore agree with numerous emerging works using mouse AD models, including the 3xTgAD mouse demonstrating a link between impaired clearance of amyloidogenic fragments (mainly Aβ) and autophagosomal/lysosomal dysfunction (Caccamo et al., 2010; Funderburk et al., 2010; Spilman et al., 2010; Butler et al., 2011; Majumder et al., 2011; Yang et al., 2011).
Although the cause for C99 accumulation remains to be established, it has been interesting to observe a paralleled increase in AICD levels in both 2xTgAD and 3xTgAD mice that display an identical C99 accumulation. Although AICD can theoretically be produced in both amyloidogenic and nonamyloidogenic pathways (Konietzko, 2012; Pardossi-Piquard and Checler, 2012), we and others have shown that AICD is preferentially derived from C99 in the amyloidogenic pathway (Goodger et al., 2009; Belyaev et al., 2010; Flammang et al., 2012). This fragment has been reported to translocate into the nucleus and regulate transcription and expression of a series of gene products involved in cellular processes linked to or altered in AD (Beckett et al., 2012; Konietzko, 2012; Pardossi-Piquard and Checler, 2012). In addition to the increases in C99 and AICD levels, we also observed a concomitant enhancement of C83 expression. We recently showed (Flammang et al., 2012), in agreement with others (Jäger et al., 2009; Portelius et al., 2009), that C83 could derive from C99 by an α-secretase-mediated process, suggesting that the increased C83 levels in the 3xTgAD mouse correspond to a secondary biogenesis event directly linked to prior C99 accumulation. Therefore, the accumulation of C83 observed in ELND006-treated animals could be due to the blockade of AICD production or to the accumulation of its precursor C99.
Together, our work shows an early and brain-specific accumulation of C99 in the hippocampus of the 3xTgAD mouse. This brain region is the first to present both extracellular deposits and aggregated intraneuronal Tau in older mice, suggesting a putative correlation between C99 levels and the development of these anatomical lesions. Apart from being the substrate for the generation of amyloidogenic fragments, Aβ and AICD, C99 may also have a more direct role in AD pathogenesis (Oster-Granite et al., 1996; Nalbantoglu et al., 1997; Suh, 1997; Choi et al., 2001; Bittner et al., 2009; Mitani et al., 2012; Tamayev et al., 2012). Very interestingly, a recent paper documents that sAPPβ and/or C99, rather than Aβ, causes memory deficits in a mouse model of dementia (Tamayev et al., 2012). Moreover, although treatment with γ-secretase inhibitors have been shown to rescue memory deficits in mouse models of Alzheimer's disease (Comery et al., 2005; Martone et al., 2009) and Down's syndrome (Netzer et al., 2010), other studies have shown that γ-secretase inhibitors leading to strong accumulation of C-terminal fragments (C83 or C99) have undesirable effects on synapses (Bittner et al., 2009) and negatively affect cognitive function in Tg2576 and wild-type mice, when administrated subchronically (Mitani et al., 2012). Although the exact role of C99 in AD pathogenesis remains to be elucidated, our work suggests that C99 should be considered as an early and key contributor to anatomical lesions in the 3xTgAD mouse.
Footnotes
This work was supported by “Fondation de Coopération Scientifique du Plan Alzheimer,” “Fondation pour la Recherche Médicale.” This work has been developed and supported through the LABEX “Excellence Laboratory Program Investment for the Future” Development of Innovative Strategies for Transdisciplinary Approach to Alzheimer's Disease. We thank Dr. F. LaFerla for providing 3xTgAD and nontransgenic mice and Dr. H. Marie for giving Tg2575 animals. We also thank Dr. D. Schenk and Dr. M. Goedert for providing antibodies and Franck Aguila for artwork.
References
- Andrau D, Dumanchin-Njock C, Ayral E, Vizzavona J, Farzan M, Boisbrun M, Fulcrand P, Hernandez JF, Martinez J, Lefranc-Jullien S, Checler F. BACE1- and BACE2-expressing human cells: characterization of beta-amyloid precursor protein-derived catabolites, design of a novel fluorimetric assay, and identification of new in vitro inhibitors. J Biol Chem. 2003;278:25859–25866. doi: 10.1074/jbc.M302622200. [DOI] [PubMed] [Google Scholar]
- Asai M, Yagishita S, Iwata N, Saido TC, Ishiura S, Maruyama K. An alternative metabolic pathway of amyloid precursor protein C-terminal fragments via cathepsin B in a human neuroglioma model. FASEB J. 2011;25:3720–3730. doi: 10.1096/fj.11-182154. [DOI] [PubMed] [Google Scholar]
- Barelli H, Lebeau A, Vizzavona J, Delaere P, Chevallier N, Drouot C, Marambaud P, Ancolio K, Buxbaum JD, Khorkova O, Heroux J, Sahasrabudhe S, Martinez J, Warter JM, Mohr M, Checler F. Characterization of new polyclonal antibodies specific for 40 and 42 amino acid-long amyloid beta peptides: their use to examine the cell biology of presenilins and the immunohistochemistry of sporadic Alzheimer's disease and cerebral amyloid angiopathy cases. Mol Med. 1997;3:695–707. [PMC free article] [PubMed] [Google Scholar]
- Bayer TA, Wirths O. Intracellular accumulation of amyloid-beta—a predictor for synaptic dysfunction and neuron loss in Alzheimer's disease. Front Aging Neurosci. 2010;2:8. doi: 10.3389/fnagi.2010.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckett C, Nalivaeva NN, Belyaev ND, Turner AJ. Nuclear signalling by membrane protein intracellular domains: the AICD enigma. Cell Signal. 2012;24:402–409. doi: 10.1016/j.cellsig.2011.10.007. [DOI] [PubMed] [Google Scholar]
- Belyaev ND, Kellett KA, Beckett C, Makova NZ, Revett TJ, Nalivaeva NN, Hooper NM, Turner AJ. The transcriptionally active amyloid precursor protein (APP) intracellular domain is preferentially produced from the 695 isoform of APP in a β-secretase-dependent pathway. J Biol Chem. 2010;285:41443–41454. doi: 10.1074/jbc.M110.141390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Bittner T, Fuhrmann M, Burgold S, Jung CK, Volbracht C, Steiner H, Mitteregger G, Kretzschmar HA, Haass C, Herms J. γ-Secretase inhibition reduces spine density in vivo via an amyloid precursor protein-dependent pathway. J Neurosci. 2009;29:10405–10409. doi: 10.1523/JNEUROSCI.2288-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigham E, Quinn K, Kwong G, Willits C, Goldbach E, Motter R, Lee M, Hu K, Wallace W, Kholodenko D, Tang-Tanaka P, Ni H, Hemphill S, Chen X, Eichenbaum T, Ruslim L, Nguyen L, Santiago P, Liao A, Bova M, et al. Pharmacokinetic and pharmacodynamic investigation of ELND006, a novel APP-selective gamma-secretase inhibitor, on amyloid concentrations in the brain, CSF and plasma of multiple nonclinical species following oral administration. Paper presented at International Conference on Alzheimer's Disease; July; Honolulu. 2010. [Google Scholar]
- Butler D, Hwang J, Estick C, Nishiyama A, Kumar SS, Baveghems C, Young-Oxendine HB, Wisniewski ML, Charalambides A, Bahr BA. Protective effects of positive lysosomal modulation in Alzheimer's disease transgenic mouse models. PLoS One. 2011;6:e20501. doi: 10.1371/journal.pone.0020501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–13120. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Zhao B, Li K, Zhang L, Li C, Quazi SH, Tan Y. Mammalian target of rapamycin: a valid therapeutic target through the autophagy pathway for Alzheimer's disease? J Neurosci Res. 2012;90:1105–1118. doi: 10.1002/jnr.23011. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Nixon RA. Enzymatically active lysosomal proteases are associated with amyloid deposits in Alzheimer brain. Proc Natl Acad Sci U S A. 1990;87:3861–3865. doi: 10.1073/pnas.87.10.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157:277–286. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checler F. Processing of the beta-amyloid precursor protein and its regulation in Alzheimer's disease. J Neurochem. 1995;65:1431–1444. doi: 10.1046/j.1471-4159.1995.65041431.x. [DOI] [PubMed] [Google Scholar]
- Chen F, Yang DS, Petanceska S, Yang A, Tandon A, Yu G, Rozmahel R, Ghiso J, Nishimura M, Zhang DM, Kawarai T, Levesque G, Mills J, Levesque L, Song YQ, Rogaeva E, Westaway D, Mount H, Gandy S, St George-Hyslop P, et al. Carboxyl-terminal fragments of Alzheimer beta-amyloid precursor protein accumulate in restricted and unpredicted intracellular compartments in presenilin 1-deficient cells. J Biol Chem. 2000;275:36794–36802. doi: 10.1074/jbc.M006986200. [DOI] [PubMed] [Google Scholar]
- Chen F, Gu Y, Hasegawa H, Ruan X, Arawaka S, Fraser P, Westaway D, Mount H, St George-Hyslop P. Presenilin 1 mutations activate gamma 42-secretase but reciprocally inhibit epsilon-secretase cleavage of amyloid precursor protein (APP) and S3-cleavage of notch. J Biol Chem. 2002;277:36521–36526. doi: 10.1074/jbc.M205093200. [DOI] [PubMed] [Google Scholar]
- Choi SH, Park CH, Koo JW, Seo JH, Kim HS, Jeong SJ, Lee JH, Kim SS, Suh YH. Memory impairment and cholinergic dysfunction by centrally administered Abeta and carboxyl-terminal fragment of Alzheimer's APP in mice. FASEB J. 2001;15:1816–1818. doi: 10.1096/fj.00-0859fje. [DOI] [PubMed] [Google Scholar]
- Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- Comery TA, Martone RL, Aschmies S, Atchison KP, Diamantidis G, Gong X, Zhou H, Kreft AF, Pangalos MN, Sonnenberg-Reines J, Jacobsen JS, Marquis KL. Acute gamma-secretase inhibition improves contextual fear conditioning in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2005;25:8898–8902. doi: 10.1523/JNEUROSCI.2693-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flammang B, Pardossi-Piquard R, Sevalle J, Debayle D, Dabert-Gay AS, Thévenet A, Lauritzen I, Checler F. Evidence that the amyloid-beta protein precursor intracellular domain, AICD, derives from beta-secretase-generated C-terminal fragment. J Alzheimers Dis. 2012;30:145–153. doi: 10.3233/JAD-2012-112186. [DOI] [PubMed] [Google Scholar]
- Funderburk SF, Marcellino BK, Yue Z. Cell “self-eating” (autophagy) mechanism in Alzheimer's disease. Mt Sinai J Med. 2010;77:59–68. doi: 10.1002/msj.20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122:1131–1135. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- Golde TE, Estus S, Younkin LH, Selkoe DJ, Younkin SG. Processing of the amyloid protein precursor to potentially amyloidogenic derivatives. Science. 1992;255:728–730. doi: 10.1126/science.1738847. [DOI] [PubMed] [Google Scholar]
- Goodger ZV, Rajendran L, Trutzel A, Kohli BM, Nitsch RM, Konietzko U. Nuclear signaling by the APP intracellular domain occurs predominantly through the amyloidogenic processing pathway. J Cell Sci. 2009;122:3703–3714. doi: 10.1242/jcs.048090. [DOI] [PubMed] [Google Scholar]
- Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer's disease. Neurobiol Aging. 2005;26:1235–1244. doi: 10.1016/j.neurobiolaging.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Koo EH, Mellon A, Hung AY, Selkoe DJ. Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature. 1992;357:500–503. doi: 10.1038/357500a0. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Hirata-Fukae C, Li HF, Hoe HS, Gray AJ, Minami SS, Hamada K, Niikura T, Hua F, Tsukagoshi-Nagai H, Horikoshi-Sakuraba Y, Mughal M, Rebeck GW, LaFerla FM, Mattson MP, Iwata N, Saido TC, Klein WL, Duff KE, Aisen PS, Matsuoka Y. Females exhibit more extensive amyloid, but not tau, pathology in an Alzheimer transgenic model. Brain Res. 2008;1216:92–103. doi: 10.1016/j.brainres.2008.03.079. [DOI] [PubMed] [Google Scholar]
- Horikoshi Y, Sakaguchi G, Becker AG, Gray AJ, Duff K, Aisen PS, Yamaguchi H, Maeda M, Kinoshita N, Matsuoka Y. Development of Abeta terminal end-specific antibodies and sensitive ELISA for Abeta variant. Biochem Biophys Res Commun. 2004;319:733–737. doi: 10.1016/j.bbrc.2004.05.051. [DOI] [PubMed] [Google Scholar]
- Jäger S, Leuchtenberger S, Martin A, Czirr E, Wesselowski J, Dieckmann M, Waldron E, Korth C, Koo EH, Heneka M, Weggen S, Pietrzik CU. alpha-secretase mediated conversion of the amyloid precursor protein derived membrane stub C99 to C83 limits Abeta generation. J Neurochem. 2009;111:1369–1382. doi: 10.1111/j.1471-4159.2009.06420.x. [DOI] [PubMed] [Google Scholar]
- Jarrett JT, Berger EP, Lansbury PT., Jr The C-terminus of the beta protein is critical in amyloidogenesis. Ann N Y Acad Sci. 1993;695:144–148. doi: 10.1111/j.1749-6632.1993.tb23043.x. [DOI] [PubMed] [Google Scholar]
- Konietzko U. AICD Nuclear signaling and its possible contribution to Alzheimer's disease. Curr Alzheimer Res. 2012;9:200–216. doi: 10.2174/156720512799361673. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer's disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- Lefranc-Jullien S, Sunyach C, Checler F. APPepsilon, the epsilon-secretase-derived N-terminal product of the beta-amyloid precursor protein, behaves as a type I protein and undergoes alpha-, beta-, and gamma-secretase cleavages. J Neurochem. 2006;97:807–817. doi: 10.1111/j.1471-4159.2006.03748.x. [DOI] [PubMed] [Google Scholar]
- Majumder S, Richardson A, Strong R, Oddo S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One. 2011;6:e25416. doi: 10.1371/journal.pone.0025416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martone RL, Zhou H, Atchison K, Comery T, Xu JZ, Huang X, Gong X, Jin M, Kreft A, Harrison B, Mayer SC, Aschmies S, Gonzales C, Zaleska MM, Riddell DR, Wagner E, Lu P, Sun SC, Sonnenberg-Reines J, Oganesian A, et al. Begacestat (GSI-953): a novel, selective thiophene sulfonamide inhibitor of amyloid precursor protein gamma-secretase for the treatment of Alzheimer's disease. J Pharmacol Exp Ther. 2009;331:598–608. doi: 10.1124/jpet.109.152975. [DOI] [PubMed] [Google Scholar]
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrangelo MA, Bowers WJ. Detailed immunohistochemical characterization of temporal and spatial progression of Alzheimer's disease-related pathologies in male triple-transgenic mice. BMC Neurosci. 2008;9:81. doi: 10.1186/1471-2202-9-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitani Y, Yarimizu J, Saita K, Uchino H, Akashiba H, Shitaka Y, Ni K, Matsuoka N. Differential effects between gamma-secretase inhibitors and modulators on cognitive function in amyloid precursor protein-transgenic and nontransgenic mice. J Neurosci. 2012;32:2037–2050. doi: 10.1523/JNEUROSCI.4264-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller-Steiner S, Zhou Y, Arai H, Roberson ED, Sun B, Chen J, Wang X, Yu G, Esposito L, Mucke L, Gan L. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer's disease. Neuron. 2006;51:703–714. doi: 10.1016/j.neuron.2006.07.027. [DOI] [PubMed] [Google Scholar]
- Nalbantoglu J, Tirado-Santiago G, Lahsaïni A, Poirier J, Goncalves O, Verge G, Momoli F, Welner SA, Massicotte G, Julien JP, Shapiro ML. Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature. 1997;387:500–505. doi: 10.1038/387500a0. [DOI] [PubMed] [Google Scholar]
- Netzer WJ, Powell C, Nong Y, Blundell J, Wong L, Duff K, Flajolet M, Greengard P. Lowering beta-amyloid levels rescues learning and memory in a Down syndrome mouse model. PLoS One. 2010;5:e10943. doi: 10.1371/journal.pone.0010943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA. Autophagy in neurodegenerative disease: friend, foe or turncoat? Trends Neurosci. 2006;29:528–535. doi: 10.1016/j.tins.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Yang DS. Autophagy failure in Alzheimer's disease—locating the primary defect. Neurobiol Dis. 2011;43:38–45. doi: 10.1016/j.nbd.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Cataldo AM, Mathews PM. The endosomal-lysosomal system of neurons in Alzheimer's disease pathogenesis: a review. Neurochem Res. 2000;25:1161–1172. doi: 10.1023/a:1007675508413. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Tseng B, Cheng D, Vasilevko V, Cribbs DH, LaFerla FM. Blocking Abeta42 accumulation delays the onset and progression of tau pathology via the C terminus of heat shock protein70-interacting protein: a mechanistic link between Abeta and tau pathology. J Neurosci. 2008;28:12163–12175. doi: 10.1523/JNEUROSCI.2464-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oster-Granite ML, McPhie DL, Greenan J, Neve RL. Age-dependent neuronal and synaptic degeneration in mice transgenic for the C terminus of the amyloid precursor protein. J Neurosci. 1996;16:6732–6741. doi: 10.1523/JNEUROSCI.16-21-06732.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardossi-Piquard R, Checler F. The physiology of the beta-amyloid precursor protein intracellular domain AICD. J Neurochem. 2012;120(Suppl 1):109–124. doi: 10.1111/j.1471-4159.2011.07475.x. [DOI] [PubMed] [Google Scholar]
- Pardossi-Piquard R, Böhm C, Chen F, Kanemoto S, Checler F, Schmitt-Ulms G, St George-Hyslop P, Fraser PE. TMP21 transmembrane domain regulates gamma-secretase cleavage. J Biol Chem. 2009;284:28634–28641. doi: 10.1074/jbc.M109.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak SH, Callahan JW, Mahuran DJ. The role of the endosomal/lysosomal system in amyloid-beta production and the pathophysiology of Alzheimer's disease: reexamining the spatial paradox from a lysosomal perspective. J Alzheimers Dis. 2004;6:53–65. doi: 10.3233/jad-2004-6107. [DOI] [PubMed] [Google Scholar]
- Portelius E, Brinkmalm G, Tran AJ, Zetterberg H, Westman-Brinkmalm A, Blennow K. Identification of novel APP/Abeta isoforms in human cerebrospinal fluid. Neurodegener Dis. 2009;6:87–94. doi: 10.1159/000203774. [DOI] [PubMed] [Google Scholar]
- Schroeter S, Brigham E, Motter R, Nishioka C, Guido T, Khan K, Kholodenko D, Tanaka P, Soriano F, Quinn K, Goldbach E, Games D, Ness D. APP-selective gamma secretase inhibitor ELND006 effects on brain parenchymal and vascular amyloid beta. Paper presented at International Conference on Alzheimer's Disease; July; Honolulu. 2010. [Google Scholar]
- Sevalle J, Ayral E, Hernandez JF, Martinez J, Checler F. Pharmacological evidences for DFK167-sensitive presenilin-independent gamma-secretase-like activity. J Neurochem. 2009;110:275–283. doi: 10.1111/j.1471-4159.2009.06131.x. [DOI] [PubMed] [Google Scholar]
- Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS One. 2010;5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh YH. An etiological role of amyloidogenic carboxyl-terminal fragments of the beta-amyloid precursor protein in Alzheimer's disease. J Neurochem. 1997;68:1781–1791. doi: 10.1046/j.1471-4159.1997.68051781.x. [DOI] [PubMed] [Google Scholar]
- Tamayev R, Matsuda S, Arancio O, D'Adamio L. beta- but not gamma-secretase proteolysis of APP causes synaptic and memory deficits in a mouse model of dementia. EMBO Mol Med. 2012;4:171–179. doi: 10.1002/emmm.201100195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Kovacs DM, Yan R, Wong PC. The beta-secretase enzyme BACE in health and Alzheimer's disease: regulation, cell biology, function, and therapeutic potential. J Neurosci. 2009;29:12787–12794. doi: 10.1523/JNEUROSCI.3657-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vingtdeux V, Hamdane M, Bégard S, Loyens A, Delacourte A, Beauvillain JC, Buée L, Marambaud P, Sergeant N. Intracellular pH regulates amyloid precursor protein intracellular domain accumulation. Neurobiol Dis. 2007a;25:686–696. doi: 10.1016/j.nbd.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Vingtdeux V, Hamdane M, Loyens A, Gelé P, Drobeck H, Bégard S, Galas MC, Delacourte A, Beauvillain JC, Buée L, Sergeant N. Alkalizing drugs induce accumulation of amyloid precursor protein by-products in luminal vesicles of multivesicular bodies. J Biol Chem. 2007b;282:18197–18205. doi: 10.1074/jbc.M609475200. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Teplow DB. Alzheimer's disease and the amyloid beta-protein. Prog Mol Biol Transl Sci. 2012;107:101–124. doi: 10.1016/B978-0-12-385883-2.00012-6. [DOI] [PubMed] [Google Scholar]
- Winton MJ, Lee EB, Sun E, Wong MM, Leight S, Zhang B, Trojanowski JQ, Lee VM. Intraneuronal APP, not free Abeta peptides in 3xTg-AD mice: implications for tau versus Abeta-mediated Alzheimer neurodegeneration. J Neurosci. 2011;31:7691–7699. doi: 10.1523/JNEUROSCI.6637-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wirths O, Dins A, Bayer TA. AbetaPP accumulation and/or intraneuronal amyloid-beta accumulation? The 3xTg-AD mouse model revisited. J Alzheimers Dis. 2012;28:897–904. doi: 10.3233/JAD-2011-111529. [DOI] [PubMed] [Google Scholar]
- Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson D, Bandyopadhyay U, Jiang Y, Pawlik M, Peterhoff CM, Yang AJ, Wilson DA, St George-Hyslop P, Westaway D, Mathews PM, Levy E, Cuervo AM, Nixon RA. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain. 2011;134:258–277. doi: 10.1093/brain/awq341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WH, Kumar A, Peterhoff C, Shapiro Kulnane L, Uchiyama Y, Lamb BT, Cuervo AM, Nixon RA. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for beta-amyloid peptide over-production and localization in Alzheimer's disease. Int J Biochem Cell Biol. 2004;36:2531–2540. doi: 10.1016/j.biocel.2004.05.010. [DOI] [PubMed] [Google Scholar]