

Abstract
Four novel chelators (L1–L4) and their 89zirconium complexes were prepared and compared with the 89zirconium desferrioxamine B (DFO) complex. The new chelates are based on 1,4,7,10-tetraazacyclododecane (cyclen) and 1,4,8,11-tetraazacyclotetradecane (cyclam) scaffolds and present either three or four hydroxamate arms for coordination with Zr4+ ions with coordination numbers between six and eight. The 89Zr–L4 complex showed similar stability to that of 89Zr–DFO when incubated in either rat blood plasma or ethylenediaminetetraacetic acid challenge experiments. Positron imaging and biodistribution studies in mice showed that 89Zr–L4 had similar pharmacokinetic behavior to that of 89Zr–DFO, with rapid renal elimination and low residual activity in background tissues. A bifunctional version of L4 (L5) was synthesized and conjugated to trastuzumab; an anti-HER2/neu antibody. Immunopositron emission tomography imaging and biodistribution with 89Zr–L5–trastuzumab revealed high tumor to background ratios (tumor/blood ratio: 14.2 ± 2.25) and a high tumor specificity that was comparable to the performance of 89Zr–DFO–trastuzumab.
Keywords: antitumor agents, imaging agents, macrocycles, radiopharmaceuticals, zirconium
Graphical abstract
Targeting tumors: The preparation of four chelators and their 89Zr complexes is described and their activity compared with the 89Zr–desferrioxamine B complex. A bifunctional version of one chelator has also been synthesized and conjugated to the anti-HER2/neu antibody trastuzumab for imaging in xenograft-bearing mice (see figure).

Introduction
Immunopositron emission tomography (immunoPET) is a rapidly advancing method for noninvasively tracking and quantifying the distribution of monoclonal antibodies (mAbs) in the body by using positron emission tomography (PET).[1] Immuno-PET is increasingly used as a precursor to enrolling patients for immunotherapy and radioimmunotherapy.[2]
89Zirconium has suitable emission properties for immunoPET (β+ =23%, E=avg. 395.5 keV)[3] and its half-life of 78.4 h is an optimal match for the pharmacokinetics of most mAbs.[4] The aqueous chemistry of zirconium is dominated by the 4+ oxidation state. Zr4+ is a hard, oxophilic cation that exhibits a preference for the coordination of hard, class a donors, including oxalate and polyhydroxamates such as desferrioxamine B (DFO; Figure 1). Notably, Zr4+ ions form complexes with coordination numbers of six to eight.[5]
Figure 1.

Chemical structures of DFO and azamacrocycle tethered polyhydroxamates L1, L2, L3, L4, and L5.
DFO is currently the chelator of choice when developing 89Zr PET probes. The tris-hydroxamate donor set and sterically flexible coordination environment result in a thermodynamically and kinetically stable complex, and the free terminal primary amine provides a handle for functionalization.[6] However, despite many preclinical and clinical applications of 89Zr–DFO–mAb, DFO remains suboptimal as a chelator for Zr4+.[5d,7] Most prominently, 89Zr deposition in bone (typically ≈5% of the injected dose per gram of bone (%ID g−1) in mice) has been observed at prolonged imaging times.[6a,8] High accumulation of 89Zr radioactivity in bone raises dosimetry concerns for human use.[6g,9] These shortcomings have stimulated research toward the development of new Zr4+ chelators with increased kinetic stability in vivo.[5e,7a–c,10]
To address the challenge of improving the Zr4+ complex stability and simplifying the conjugation chemistry, we are developing new chelates. Herein, the synthesis and characterization of four novel zirconium chelators based on hydroxamate-functionalized macrocycles is presented. The most promising chelate, a hexadentate donor (L4; Figure 1) was subsequently developed into a bifunctional analogue (L5) and characterized in vitro and in vivo.
Results and Discussion
Screening of ligands
Because Zr4+ exhibits hexa- to octadentate coordination, we selected 1,4,7,10-tetraazacyclododecane (cyclen) and 1,4,8,11-tetraazacyclotetradecane (cyclam) as macrocyclic scaffolds for the attachment of either three or four hydroxamate donor groups. The four secondary amines provide a facile handle for functionalization through alkylation or acylation with three to four arms containing hydroxamate groups. Initially, N-(benzyloxy)-2-bromo-N-methylacetamide[11] was used as an alkylating agent (see Scheme S1 in the Supporting Information for the synthetic procedure). Subsequent alkylation to produce compounds 1, 2, and 3 proceeded rapidly with both cyclen and cyclam, resulting in products that were isolated by preparative HPLC. Debenzylation with Pd/C-catalyzed hydrogenation provided ligands L1, L2, and L3 in overall yields of 70, 64, and 17%, respectively. Radiolabeling experiments were performed under typical conditions used for the labeling of mAb with 89Zr.[6a] Reactions were monitored by means of radio-thin layer chromatography (radioTLC; see Figure S2 in the Supporting Information), with an aqueous solution of 50 mm ethylenediaminetetraacetic acid (EDTA; pH 7) as the mobile phase. Under these conditions, 89zirconium labeling was comparable to that of DFO and ligands L1, L2, and L3 gave quantitative radiochemical yields (RCYs) of the corresponding 89Zr–L1, 89Zr–L2, and 89Zr–L3 complexes after 5 min of incubation at room temperature.
The stability of the 89zirconium complexes was assessed in vitro by using challenge experiments with excess EDTA (pH 7, 50 mM; Figure 2) at 37°C for 6 days. Accelerated transchelation can occur under these forcing conditions. Under these conditions, the order of stability was 89Zr–DFO> 89Zr–L2> 89Zr–L3> 89Zr–L1. We also measured the stability of these complexes in rat plasma at 37°C. Complex stability was monitored by removal of small aliquots at selected time points and analysis by size exclusion chromatography (SEC). The intact complex eluted in the low-molecular-weight fraction, whereas 89Zr radioactivity associated with plasma components eluted in the macromolecular fraction (Figure S2 in the Supporting Information). In this assay, 89Zr–DFO was again the most inert complex with an order of stability of 89Zr–DFO> 89Zr–L3> 89Zr–L2> 89Zr–L1.
Figure 2.
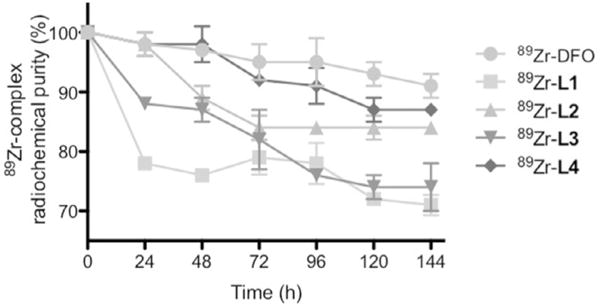
The percentage of intact complex at selected time points (between 0 and 144 h during incubation in EDTA (pH 7), concentration 55 mM, 37°C) shows comparable transchelation for 89ZrDFO and 89Zr–L4 with EDTA.
To investigate the electronic origins of the complex stability, DFT calculations were used to optimize the structures of complexes Zr–L1, Zr–L2, and Zr–L3, and the bonding interactions between the donor oxygen atoms and Zr4+ metal ion were studied by means of natural bond order (NBO) analysis. The optimized structures revealed that both Zr–L1 and Zr–L3 complexes formed seven-coordinate species, instead of the expected eight-coordinate complexes. Steric constraints imposed by the macrocyclic scaffold and the relatively short linker arms resulted in ligands that were unable to coordinate the Zr4+ ion in an octadentate fashion, despite the presence of eight potential donor atoms. Notably, as with the coordinatively unsaturated Zr–DFO complex, vacant coordination sites were potentially occupied (albeit transiently) by water ligands.[6a,7b]
As anticipated, DFT calculations indicated that the Zr–L2 complex formed a six-coordinate species, but this complex also resulted in considerable ligand strain. In an attempt to identify alternative chelators with reduced strain about the ligand backbone and first coordination sphere of the Zr4+ ions, we evaluated different chelators with increased flexibility and lengths of the hydroxamate donor arms. DFT calculations predicted that cyclam acylated with longer hydroxamate arms (L4; Figure 1) could result in a more favorable arrangement of the donor hydroxamate groups, potentially giving a more stable zirconium complex (Tables S3 and S4 and Figures S6–S8 in the Supporting Information). The optimized structure of Zr–L4 revealed that, in comparison to complex Zr–L2, the ligand was capable of adopting a more ideal geometry in the first coordination sphere (Figure 3) with decreased ligand strain. Overall, structural and bonding analysis predicted that Zr–L4 was thermodynamically more stable than Zr–L2 (see the Supporting Information). Although L4 presents only six donor atoms, and hence, is potentially (similar to DFO) coordinatively unsaturated about the Zr4+ ion, DFT calculations indicate that extension of the alkyl chain length leads to an almost ideal hexadentate coordination geometry. Relief of strain energy in the ligand backbone and first coordination sphere is likely to enhance the thermodynamic (and potentially kinetic) stability of Zr–L4. In addition, the use of only three of the macrocylic amine groups for coupling to hydroxamic acid groups leaves one free amine, which presents a handle for facile derivatization.
Figure 3.

DFT-optimized structure of Zr–L4. Hydrogen atoms have been omitted for clarity.
To obtain L4, the protected hydroxamate reagent 4-[(benzyloxy)(methyl)amino]-4-oxobutanoic acid[12] was synthesized to introduce a longer hydroxamate arm by amidation. The tri-amidated product 4 was isolated from the crude mixture after preparative HPLC and debenzylation gave L4 (6% overall yield). Interestingly, although isolation of a protected, octadentate version of compound L4 was feasible, debenzylation led to decomposition (in contrast to L4, which could be isolated without difficulty). Quantitative 89zirconium radiolabeling of L4 was attained in 5 min at room temperature.
As predicted by calculations, experimental studies revealed that the stability of 89Zr–L4 was greatly improved compared with our first-generation chelators. In the EDTA challenge, 89Zr–DFO and 89Zr–L4 performed equally well with 91 and 87% intact complex, respectively, after 6 days. In the rat plasma assay, 89Zr–L4 showed improved performance compared with that of 89Zr–DFO.
For example, the 89Zr–DFO samples showed substantial association of 89Zr to plasma proteins after 72 h (only 53% complex intact), whereas 89Zr–L4 showed less plasma association than that of 89Zr–DFO at all time points and was (94 ± 5)% intact after 72 h.
The lipophilicity (logD(n-octanol/phosphate-buffered saline (PBS) pH 7.4)) of the four novel complexes and 89Zr–DFO was measured (Table 1). As expected, the neutral complexes 89Zr–L1 (logD= −1.4) and 89Zr–L3 (logD= −2.1) were less hydrophilic than their cationic counterparts 89Zr–L2 (logD= −2.3), 89Zr–L4 (logD= −3.4) and 89Zr–DFO (logD= −3.1). The hydrophilicity of 89Zr–L4 is comparable to that of 89Zr–DFO.
Table 1.
The logD values obtained for the 89Zr complexes investigated herein (n=3).
| Complex | logD |
|---|---|
| 89Zr–L1 | −1.4±0.0 |
| 89Zr–L2 | −2.3±0.2 |
| 89Zr–L3 | −2.1±0.0 |
| 89Zr–L4 | −3.4±0.1 |
| 89Zr–DFO | −3.1±0.1 |
Having established that 89Zr–L4 displayed comparable stability to that of 89Zr–DFO in vitro, we next compared the pharmacokinetic behavior of 89Zr–L4 and 89Zr–DFO in mice by means of dynamic PET imaging and biodistribution. Tumor-naïve Balb/C mice were injected with 15–20 μCi of 89Zr–L4 or 89Zr–DFO and imaged from 0 to 30 min by PET. Complexes 89Zr–L4 and 89Zr–DFO behaved in an analogous manner during the dynamic imaging experiment, and the pharmacokinetic profiles were equivalent to previously reported experiments on 89Zr–DFO.[13] Rapid renal clearance was observed and >98% of the administered 89Zr activity was excreted into the bladder after 30 min (Figure S4 in the Supporting Information). Biodistribution data obtained 24 h postinjection revealed minimal differences in uptake/retention of 89Zr radioactivity in normal tissues between 89Zr–L4 and 89Zr–DFO (Table 2). Notably, complex 89Zr–L4 displayed slightly higher bone uptake ((0.60 ± 0.19)%IDg−1) than that of 89Zr–DFO ((0.05±0.02)%IDg−1), but also showed statistically significant lower retention in the kidneys ((2.76±0.40) % IDg−1 versus (3.50±0.08)%IDg−1, respectively; Student’s t-test P value <0.05). At 24 h postadministration, the 89Zr radioactivity circulating in the blood was very low for both 89Zr–L4 ((0.09±0.01)%IDg−1) and 89Zr–DFO ((0.06±0.02)%IDg−1). Higher accumulation of 89Zr radioactivity in the bone for 89Zr–L4 versus 89Zr–DFO was not encouraging; however, metabolism in vivo is also dependent on the nature of the biologically targeting vector (i.e., the entire radiotracer) and not simply the zirconium complex stability. Therefore, we designed and synthesized a bifunctional version of L4 (L5, see below) to evaluate if this ligand construct could still be useful in the development of radiotracers for immunoPET.
Table 2.
Biodistribution of 89Zr–DFO and 89Zr–L4 at 24 h postinjection in Balb/C mice (n=4). Values are given in %IDg−1.
| Organ | Accumulated 89Zr radioactivity | |
|---|---|---|
| 89Zr–DFO | 89Zr–L4 | |
| heart | 0.01±0.00 | 0.03±0.02 |
| lung | 0.02±0.02 | 0.03±0.02 |
| liver | 0.31±0.16 | 0.40±0.14 |
| spleen | 0.38±0.17 | 0.28±0.12 |
| kidney | 3.50±0.08 | 2.76±0.40 |
| small intestine | 0.06±0.04 | 0.11±0.05 |
| muscle | 0.00±0.00 | 0.00±0.00 |
| bone | 0.05±0.02 | 0.60±0.19 |
| blood | 0.06±0.02 | 0.09±0.01 |
Bifunctional ligand synthesis and bioconjugation
A bifunctional version of L4 was obtained by alkylation with benzyl (2-bromoacetyl)glycinate to afford compound 5, which was debenzylated to afford L5 (27% yield). The structure of L5 is analogous to that of DFO–succinate; a common intermediate used to synthesize DFO–mAb.[6a,f] Attempts to synthesize an isothiocyanato benzyl backbone-functionalized cyclam derivative were not successful because we found that the compound was prone to hydrolysis of the hydroxamate portion.
To evaluate the potential of L5 as a chelate for 89Zr immuno-PET, we conjugated and radiolabeled trastuzumab, a mAb targeting human epidermal growth factor 2 (HER2), which is overexpressed in a wide range of pathologies, including human breast, colon, lung, and ovarian cancers.[14] Trastuzumab (tz) represents one of the most thoroughly studied mAbs in the context of 89Zr immunoPET in both preclinical and clinical scenarios.[15] Conjugation of L5 to tz involved initial complexation with FeIII, followed by in situ activation of the carboxylate group with 2,3,5,6-tetrafluorophenol (TFP) by following similar protocols to those used to couple DFO–succinate to mAbs.[6f,15c] The reactions were quenched with excess EDTA (100-fold excess) and L5–tz was isolated by SEC. Conjugation was confirmed by ESI-MS. Radiolabeling with 89Zr was performed under identical conditions to those used for DFO-succinate–tz, with essentially quantitative RCY and a specific activity of 1.5–2.0 mCimg−1mAb (0.17–0.45 chelate mAb−1). Control experiments were performed by using radioTLC, followed by gel electrophoresis and autoradiography with the purified, radiolabeled conjugates (Figure 4). Cellular binding assays in vitro as well as PET and biodistribution studies in mice bearing either BT20 (HER2/neu negative) or BT474 (HER2/neu positive) subcutaneous tumors were used to assess the relative performance of 89Zr–L5–tz with 89Zr–DFO–tz.
Figure 4.
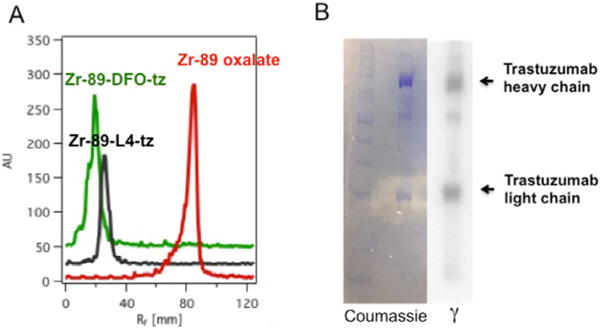
A) RadioTLC traces of 89Zr-labeled tz conjugates, with 89Zr–oxalate shown as a reference. B) Gel electrophoresis results for 89Zr–L5–tz, showing heavy and light chains in both the photograph obtained after Coumassie protein staining and an image obtained from autoradiography of an identical gel; these results show clear association of the radioactive label with the mAb fragments.
The specificity of 89Zr–L5–tz and 89Zr–DFO–tz to the HER2/neu target was assessed by using cellular binding assays with either BT20 (HER2−) and BT474 (HER2+) cells.[16] Both compounds maintained binding to HER2+ cells (89Zr–L5–tz: (4.3±0.5)%; 89Zr–DFO–tz: (5.5±0.5) % of total activity incubated) and significantly higher binding than nonspecific binding to HER2− cells (89Zr–L5–tz: (0.8±0.2) %; 89Zr–DFO–tz: (0.6±0.3)%, P<0.001 for both data sets for comparison of HER2 + and HER2− cells with each compound; Figure S5 in the Supporting Information). These data confirmed that, for both 89Zr–L5–tz and 89Zr–DFO–tz, the functionalization and 89Zr radiolabeling steps did not impinge on the HER2/neu binding specificity. Furthermore, in the same assays, both radiotracers showed equivalent total cellular binding, which indicated equivalent affinity and immunoreactivity.
Characterization of L5–tz in vivo
Next we compared the tumor uptake of 89Zr–L5–tz and 89Zr–DFO–tz in mice bearing subcutaneous HER2+ or HER2− tumors. Four groups of mice (HER2+, 89Zr–L5–tz; HER2−, 89Zr–L5–tz n=4 per group; HER2+, 89Zr–DFO–tz; HER2−, 89Zr–DFO–tz; n=5 per group) were imaged at 24, 48, 72, and 96 h postadministration of 89Zr antibody (Figure 5). After the final imaging time point, the biodistribution of 89Zr radioactivity was measured by tissue harvest and gamma counting ex vivo (Figure 6 and Table S1 in the Supporting Information). Regio-of-interest (ROI) analysis of blood (from heart volume), kidneys, liver, tumor, and bone was performed for each imaging time point to evaluate the time-dependent tissue uptake of each probe (Figure S6 in the Supporting Information). ROI analysis indicated that initial probe uptake in nontarget organs, such as blood, kidneys, and liver, peaked early and decreased over time. For mice with HER2– tumors, nonspecific tumor uptake of 89Zr activity with 89Zr–L5–tz was sevenfold lower than that with 89Zr–DFO–tz. PET quantification also showed that 89Zr radioactivity accumulation in bone increased over time in animals that received 89Zr–L5–tz. These results were confirmed by biodistribution and ex vivo counting. In HER2+ mice, 89Zr radioactivity uptake in the tumor increased over time and was similarly high for both radiotracers. Biodistribution data further confirmed that the uptake of 89Zr–L5–tz in HER2+ tumors was (65.7±7.7) % IDg−1 compared with (2.0 ± 0.9) % IDg−1 in HER2– tumors. With the exception of bone uptake, no statistically significant differences in the accumulation of 89Zr radioactivity between 89Zr–L5–tz and 89Zr–DFO–tz were found in off-target tissues in the groups of mice bearing HER2+ tumors. Complex 89Zr–L5–tz showed considerably higher bone uptake than that of 89Zr–DFO–tz ((18.9±1.1) % IDg−1 versus (2.9±1.0) % IDg−1, respectively; P<0.0001).
Figure 5.
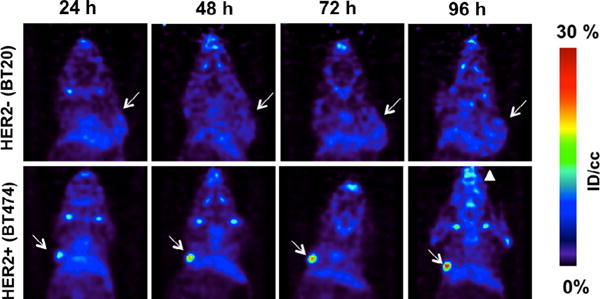
Coronal 89Zr–L5–tz PET images of mice bearing HER2− BT20 (top row) and HER2+ BT474 (bottom row) tumors. White arrows indicate tumors, showing low uptake of probe in the BT20 tumor, but high uptake in the BT474 tumor. The white triangle indicates enhanced uptake in the jaw at 96 h in animals with HER2+ xenografts.
Figure 6.

Biodistribution 96 h postinjection shows high specific uptake in HER2+ xenografts for both probes investigated in relevant organs. See the Supporting Information for data on additional organs.
Data from the animal experiments lead to the conclusion that L5 is less stable in vivo than DFO. To date, the chemical nature of the bone-seeking 89Zr species remains unknown. Although experimental data indicate that L5 is not a suitable replacement for DFO, these data provide valuable information from which we aim to improve the design of our next-generation chelates.
Conclusion
Four azamacrocycle-based hydroxamate chelators were synthesized for the complexation of 89zirconium, and their chemical and biochemical properties (radiolabeling, lipophilicity, in vitro complex stability, and behavior in mice) were examined. The stability of complex 89Zr–L4 was comparable to that of 89Zr–DFO in plasma and EDTA challenge experiments. In addition, initial studies in mice demonstrated that 89Zr–L4 had similar pharmacokinetics to that of 89Zr–DFO, with equivalent blood pool clearance and renal excretion profiles. The corresponding bifunctional version, L5, was synthesized, conjugated to tz, and radiolabeled to yield 89Zr–L5–tz. This radiotracer was shown to maintain specific targeting in vivo in HER2+ tumors, although uptake in nontarget organs and HER2− tumors remained low. Bone uptake was found to increase over time and was considerably higher than that in equivalent control experiments with 89Zr–DFO–tz. Although the zirconium complex of L5 may not be sufficiently stable to warrant continued development of this chelate, the results obtained herein represent an important step toward building a more stable chelator for imaging with the long-lived radionuclide 89zirconium.
Experimental Section
General methods and procedures
1H and 13C NMR spectra were recorded on a Varian 500 NMR system. LC-MS was performed by using an Agilent 1100 Series apparatus with an LC/MSD trap and Daly conversion dynode detector with UV detection at λ=220, 254, and 280 nm. Chemicals were supplied by Aldrich Chemical, TCI America, and Acros, and were used without further purification. Solvents (HPLC grade) were purchased from various commercial suppliers and used as received. Cyclam was received from Alfa Aesar; cyclen was obtained from Strem Chemicals. Purification of intermediates by HPLC was performed by using the HPLC method after injection of the filtered, crude mixture onto preparative HPLC (Rainin, Dynamax, column: 250 mm Phenomenex C18).
Method A
Solvent A: 0.1% trifluoroacetic acid in water; solvent B: 0.1% trifluoroacetic acid in MeCN; flow rate 15 mLmin−1, 0–1 min: 5% B, 1–3 min: 5–40% B, 3–15 min: 40–80% B, 15–16 min: 80–95% B, 16–20 min: 95% B, 20–21 min: 95–5% B, 21–23 min: 5% B. HPLC purity analysis (both UV and MS detection) of intermediates, final ligands, and corresponding FeIII complexes was performed on an Agilent 1260 system (column: Phenomenex Luna, C18(2) 100/2 mm) with UV detection at λ=220, 254 and 280 nm by using method B.
Method B
A gradient of A (0.1% trifluoroacetic acid in water) to 95% B(0.1% trifluoroacetic acid in MeCN), flow rate 0.8 mLmin−1. 0–1 min: 5% B, 1–10 min: 5–95% B, 10–12 min: 95% B, 12–12.5 min: 95–5% B, 12.5–15 min: 5% B.
Intermediate 1 was synthesized as reported by Esteves et al.[20] Intermediate 4 was synthesized as reported by Sharma et al.[12]
General procedure 1
A typical procedure for alkylation with N-(benzyloxy)-2-bromo-N-methylacetamide was performed by dissolving the azamacrocycle (0.07 mmol) in MeCN (5 mL). K2CO3 was added (0.08 g, 0.57 mmol). N-(Benzyloxy)-2-bromo-N-methylacetamide (0.1 g, 0.38 mmol) was dissolved in MeCN (1 mL) and added to the slurry, which was subsequently stirred for 18 h at room temperature. Reaction monitoring was performed by analysis of small aliquots of the reaction by LC-MS. The slurry was filtered and the resulting filtrate was concentrated in vacuo. The crude reaction filtrate was purified by preparative HPLC, method A.
General procedure 2
A typical procedure involving the removal of benzyl groups from precursors 1–4 was performed by dissolution of the starting material in methanol (5 mL). Pd/C (5% w/w matching the weight of the reactant used) was slowly added to the reaction mixture. The opaque slurry was transferred to a round-bottomed flask, which was sealed with a septum. The flask was evacuated and purged with H2 twice before charging with H2 (1 atm). The reaction was stirred for 6 h while being monitored for progression of the debenzylation through analysis of small aliquots of the reaction mixture by LC-MS. After the reaction was found to be complete, Pd/C was removed by filtration. The filtrate was collected and MeOH was removed in vacuo to afford the clean product.
Compound 1
Compound 1 was synthesized by using general procedure 1 with cyclen as the azamacrocycle. The product was isolated as a colorless solid after lyophilization (0.047 g, 0.053 mmol, 76%). 1H NMR (CD3OD, 500 MHz): δ=7.46–7.42 (m, 20H), 4.99 (s, 12H), 3.90 (br s, 8H), 3.45–2.81 ppm (m, 28H); 13C NMR (CD3OD, 125 MHz): δ= 160.7, 160.4, 160.1, 159.8, 134.5, 129.7, 128.9, 128.4, 119.7, 117.3, 115.0, 121.7, 75.7, 51.6, 32.5 ppm; LC-MS (ESI): m/z: 881.4 [M+H]+, Rt=8.7 min (method B).
Compound 2
Compound 2 was synthesized by using general procedure 1 with cyclam as the azamacrocycle. The product was isolated as a colorless solid after lyophilization (0.045 g, 0.061 mmol, 87%). 1H NMR (CD3OD, 500 MHz): δ=7.47–7.42 (m, 15H), 4.97–4.90 (m, 12H), 3.88 (br s, 2H), 3.36–2.76 (m, 23 H), 1.53 ppm (br m, 4H); 13C NMR (CD3OD, 125 MHz): δ=173.0, 165.7, 160.8, 160.5, 160.2, 159.9, 134.5, 134.4, 134.2, 130.0, 129.8, 129.7, 129.2, 129.0, 128.7, 128.5, 119.7, 117.3, 115.0, 112.7, 75.4, 53.9, 53.1, 51.2, 44.9, 32.3, 32.2, 32.0, 22.9, 22.1 ppm; LC-MS (ESI): m/z: 732.4 [M+H]+; Rt=7.8 min (method B).
Compound 3
Compound 3 was synthesized by using general procedure 1 with cyclam as the azamacrocycle. The product was isolated as a colorless solid after lyophilization (0.012 g, 0.013 mmol, 18%). 1H NMR (CD3OD, 500 MHz): δ=7.48–7.42 (m, 20 H), 4.96 (s, 8 H), 4.87 (s, 8 H) 3.62 (br s, 12 H), 3.36 (s, 8 H), 2.92 (br s, 8 H), 1.58 ppm (br m, 4H); 13C NMR (CD3OD, 125 MHz): δ=169.5, 160.5, 160.2, 134.3, 129.8, 129.1, 128.6, 75.4, 53.8 (br m), 48.2, 32.2, 22.0 ppm; LC-MS (ESI): m/z: [M+H]+; Rt=8.7 min (method B).
Compound 4
Compound 4 was synthesized by means of an amide bond-formation reaction. Cyclam (0.017 g, 0.089 mmol) was dissolved in DMF (5 mL). In a separate vial, 4-[(benzyloxy)(methyl)amino]-4-oxobutanoic acid (0.103 g, 0.43 mmol, 4.8 equiv) was dissolved in DMF with 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU; 0.196 g, 0.51 mmol) in presence of N,N-diisopropylethylamine (DIPEA; 0.067 mL, 0.09 g, 0.7 mmol) and stirred for 2 min before mixing with the solution containing cyclam. The reaction was allowed to proceed for 2.5 h, after which time the reaction appeared to be complete according to LC-MS analysis. The product was separated from side products by using preparative HPLC, method A and was isolated as a colorless solid after lyophilization (0.005 g, 0.006 mmol, 7%). 1H NMR (CD3OD, 500 MHz): δ=7.45–7.38 (m, 15 H), 4.95 (s, 6 H), 4.79 (s, 2 H) 3.60 (m, 10 H), 3.31 (m, 4 H), 3.21 (m, 10 H), 3.82–3.56 (m, 12 H), 2.12 ppm (m, 4 H); 13C NMR (CD3OD, 125 MHz): δ=174.3, 172.1, 134.8, 129.3, 129.2, 128.6, 28.3, 75.8, 46.7,45.7, 45.2, 32.4, 28.0, 27.2 ppm; LC-MS (ESI): m/z: 858.4 [M+H]+; Rt=8.1 min (method B).
Compound 5
Compound 5 was synthesized by dissolution of 4 (0.032 g, 0.038 mmol) and benzyl (2-bromoacetyl)glycinate (0.022 g, 0.077 mmol, 2 equiv) in MeCN (5 mL). KI (0.025 g, 0.15 mmol) and K2CO3 (0.03 g, 0.21 mmol) were added and the slurry was stirred for 48 h at room temperature, followed by filtration and reduction of the volume of the filtrate, which contained the product. The crude was purified by using HPLC, method A, and was isolated as a colorless solid after lyophilization (0.012 g, 0.011 mmol, 29%). 1H NMR (CD3OD, 500 MHz): 7.36–7.19 (m, 20 H), 5.08–4.79 (m, 16 H), 3.94–3.07 (m, 14 H), 2.71–2.39 (dd, 8 H), 1.83 ppm (m, 4 H); 13C NMR (CD3OD, 125 MHz): δ=174.4, 174.0, 173.5, 172.9, 170.9, 169.4, 169.3, 135.6, 135.1, 134.8, 129.3–127.8 (11 signals), 75.8, 66.7, 66.3, 56.3, 46.3–40.8 (5 signals), 32.4, 27.3 ppm; LC-MS (ESI): m/z: 1063.4 [M+H]+; Rt=8.4 min (method B).
Ligand L1
Ligand L1 was synthesized by using general procedure 2 with 1 as the starting material. The product was isolated as a colorless solid after lyophilization (0.025 g, 0.047 mmol, 89%). 1H NMR (CD3OD, 500 MHz): δ=4.87 (br s, 12 H), 4.11 (br s, 8 H), 3.35–3.21 ppm (m, 16 H); 13C NMR (CD3OD, 125 MHz): δ=161.4, 161.2, 160.9, 160.6, 53.2, 50.2, 48.5, 34.9 ppm; LC-MS (ESI): m/z: 521.2 [M+H]+; Rt=1.4 min (method B).
Ligand L2
Ligand L2 was synthesized by using general procedure 2 with 1 as the starting material. The product was isolated as a colorless solid after lyophilization (0.021 g, 0.048 mmol, 73%). 1H NMR (CD3OD, 500 MHz): δ=4.88 (m, 6 H), 4.21 (br s, 2 H), 3.61–2.68 (m, 26 H), 1.90 ppm (br s, 4 H); 13C NMR (CD3OD, 125 MHz): δ=174.0, 55.9, 51.1, 34.9, 33.5, 24.1, 22.8 ppm; LC-MS (ESI): m/z: 462.3 [M+H]+; Rt=1.2 min (method B).
Ligand L3
Ligand L3 was synthesized by using general procedure 2 with 1 as the starting material. The product was isolated as a colorless solid after lyophilization (0.007 g, 0.012 mmol, 92%). 1H NMR (CD3OD, 500 MHz): δ=3.94 (m, 8 H), 3.38–3.10 (m, 32 H), 1.99 ppm (br s, 4H); 13C NMR (CD3OD, 125 MHz): δ=168.3, 161.2, 160.9, 53.4 (m), 35.1, 22.4 ppm; LC-MS (ESI): m/z: 549.3 [M+H]+; Rt=1.4 min (method B).
Ligand L4
Ligand L4 was synthesized by using general procedure 2 with 4 as the starting material. The product was isolated as a colorless solid after lyophilization (0.008 g, 0.013 mmol, 81%). 1H NMR (CD3OD, 500 MHz): δ=3.83–3.56 (m, 12 H), 3.37 (m, 2 H), 3.19 (m, 9 H), 2.93 (br s, 2 H), 2.81–2.57 (s, 10 H), 2.02 (m, 2 H), 2.00 ppm (m, 2 H); 13C NMR (CD3OD, 125 MHz): 173.7, 173.3, 69.9, 69.8, 44.7, 44.6, 44.1, 40.7, 35.2, 34.9, 29.3, 29.0, 28.4, 28.2, 27.6, 27.2, 27.0, 26.9, 24.9, 22.8, 20.9 ppm; LC-MS (ESI): m/z: 588.3 [M+H]+; Rt=1.8 min (method B).
Ligand L5
Ligand L5 was synthesized by using general procedure 2 with 5 as the starting material. The product was isolated as a colorless solid after lyophilization (0.008 g, 0.012 mmol, 95%). 1H NMR (CD3OD, 500 MHz): 3.92–3.79 (m, 7 H), 3.77–3.55, 3.25–3.00 (m, 16 H), 2.71–2.44 (m, 23 H), 1.89 ppm (m, 4 H); 13C NMR (CD3OD, 125 MHz): δ= 173.7, 173.3, 172.7, 171.2, 171.0, 56.7, 47.7–46.2 (8 signals), 41.1, 40.4, 34.9, 27.1, 26.8, 26.9, 24.9 ppm; LC-MS (ESI): m/z: 703.3 [M+H]+; Rt=1.9 min (method B).
FeIII complexes
FeCl3 was dissolved in H2O (1 mL) and mixed with ligand (1 equiv, in H2O). The light-yellow solution changed color instantaneously upon addition of the ligand to red. The corresponding FeIII complexes were detected by LC-MS. FeL1: LC-MS (ESI): 574.1 [M+2 H]+; FeL2: LC-MS (ESI): 515.1 [M+H]+; FeL3: LC-MS (ESI): 602.2 [M+2 H]+; FeL4: LC-MS (ESI): 641.2 [M+H]+; FeL5: LC-MS (ESI): 378.6 [M+2 H]2+.
ZrIV complexes
A solution of L4 (1 equiv, 0.001 g, 0.0017 mmol) in H2O (50 μL) was added to a solution of [Zr(acac)4] (acac=acetylacetonate; 0.0008 g, 0.0017 mmol) in MeOH (1 mL). The reaction was stirred at room temperature. The product could be detected by LC-MS after just 30 min of reaction time. ZrL4: LC-MS (ESI): 674.2 [M]+; ZrL5: LC-MS (ESI): 789.2 [M]+.
Radiolabeling protocol
89Zr–oxalate was received from Washington University at an average specific activity of (15 mCimL−1). For a typical radiolabeling procedure, an aliquot (100 μL) was removed from the stock solution and transferred into a separate vial. The pH of the solution was adjusted with 1m Na2CO3 (105 μL) to 7.4–7.6. The aliquot was mixed with ligand L1–L4 (10 μg) or DFO (in 100 μL solution) and reacted under slow mixing for 1 h. The reaction was monitored by using radioTLC (solid phase: Sigma–Aldrich, TLC on silica gel on aluminum foils, 10×70 mm; mobile phase; 50 mm EDTA, pH 7). Under these conditions, 89Zr–oxalate migrates with the solvent front, whereas chelated 89Zr remains at the origin (Figure S1 in the Supporting Information). Complex solutions for in vivo administration were diluted with PBS (1×) for an oxalate content of <5%.
LogD measurements
Aliquots (50 μL) of 89Zr complex were diluted in PBS (450 μL). Then octanol (500 μL) was added and the mixture was vortexed for 15 min at 2000 rpm and centrifuged for 2 min. Subsequently, aliquots (50 μL) of the aqueous and octanol layers were collected and measured in the gamma counter.
EDTA stability protocol
An aliquot (10 μL) of quantitatively chelated 89Zr was mixed with 50 mm EDTA (290 μL) and kept at 37°C or room temperature. Aliquots (10 μL) were removed at select time points and analyzed by radioTLC. Under these conditions, 89Zr–EDTA migrates with the solvent front, whereas 89ZrL remains at the origin.
Computational details
All calculations were conducted by using DFT, as implemented in the Gaussian 03W suite of ab initio quantum chemistry programs.[17] Normal self-consistent field (SCF) and geometry convergence criteria were employed throughout. Structures were optimized in the gas phase without the use of symmetry constraints. Harmonic frequency analysis based on calculated analytical second derivatives was used to characterize optimized structures as local minima. All calculations employed the B3LYP[18] exchange-correlation functionals in combination with the LANL2DZ[19] basis sets. NBO analysis was performed by using default parameters. Optimized structures and molecular orbitals were analyzed by using Chemcraft (version 1.7, build 382).
In vivo imaging and biodistribution (healthy animals)
All animal experiments were conducted according to the guidelines of the Institutional Animal Care and Use Committee (IACUC). For the evaluation of in vivo behavior through imaging and biodistribution, healthy male BALB/c mice (Charles River Laboratories, Cambridge, MA) were intravenously injected with 15 μCi of 89Zr–L4 (n=4) or 89Zr–DFO (n=5) through a tail vein catheter. The micro-PET scans were analyzed with AMIDE (version 1.0.5 for OS.X; Stanford, CA, USA). PET images were obtained by using a small animal PET/X-ray scanner (Sofie Biosciences G4 PET scanner) with a peak resolution of 1.4 mm, equipped with anesthesia and a bed heating system. The field of view was 9.0 cm and covered the nose to the base of the tail in each mouse. Animals were scanned for 30 min immediately following injection. The 30 min scans were reconstructed into dynamic sequences of 11 frames of 1 min, 3 frames of 3 min, and 2 frames of 5 min. All reconstructions were automatically performed by using a 3D maximum-likelihood expectation maximization (MLEM) algorithm. Reconstructed data were evaluated by using the AMIDE software package. ROI were drawn in kidney, heart, and bladder. Results were expressed as Becquerel per cubic centimeter of tissue or as % IDcm−3 of tissue. Mice were sacrificed at 24 h postinjection. Select organs were harvested and collected; radioactivity was counted by using a gamma counter.
Conjugation, radiolabeling, and purification of L5–tz
Ligand L5 (1.0 mg, 1.4 μmol) was suspended in H2O (0.5 mL, chelex treated). Then a solution of FeCl3·6H2O (16.6 μL of a 10 mm solution in 0.1M HClaq) was added, providing a bright-orange solution. A solution of TFP (30 μL of 1.2 mm solution) was added to the reaction followed by the addition of N′-(3-dimethylaminopropyl)-N-ethylcarbodiimide (EDC; 13 mg, 0.068 mmol, Sigma Aldrich). The reaction mixture (pH 6.5) was then stirred at room temperature for 1 h before purification of the activated ester product by use of a C-18 Light Sep-pak cartridge (Waters, Milford, MA). The obtained activated ester TFP–L5 was used immediately for the subsequent coupling step. Tz (2 mg, in 220 μL in chelex treated water, pH adjusted to 9.5) and TFP–L5 (in MeCN) were reacted under continuous slow mixing for 2 h. This was followed by the addition of EDTA (0.075 mL of a 100 mm aqueous solution), decrease of the pH to 4 by using 0.25 mL H2SO4 (0.1 M aqueous solution) and incubation of the crude reaction mixture at 40 °C for 1 h to remove FeIII by transchelation with EDTA. The reaction was monitored by visual inspection of the disappearance of the color of the initially yellow–orange solution. Complex L5–tz was purified by SEC. Final product fractions were analyzed by gel electrophoresis and TOF-MS-ES. On average, 0.17 equivalents of L5 were covalently associated with tz (whereas 0.45 equivalents of DFO–succinate were conjugated under equivalent reaction conditions). Radiolabeling was performed by using the protocol described above, followed by purification by SEC. The labeled product was characterized by radioTLC and gel electrophoresis.
In vivo imaging and biodistribution (xenograft model)
All animal experiments were conducted according to the guidelines of the IACUC. In vivo PET imaging and biodistribution studies were conducted in nude mice (Charles River Laboratories, MA). Mice were inoculated with 106 cells (100 μL; BT-474 or BT-20) suspended in 1:1 saline and matrigel with no artificial stimuli. Tumors were allowed to grow for three to four weeks until tumors were palpable. Tumor sizes ranged from 10 to 70 mm3. Mice were injected through the tail vein with 10–20 μCi of 89Zr–L5–tz or 89Zr–DFO–tz (100 μL; specific activity 1.5–2.0 mCimg−1mAb) in saline. Micro-PET/CT imaging experiments were conducted on a Sofie Biosciences G4 PET scanner (Culver City, CA, USA), 10 min static microPET images were acquired under general anesthesia (isoflurane/O2) at 24, 48, 72, and 96 h postinjection. Image data was automatically reconstructed by using a 3D MLEM algorithm, which was evaluated by ROI analysis with the AMIDE software package. ROI were drawn in the tumor, heart, liver, kidney, and bone, with results expressed as % IDcm−3 of tissue. Mice were sacrificed and organs were harvested, weighed, and assayed in the gamma counter for biodistribution studies. Radioactivity associated with each organ was expressed as % IDg−1. Biodistribution data were assessed by unpaired t tests by using GraphPad Prism (version 6.02 for Windows Graph-Pad Software, San Diego, CA, U.S.) to determine if differences between groups were statistically significant (p<0.05).
Supplementary Material
Acknowledgments
Fang Qian, Dingyi Wen, Yuting Huang, Susan Foley, and Paul Weinreb of Biogen Inc. are acknowledged for MS analysis of antibody conjugates. Chongzhao Ran and Anna Moore are acknowledged for providing access to the Sofie Biosciences G4 PET scanner. Shadi Esfahani and Umar Mahmood are acknowledged for a donation of BT474 cells and providing guidance on tumor implantation. Neil Vasdev is acknowledged for providing 89Zr(oxalate) in house for early experiments. The NIH is acknowledged for funding support to E.B. (K99HL125728) and P.C. (R01EB009062).
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cplu.201600003.
References
- 1.a) Holland JP, Cumming P, Vasdev N. J Nucl Med. 2012;53:1333. doi: 10.2967/jnumed.112.105387. [DOI] [PubMed] [Google Scholar]; b) Sawyers CL. Nature. 2008;452:548–552. doi: 10.1038/nature06913. [DOI] [PubMed] [Google Scholar]
- 2.a) Van Dongen GA, Visser GW, Lub-de Hooge MN, De Vries EG, Perk LR. Oncologist. 2007;12:1379–1389. doi: 10.1634/theoncologist.12-12-1379. [DOI] [PubMed] [Google Scholar]; b) Wu AM. J Nucl Med. 2009;50:2–5. doi: 10.2967/jnumed.108.056887. [DOI] [PubMed] [Google Scholar]
- 3.Pandey MK, Engelbrecht HP, Byrne JF, Packard AB, DeGrado TR. Nucl Med Biol. 2014;41:309–316. doi: 10.1016/j.nucmedbio.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 4.a) Zhang Y, Hong H, Cai W. Curr Radiopharm. 2011;4:131. doi: 10.2174/1874471011104020131. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Holland JP, Williamson MJ, Lewis JS. Mol Imaging. 2010;9:1–20. [PMC free article] [PubMed] [Google Scholar]
- 5.a) Wadas TJ, Wong EH, Weisman GR, Anderson CJ. Chem Rev. 2010;110:2858–2902. doi: 10.1021/cr900325h. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zeglis BM, Lewis JS. Dalton Trans. 2011;40:6168–6195. doi: 10.1039/c0dt01595d. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Price EW, Orvig C. Chem Soc Rev. 2014;43:260–290. doi: 10.1039/c3cs60304k. [DOI] [PubMed] [Google Scholar]; d) Holland JP, Vasdev N. Dalton Trans. 2014;43:9872–9884. doi: 10.1039/c4dt00733f. [DOI] [PubMed] [Google Scholar]; e) Pandya DN, Pailloux S, Tatum D, Magda D, Wadas TJ. Chem Commun. 2015;51:2301–2303. doi: 10.1039/c4cc09256b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Holland JP, Divilov V, Bander NH, Smith-Jones PM, Larson SM, Lewis JS. J Nucl Med. 2010;51:1293–1300. doi: 10.2967/jnumed.110.076174. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Knowles SM, Zettlitz KA, Tavaré R, Rochefort MM, Salazar FB, Stout DB, Yazaki PJ, Reiter RE, Wu AM. J Nucl Med. 2014;55:452–459. doi: 10.2967/jnumed.113.120873. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ikotun OF, Marquez BV, Huang C, Masuko K, Daiji M, Masuko T, McConathy J, Lapi SE. PloS One. 2013;8:e77476. doi: 10.1371/journal.pone.0077476. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Deri MA, Zeglis BM, Francesconi LC, Lewis JS. Nucl Med Biol. 2013;40:3–14. doi: 10.1016/j.nucmedbio.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Bhattacharyya S, Kurdziel K, Wei L, Riffle L, Kaur G, Hill GC, Jacobs PM, Tatum JL, Doroshow JH, Kalen JD. Nucl Med Biol. 2013;40:451–457. doi: 10.1016/j.nucmedbio.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Verel I, Visser GWM, Boellaard R, Stigter-van Walsum M, Snow GB, van Dongen GAMS. J Nucl Med. 2003;44:1271–1281. [PubMed] [Google Scholar]; g) Vosjan MJ, Perk LR, Visser GW, Budde M, Jurek P, Kiefer GE, van Dongen GA. Nat Protoc. 2010;5:739–743. doi: 10.1038/nprot.2010.13. [DOI] [PubMed] [Google Scholar]
- 7.a) Patra M, Bauman A, Mari C, Fischer CA, Blacque O, Häussinger D, Gasser G, Mindt TL. Chem Commun. 2014;50:11523–11525. doi: 10.1039/c4cc05558f. [DOI] [PubMed] [Google Scholar]; b) Deri MA, Ponnala S, Zeglis BM, Pohl G, Dannenberg J, Lewis JS, Francesconi LC. J Med Chem. 2014;57:4849–4860. doi: 10.1021/jm500389b. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Guérard F, Lee YS, Tripier R, Szajek LP, Deschamps JR, Brechbiel MW. Chem Commun. 2013;49:1002–1004. doi: 10.1039/c2cc37549d. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Guérard F, Lee YS, Brechbiel MW. Chem Eur J. 2014;20:5584–5591. doi: 10.1002/chem.201304115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abou DS, Ku T, Smith-Jones PM. Nucl Med Biol. 2011;38:675–681. doi: 10.1016/j.nucmedbio.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perk LR, Vosjan MJ, Visser GW, Budde M, Jurek P, Kiefer GE, van Dongen GA. Eur J Nucl Med Mol Imaging. 2010;37:250–259. doi: 10.1007/s00259-009-1263-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Zhai C, Summer D, Rangger C, Franssen GM, Laverman P, Haas H, Petrik M, Haubner R, Decristoforo C. Mol Pharm. 2015;12:2142–2150. doi: 10.1021/acs.molpharmaceut.5b00128. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ma MT, Meszaros LK, Paterson BM, Berry DJ, Cooper MS, Ma Y, Hider RC, Blower PJ. Dalton Trans. 2015;44:4884–4900. doi: 10.1039/c4dt02978j. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Deri M, Ponnala S, Kozlowski P, Burton-Pye BP, Cicek H, Hu C, Lewis JS, Francesconi LC. Bioconjugate Chem. 2015;26:2579–2259. doi: 10.1021/acs.bioconjchem.5b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaspar M, Grazina R, Bodor A, Farkas E, Santos MA. Dalton Trans. 1999:799–806. [Google Scholar]
- 12.Sharma SK, Miller MJ, Payne SM. J Med Chem. 1989;32:357–367. doi: 10.1021/jm00122a013. [DOI] [PubMed] [Google Scholar]
- 13.Holland JP, Sheh Y, Lewis JS. Nucl Med Biol. 2009;36:729–739. doi: 10.1016/j.nucmedbio.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arteaga CL, Sliwkowski MX, Osborne CKent, Perez EA, Puglisi F, Gianni L. Nat Rev Clin Oncol. 2012;9:16–32. doi: 10.1038/nrclinonc.2011.177. [DOI] [PubMed] [Google Scholar]
- 15.a) Chang AJ, DeSilva R, Jain S, Lears K, Rogers B, Lapi S. Pharmaceuticals. 2012;5:79–93. doi: 10.3390/ph5010079. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Janjigian YY, Viola-Villegas N, Holland JP, Divilov V, Carlin SD, Gomes-DaGama EM, Chiosis G, Carbonetti G, de Stanchina E, Lewis JS. J Nucl Med. 2013;54:936–943. doi: 10.2967/jnumed.112.110239. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dijkers EC, Kosterink JG, Rademaker AP, Perk LR, van Dongen GA, Bart J, Jong JRde, de Vries EG, Lub-de Hooge MN. J Nucl Med. 2009;50:974–981. doi: 10.2967/jnumed.108.060392. [DOI] [PubMed] [Google Scholar]; d) Dijkers E, Oude Munnink T, Kosterink J, Brouwers A, Jager P, Jong J, Dongen G, Schrçder C, Lub-de Hooge M, Vries E. Clin Pharmacol Ther. 2010;87:586–592. doi: 10.1038/clpt.2010.12. [DOI] [PubMed] [Google Scholar]
- 16.Subik K, Lee JF, Baxter L, Strzepek T, Costello D, Crowley P, Xing L, Hung MC, Bonfiglio T, Hicks DG, Tang P. Breast Cancer: Basic Clin Res. 2010;4:35. [PMC free article] [PubMed] [Google Scholar]
- 17.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven T, Jr, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03W, Revision B.04. Gaussian, Inc; Wallingford CT: 2004. [Google Scholar]
- 18.a) Becke AD. Phys Rev A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]; b) Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; c) Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. J Phys Chem. 1994;98:11623–11627. [Google Scholar]
- 19.a) Dunning THJ, Hay PJ. In: Modern Theoretical Chemistry. Schaefer HF III, editor. Vol. 3. Plenum; New York: 1976. pp. 1–28. [Google Scholar]; b) Hay PJ, Wadt WR. J Chem Phys. 1985;82:299–310. [Google Scholar]; c) Hay PJ, Wadt WR. J Chem Phys. 1985;82:270–283. [Google Scholar]; d) Wadt WR, Hay PJ. J Chem Phys. 1985;82:284–298. [Google Scholar]
- 20.Esteves MA, Vaz MCT, Gonçalves MLSS, Farkas E, Santos MA. J Chem Soc Dalton. 1995:2565–2573. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.