

Abstract
Enveloped virus entry occurs when viral and cellular membranes fuse releasing particle contents into the target cell. Human immunodeficiency virus (HIV) entry occurs by cell-free virus or virus transferred between infected and uninfected cells through structures called virological synapses. We developed a high-throughput cell-based assay to identify small molecule inhibitors of cell-free or virological synapse-mediated entry. An HIV clone carrying Cre recombinase as a Gag-internal gene fusion releases active Cre into cells upon viral entry activating a recombinatorial gene switch changing dsRed to GFP-expression. A screen of a 1998 known-biological profile small molecule library identified pharmacological HIV entry inhibitors that block both cell-free and cell-to-cell infection. Many top hits were noted as HIV inhibitors in prior studies, but not previously recognized as entry antagonists. Modest therapeutic indices for simvastatin and nigericin were observed in confirmatory HIV infection assays. This robust assay is adaptable to study HIV and heterologous viral pseudotypes.
Keywords: HIV-1, Gag, Cre recombinase, Drug screening, Virus entry, Env, Viral membrane fusion
Introduction
In order to enter the host cell, enveloped HIV particles must undergo membrane fusion with the host cell membrane. Many assays have been developed to interrogate this process in vitro, which can be categorized in two broad categories. The first category is cell-based fusion assays, in which viral envelope proteins are expressed on the surface of cells which are co-cultured with receptor-expressing cells; syncytia formation between the two cell types is detected via activation of a reporter gene expression (Herschhorn et al., 2011; Huerta et al., 2002; Sakamoto et al., 2003). These assays may not fully recapitulate the unique composition of lipids and proteins found on virus particles. The second category of fusion assay is virus particle based, where lipid membrane probes act as sensors of membrane mixing (Lowy et al., 1990; Raviv et al., 2002) or viral contents release into a target cell serve as measures of viral entry. A practical example of this type of assay utilizes the beta-lactamase (Blam) reporter gene fused to HIV Vpr, which is efficiently packaged into virus particles and then delivered into target cells upon virus fusion (Cavrois et al., 2002). Fusion activity is measured by cleavage of the fluorescent reporter substrate CCF2-AM that is loaded into the target cell cytoplasm, which causes in a shift in its fluorescent emission. This assay can provide a reliable indicator for HIV entry, however it is not ideal for high throughput screens due to the high cost of the CCF2-AM substrate and a complex assay protocol that requires numerous wash and incubation steps.
Viral membrane fusion assays have primarily been examined in the context of cell-free HIV entry. HIV particles infect CD4+ T cells after exposure to cell-free virions, yet studies have revealed that infections mediated by cell–cell contact can greatly enhance the efficiency of viral dissemination in vitro (Dimitrov et al., 1993; Sourisseau et al., 2007). An adhesive junction between infected and uninfected T cells has been called the virological synapse (VS) and is defined by the localization of viral proteins Gag and Env and cellular receptor, CD4 to the site of cell–cell contact (Jolly et al., 2007). Live confocal imaging of HIV virological synapses revealed that the viral Gag protein was dynamically recruited to the site of cell–cell adhesion where newly formed viruses are translocated into the target cell through an endocytic process (Hubner et al., 2007). The transfer of virus from cell-to-cell requires active targeting of viral proteins to the VS in the donor cell, and the endocytic internalization process also appeared to require active signaling. Cellular inhibitors such as actin antagonists or certain antibodies have differential abilities to inhibit HIV infection via VS and cell-free virus (Durham et al., 2012). Recent studies have revealed that the high copy number of incoming virus during VS require higher concentrations of reverse transcriptase inhibitors to block and thus may contribute to reservoirs (Sigal et al., 2011). Because of the aspects of viral entry that may be unique to virological synapses, we set out to develop a cellular assay that can examine the steps of cell-to-cell infection in T cells to identify therapeutics that are efficient in preventing this mode of transmission.
VS formation utilizes many cellular signaling and vesicular transport processes that could be targeted to inhibit infection. To screen for inhibitors, a desirable assay would measure the ability of HIV to engage in the early steps of viral replication up to the point of viral membrane fusion to identify inhibitors of HIV cell-to-cell transmission. A challenge to developing such an assay is that the VS requires direct mixing of infected and uninfected cells, limiting the sensitivity of conventional viral detection methods that measure the viral nucleic acids or gene products in both the donor and target cells within a mixed population. HIV reporter cell lines may be used, but these generally measure many steps associated with early infection including reverse transcription, integration and gene expression. An optimal system is one in which the expression of a heterologous marker gene is induced by the initiation of infection in the target cell but not present in the infected donor cell. The desired assay must be sensitive, without a high background signal. To address these design considerations, we have devised a system that utilizes a recombinant HIV that packages large amounts of the phage recombinase, Cre, into virus particles and uses a specially designed target cell that expresses a dsRed in the absence of recombination and GFP following Cre-mediated recombination. The resultant upregulation of GFP can be observed by fluorescence microscopy or quantified by flow cytometry.
To validate this assay, we have screened a small molecule library of 1998 drugs and compounds for inhibitors of HIV fusion. This library consists of compounds that affect a variety of cellular pathways and are categorized as 60% approved drugs, 25% natural compounds and 15% other bioactive compounds (less characterized toxins and cellular process inhibitors). Given the broad range of targets of this library, it may provide valuable information to identify unique structure–activity relationships required to inhibit HIV entry.
Results
Gag-iCre produces viral particles containing Cre recombinase
We set out to make HIV particles that package stoichiometric quantities of the Cre recombinase relative to the Gag polyprotein to serve as an indicator of viral entry when this enzymatic cargo is released into a target cell. The Cre gene was inserted into the gene for the HIV structural protein Gag, to create a virus analogous to a fluorescent virus that carried GFP in the same position (Hubner et al., 2007). The resulting virus packages an estimated 3000 molecules of Cre-recombinase within each virus particle (Briggs et al., 2004). The Cre is inserted between the matrix and capsid domains of Gag and flanked by viral protease sites so that the enzyme is proteolytically excised from the Gag precursor during HIV protease-mediated viral maturation (Fig. 1A). When viral membrane fusion occurs Cre within the virus particle is liberated into the target cell where it can activate recombination of a target substrate.
Fig. 1.

HIV Gag-iCre carries the Cre gene inserted into HIV gag. (A) Insertion of Cre between the MA and CA domains allow high levels of Cre to be packaged into virus particles, with the Cre enzyme cleaved out of this precursor by the viral protease. (B) ELISA quantitation of p24 produced by Gag-iCre particles compared to NL4-3. (C) Western analysis of producer cell lysates using anti-cre antibody. (D) Western analysis of virus particles from supernatants using anti-cre antibody.
When transfected into 293T cells HIV Gag-iCre produces 60% less virus than a wild type NL4-3 HIV (Fig. 1B). Western blots on transfected 293T cells show that Cre is present both as part of a Gag precursor polyprotein and as a fully processed form in 293T producer cells (Fig. 1C Left). Western blot of the same lysates using an anti-HIV serum showed a larger p55 Gag-iCre polyprotein as well as fully processed p24 indicating that the insertion of Cre into the Gag polyprotein is compatible with processing of the Gag precursor to mature virus particles (Fig. 1C Right). In the virus particles produced by the HIV Gag-iCre virus, Cre protein was observed to be processed to lower molecular weight forms (Fig. 1D). Although HIV Gag-iCre does produce virus particles when transfected into 293T cells, we find that the HIV Gag-iCre virus is unable to mediate a spreading infection when infecting the highly permissive T cell line, MT4 (data not shown). Wild type virus showed an increase in p24 production peaking at 4 days after infection then decreasing, HIV Gag-iCre did not infect these cells when measured over the same time period (data not shown).
Gag-iCre reporter signal is blocked by inhibitors of fusion but not inhibitors of replication
A target cell line for the Cre virus called Jurkat floxRG was generated by transducing Jurkat cells with a reporter cassette containing dsRed fluorescent protein flanked by loxP recombination sites and followed by GFP (Koo et al., 2011). These cells were flow sorted for dsRed expression and expanded as individual clones to identify an optimally inducible stable cell line. In the basal state, before exposure to Cre recombinase, dsRed is constitutively expressed in Jurkat floxRG cells (Fig. 2A, upper panel). Fusion of cell-free, HIV Gag-iCre, with the target cell membrane releases sufficient recombinase to catalyze the site specific deletion of the dsRed gene, activating the expression of GFP and halting new production of dsRed. The dsRed expression in the cells remains detectable during this time period and decays as a function of the half-life of existing dsRed protein and mRNA (Fig. 2A, lower panel).
Fig. 2.

HIV Gag-iCre reporter signal is specific to fusion. (A) Jurkat floxRG target cells are transduced with the MSCV floxR-G cassette that expresses dsRed (upper) and switches to GFP expression upon fusion by recombination by Cre. (B) Fluorescent microscopy of infection of RG Jurkats incubated with Gag-iCre in the presence of fusion inhibitor (AMD3100), reverse transcription inhibitor (AZT) or untreated. (C) Red channel fluorescence of samples shown in C. (D) FACS analysis of RG Jurkats exposed to HIV Gag-iCre in the presence and absence of inhibitors.
When Jurkat floxRG cells were spinoculated with Gag-iCre viral supernatants, there was an increase in Cre-activated green cells, as visualized by fluorescence microscopy (Fig. 2B and C) and measured by FACS (Fig. 2D). This increase in green cells was inhibited by treatment with the HIV fusion inhibitor AMD3100, a CXCR4 coreceptor antagonist known to inhibit HIV fusion, but not by treatment with the HIV reverse transcriptase inhibitor AZT (Fig. 2D). Quantitative downregulation of dsRed was not observed in the GFP positive cells, which is likely related to a relatively long half-life of the protein in these cells. Additionally, to measure the contribution of syncytia and doublets to the GFP positive signal, a co-culture infection assay was performed with dye labeled donor and target cells, showing only 3.13% of the GFP signal obtained was due to syncytia and cell doublets (data not shown). These results indicate that the induction of GFP upon exposure of Jurkat floxRG cells to HIV Gag-iCre is dependent on virus fusion but not later replication steps such as reverse transcription. We conclude that the assay provides an indicator of viral membrane fusion.
Validation of the Gag-iCre assay for high throughput screening of HIV fusion inhibitors during cell-to-cell infection
We next examined whether viral membrane fusion after cell-to-cell spread of HIV could be measured quantitatively with this assay. We prepared infected donor cells by nucleofecting Jurkat cells with HIV Gag-iCre plasmid. As a parallel control, donor cells expressing NL-GI reporter virus, which carries GFP expressed in the place of the viral early gene nef, with the expression of nef controlled from an internal ribosome entry site (IRES). These infected donor cells were co-cultured with Jurkat floxRG cells for 48 h untreated and in the presence of AMD3100 or AZT. Just as in the cell-free system, coculture with cells expressing HIV Gag-iCre resulted in an increase in the percentage of GFP-expressing Jurkat floxRG cells compared to the uninfected control. The induction of GFP expression in the target cell when cocultured with HIV Gag-iCre expressing cells was indicative of virus transfer and fusion (Fig. 3A). The control virus, which requires productive infection to turn target cells green (Gag-iCre requires only fusion), showed a percentage of infection similar to the percentage of fusion seen in Gag-iCre (Fig. 3A). In the cell-to-cell based viral entry assay Gag-iCre was blocked with the fusion inhibitor AMD3100 but not the reverse transcriptase inhibitor AZT (Fig. 3A). The control infection using the NL-GI virus was susceptible to both AMD3100 as well as AZT. We conducted titrations of HIV inhibitors AMD3100, AZT, Indinivir (HIV protease inhibitor) and integrase inhibitor. Since proteolytic maturation has been shown to be required for HIV membrane fusion (Wyma et al., 2004), it was not surprising that both the fusion and protease inhibitors blocked Gag-iCre dependent reporter signal while AZT and integrase inhibitor did not (Fig. 3B). These studies demonstrate the HIV Gag-iCre assay provides an indicator for viral entry, and not reverse transcription or integration of the incoming virus.
Fig. 3.

Viral membrane fusion after cell-to-cell infection. (A) FACS analysis of RG Jurkat target cells co-cultured with indicated donor cells for 48 h with or without inhibitors. (B) Titrations of AMD3100, Indinavir, AZT and integrase inhibitors in Gag-iCre infected cells co-cultured with RG Jurkat for 48 h. Values normalized to uninfected controls (8% GFP positive).
Gag-iCre reporter signal is compares favorably to Vpr–Blam assay
To determine the sensitivity of the Gag-iCre assay relative to a well-tested HIV fusion assay, we compared Gag-iCre assays with viral fusion assays using the Vpr–Blam (Cavrois et al., 2002). Parallel infections were initiated and the activation of viral fusion measured by FACS. Signal to background ratio was greater with the Gag-iCre assay detecting fusion events in 64.7% of cells at the highest virus concentration with 0.98% background detected in uninfected control cells (Fig. 4A, upper panels). In comparison the Vpr Blam assay detected fusion in 47.2% of cells at the highest concentrations with 1.4% background (Fig. 4A, lower panel). Both assays showed comparable signal for the full titration of virus (Fig. 4B).
Fig. 4.

HIV Gag-iCre reporter signal is comparable to Vpr Blam. (A) FACS analysis of Jurkat floxRG cells spinoculated with Gag-iCre virus and Jurkat cells spinoculated with NL43 Vpr Blam 16 h after spinnoculation. (B) Dose response plot of viral input versus percent fusion evident in target cells following Gag-iCre of Vpr–Blam assays. Bars represent error from 2 replicates. (C) Z factor plotted as a function of increasing inhibitor concentration for Gag-iCre and Vpr Blam assays.
To measure the robustness of the Gag-iCre assay for high throughput screening, a Z-factor was determined as described by Zhang et al. (1999). The transfer assay was performed over several concentrations of inhibitor (AMD3100) in triplicate and a Z factor was calculated over this range to indicate the reliability of the assay at different levels of inhibition compared to the DMSO control. At lower levels of inhibition (0.125 μM AMD3100, 20% inhibition of transfer) the assay meets the requirements set forth by Zhang et al for screening (Z>0). At higher levels of inhibition (1.25 and 12.5 μM AMD3100, 83% and 92% inhibition respectively) the assay is considered excellent (Z>0.5) with a very large separation between AMD-inhibited and untreated controls and therefore very suitable for high throughput screening (Fig. 4C). The Vpr Blam assay was titrated in parallel and yielded comparable Z scores however the cost of the CCF2-AM substrate makes screening with Vpr Blam cost prohibitive.
The Gag-iCre assay identified potential inhibitors of HIV fusion
After determining that the Gag-iCre assay was sufficiently robust for high-throughput screening and had comparable or improved signal to the Vpr Blam assay, we proceeded to screen for inhibitors of HIV cell-mediated infection in the Spectrum Collection, a small molecule library of 1998 compounds (Microsource). HIV Gag-iCre producing donor cells were co-cultivated with Jurkat floxRG target cells for 48 h in the presence of each of the compounds contained in the library (Fig. 5A). Measuring GFP signal by an automated flow cytometer (Intellicyte HTFC system) served as an indicator of viral membrane fusion. Initial cell toxicity was monitored by examining major shifts in forward scatter and side scatter in the mixed donor and target cells. Of the 1998 compounds tested, 128 inhibited fusion by 30% or more and were scored as a hit (Fig. 5B). These hits were then sorted based on the ratio of inhibitory potency to toxicity (Supplementary Table S1). Of these 130, two compounds were eliminated from further analysis due to a toxicity percentage that was greater than the inhibition percentage. Many compounds with similar mechanisms of actions were observed to inhibit HIV. The largest groups of compounds were the cardiac glycosides (9 compounds), microtubule inhibitors (11 compounds), nucleosides (7 compounds), ion transport affecting compounds (6 compounds) and antiseptics (5 compounds) (Supplementary Table S1). To focus our follow up studies of the initial candidate inhibitors, we grouped compounds based on similarity of known mechanism to reduce redundancy in our follow up studies. The top 36 compounds (Supplementary Table S1, shown in bold) selected based on these criteria were retested in titrations to measure fusion inhibitory activity in both cell-mediated and cell-free viral entry and subject to more stringent viability testing utilizing the ATP assay, Celltiter glo (Promega) as an indicator of cell viability. Differences in efficacy in IC50 against cell-free or cell-mediated viral entry and LC50 values were used to determine a therapeutic index for each compound (Supplementary Table S2). Therapeutic indices based on this criteria were as high as 9.6 (nigericin), which indicated a broad range of concentrations at which the compound inhibited HIV fusion without affecting cell viability. In addition, the efficacy of each candidate HIV inhibitor was also examined for differential ability to inhibit cell-free versus cell-to-cell infection. While most compounds inhibited cell-free and cell-to-cell infection to similar extents, artenimol was 7-fold more potent against cell-free virus than cell-associated virus and dequalinium was nearly 6-fold more potent against cell-free virus than cell-associated virus. In order to further validate the putative viral inhibitors, these 36 follow up compounds were also tested for their impact on cell metabolism and viability using the stringent viability assay Celltiter-glo. The Celltiter-glo viability assay gave higher therapeutic indices for several compounds including nigericin, and simvastatin (Supplementary Table S1).
Fig. 5.

Small molecule library screening overview. (A) Nucleofected Gag-iCre donor cells were co-cultured with Jurkat floxRG target cell line in the presence of each of the 1998 library compounds, performed in duplicate. Plates were incubated for 48 h followed by fixation and quantitation of fusion by flow cytometry. (B) Percent inhibition was calculated by normalizing to DMSO treated and uninfected controls. All compounds that inhibited fusion by 30% or more were considered first round hits.
Confirmation of inhibitors of HIV infection using the TZM-Bl assay
To evaluate if hits identified in the Gag-iCre screen would inhibit viral infection, 8 compounds were selected that had the highest therapeutic indices and evaluated for their ability to inhibit infection of TZM-Bl reporter cells by NL4-3 HIV. TZM-Bl cells carry an integrated HIV LTR luciferase expression cassette that is induced when the cells are infected with HIV (Derdeyn et al., 2000). TZM-Bl cells were infected in the presence of a titration of inhibitors and luminescence was read to quantitate reporter gene activity. This data was plotted and IC50 values were calculated (Fig. 6, Table 1). While half of the compounds demonstrated therapeutic indices that were greater than 2, 2 of 8 compounds showed dose dependent inhibition of viral infection with therapeutic indices greater than 20 (Supplementary Table S2). We therefore find that a fraction of hits in the Gag-iCre can be validated in an orthogonal screen for single round viral infection.
Fig. 6.

TZM assay follow up for select compounds. TZM-bl cells were infected with NL4-3 virus particles and treated with indicated compound for 48 h. % inhibition is relative to DMSO-treated control samples. Plots on the left represent data from GagiCre titration follow up, while plots on the right represent TZM-bl assay. For both assays viability is measured by Cell-titer Glo.
Table 1.
Inhibitors therapeutic indices.
| Compound name |
TI GagiCre (uM) |
TI TZM (uM) |
||
|---|---|---|---|---|
| Nigericin |
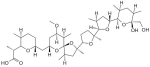
|
8.1 | > 72.2 | Antibiotic with ionophore and anti Golgi activity. Known Inhibitor of HIV |
| Simvastatin |
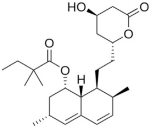
|
6.5 | > 20.9 | Statin affecting cholesterol production |
The Gag-iCre assay adapted to R5-tropic virus and Ebola pseudotyped particles
In order to see if the Gag-iCre assay has utility for R5-tropic virus a CCR5-expressing cell line A3R5 was transduced with the Cre-activated GFP expression cassette. Additionally, an HIV Gag-iCre was engineered to carry an R5-tropic JRFL Env. Gag-iCre JRFL infected donor cells were co-cultured with A3R5 RG target cells for 48 h then analyzed by flow cytometry. Compared to the Gag-iCreΔEnv negative control the Gag-iCre JRFL exposed cells upregulated GFP in a manner that could be blocked by the CCR5 antagonist TAK779 but not the CXCR4 antagonist AMD3100 (Fig. 7A). These results show that this assay can be utilized to measure fusion of other virus tropisms in alternate cell lines.
Fig. 7.

Modifications of Gag-iCre assay to study viral tropism and heterologous viral fusion glycoproteins. (A) Donor Jurkat cells were nucleofected with HIV Gag-iCre JRFL and co-cultured with the CCR5 expressing T cell Cre-responsive cell line, A3R5-RG for 48 h untreated or in presence of fusion antagonists, TAK779 or AMD3100. Entry efficiency was measured by flow cytometry. (B) Pseudotypted Gag-iCreΔEnv was produced by co transfection of HIV Gag-iCre with Ebola GP, Ebola mutant GP or VSV envelope. Cell free virus co-cultured with Hela RG target cell line for 48 hours. Entry efficiency was measured by flow cytometry.
We further examined the utility of the Gag-iCre assay to measure viral fusion mediated by heterologous viral glycoporteins by pseudotyping Gag-iCreΔEnv virus particles with Ebola GP, an Ebola GP mutant (F535R, F88A) or VSV-G. These pseudotyped particles were co-cultured with a Hela RG target cell line for 48 h and results were analyzed by FACS. The Ebola GP psuedotyped particles showed a 10-fold signal over background signal in uninfected and Gag-iCreΔEnv controls while VSV psuedotyped particles had a very robust signal (54.8% GFP positive) 548 fold over background (Fig. 7B). The Gag-iCre viral entry assay therefore has utility as a viral fusion assay for heterologous viruses.
Discussion
We have developed a T cell viral entry assay that allows us to examine the steps of viral infection up to the point of viral membrane fusion, in the context of either cell-free or cell-to-cell infection. Traditional assays that require a full replication cycle including viral gene expression for reporter activity can be very useful in screening for general inhibitors of HIV replication, yet by design these systems do not inform us about what step of replication is being inhibited. In order to identify specific inhibitors of processes up to the fusion step of HIV in a high throughput manner, we designed a reporter virus that packages high levels of the Cre-recombinase. Fusion of this virus with a target cell line harboring an integrated Cre-activated fluorescent protein expression cassette induces a genetic switch from expression of DsRed to GFP, which is measured by flow cytometry or microscopy. This system has several unique characteristics that facilitate high throughput screening compared to other fusion reporters such as BlaM–Vpr. First it does not require target cells to be loaded with substrate, a step that adds to both the cost and variability of high throughput screening. Second, due to the nature of the Cre–Lox system, only one recombination event is required for maximum reporter signal and the change is permanent for cells that fuse with virus serving as an indicator of cells exposed to virus even if productive infection is not achieved. Background enzymatic activity in the donor cells is not an issue as the substrate is only present in the target cells. The fluorescent nature of the reporter also allows for readout both via flow cytometry or fluorescence microscopy. The stable expression of red fluorescent protein in the target cells make it easy to distinguish target cells from donor cells in co-culture assays.
The Gag-iCre virus produces high levels of p24 and the Gag polyprotein is processed in a manner similar to wild type suggesting that virus assembly and release are not significantly affected by the presence of the Cre recombinase inserted into Gag. Its overall fitness is compromised in long term growth assays, but the virus exhibits high levels of fusion with the Jurkat floxRG reporter cells suggesting the impact of the Cre on viral fitness occurs after the fusion step of replication. Its utility as a measure of the early steps of the infection up to the fusion step is confirmed by its sensitivity to the fusion inhibitor AMD3100, but not to the reverse transcription inhibitor AZT. The results obtained from cell-associated virus experiments were similar to those obtained from cell-free virus experiments validating the use of the assay for studying the effect of inhibitors of cell to cell transmission of HIV. The Z-factor showed the assay was extremely robust over a range of AMD3100 concentrations further validating the assay for high throughput screening. The assay also compared favorably with the current standard for HIV fusion assays, the Vpr Blam assay.
The high throughput screen identified 136 hits that blocked HIV fusion by more than 30%. Many of these hits such as nigericin, colchicine, celastrol, phenethyl caffeate, 5-azacytidine, digoxigenin have been shown to inhibit HIV replication in vitro (Fernandez et al., 1987; Lai and Freed, 2014; Moncunill et al., 2005; Nakamura et al., 1992; Tanaka et al., 2005; Wong et al., 2013), through either known or unknown mechanisms. The confirmation of these hits validates the utility of the Gag-iCre assay to identify inhibitors of infection and can help to narrow the steps at which these identified compounds are inhibiting HIV replication as this assay does not measure steps that follow entry. Although not previously thought to be antiviral, celecoxib has been used in a clinical trial where it was found to modulate immune activation related to progression of HIV infection (Cavrois et al., 2002). Of the putative hits, several had modestly higher therapeutic indices making them possible HIV drug candidates. Several of these drugs are already in use to treat other diseases or conditions, such as celecoxib a nonsteroidal anti-inflammatory drug, artenimol, an anti-parasititic, sertraline an antidepressant. Because sertraline can accumulate at high concentrations in the brain, it may be useful to consider as an adjunct treatment for HIV associated neurocognitive disorders (HAND), which may be associated with persistent low-level replication in the brain. The putative dual activity of these drugs could be considered as therapeutically beneficial for treatment of HIV-infected patients. Some compounds identified from this screen provide a rationale for retrospective clinical analyses of observational studies or clinical trials where subsets of infected individuals may have been taking some of these commonly prescribed medications for other conditions. These studies may be reanalyzed to look for beneficial effects on HIV viral load. Repurposing these drugs or exploiting their dual activities when prescribed for their primary indications may be beneficial to consider when treating HIV-infected patients.
While the majority of compounds did not show differential inhibition between the cell-associated and cell-free infection systems the fact that artenimol, 5-azacytidine and dequalinium between 4-fold and 6-fold differences between systems suggests that there may be some therapeutic specificity between modes of viral transfer, however these compounds had low therapeutic indices (TIr1) so it is possible that the differences seen were due to differential toxicity in the two systems. The hits targeted several cellular pathways consistent with viral entry such as those involved in microtubule organization, while others target pathways not previously associated with viral entry. A large number of putative hits target Na+/K+ ATPases as well as K+ ionophores (including Nigericin) suggesting that Na+ K+ or Ca+ ion may be important for HIV entry. Future studies are needed to determine whether or not this is the case and to understand their mechanism of action.
Of the putative hits nigericin and simvastatin had broader therapeutic indices in the follow up TZM assay with cell titer glo used for viability that may make them interesting to follow up with experiments to further probe their mechanisms of action (Table 1). Both nigericin and lasalocid (which also was followed up in the TZM assay but had greater toxicity than nigericin) are polyether antibiotics which have been shown to have anti-HIV activity (Nakamura et al., 1992). Further both compounds have also been shown to affect the transport of cations such as Na+ and K+ across membranes (Alonso and Carrasco, 1981; Fernandez et al., 1987). Simvastatin, as a member of the statin family of drugs, is an inhibitor of cholesterol synthesis. It has been shown that membrane cholesterol levels can affect the conformation of the HIV fusion peptide gp41 which correlates with fusogenicity of virus particles (Lai and Freed, 2014). Additionally another study showed that Lovastatin could be reduce viral load in a HIV-infected humanized mouse model as well as in HIV-infected patients by downregulating Rho GTPase (del Real et al., 2004). Another more recent study that tested a variety of statins, including simvastatin found anti-HIV activity in vitro using PBMCs, however the same paper also tested simvastatin clinically on HIV positive patients who were not on antiretrovirals for 4 weeks and found no beneficial effect on mean viral load (Moncunill et al., 2005).
The high hit rate of the primary screen and the 50% confirmation rate in the TZM assay suggests that the Gag-iCre assay can identify inhibitors of HIV fusion that work in other infection systems. Although in their current state, most of the identified compounds are too toxic to be used clinically; it is possible that chemical derivatives may exhibit higher therapeutic indices. Additional studies to examine the possible clinical utility of these compounds are needed and will involve more extensive testing of the inhibitors on diverse HIV isolates in different replication assays. Those that succeed would be candidates for further preclinical testing the anti-viral effects of these compounds in vivo either in humanized mouse models of HIV infection or in SHIV infection in macaques.
The assay itself also has applications in screening larger libraries of compounds. Furthermore, as the VSV and Ebola psuedotyping experiments showed, this assay may be used to identify potential entry inhibitors for other enveloped viruses. The Gag-iCre entry assay will be useful for future research studying HIV fusion and identifying novel specific inhibitors of HIV fusion.
Materials and methods
Plasmids
pLM-CMV-R-Cre and pMSCV-loxp-dsRed-loxp-eGFP-Puro-WPRE were obtained from Addgene (Sadelain lab and Clevers lab). The Cre recombinase open reading frame was PCR amplified using primers 5′-CTAGTACGCGTATGTCCAATTTACTGACCGTACAC-3′ and 5′-TCATCTCTAGAATCGCCATCTTCCAGC-3′ which creates flanking MluI and XbaI sites for insertion between matrix and capsid in the previously described HIV Gag-iGFP construct (Hubner et al., 2007). In the HIV Gag-iCre construct the Cre recombinase is flanked by linker sequenced containing the HIV protease consensus sequence SQNYPIVQ.
Cells and tissue culture
Human CD4+ T Cell lines Jurkat E6.1 were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and grown RPMI 1640 containing 10% fetal bovine serum, 2 mM L-Glutamine, 100 units/ml penicillin, and 100 mg/ml streptomycin. Jurkat cells were nucleofected with 4.5 ug of plasmid DNA using Lonza Cell Line Nucleofector Kit V and program S-18. Stable Jurkat floxRG cells were made by retroviral transduction of pMSCV-loxp-dsRed-loxp-eGFP-Puro-WPRE packaged with pCL-10A1 retrovirus packaging vector. Viral supernatant was harvested and used to infect Jurkat cells that were then selected for puromycin resistance and single cell cloned by limiting dilution. The final clonal population of Jurkat floxRG cells with low EGFP background and high sensitivity to HIV Gag-iCre virus was selected by screening 12 individual clones.
Virus production
Cell-free virus was produced by transfection of 293T cells using calcium phosphate method with 20 μg of viral plasmid per 10 cm plate. Media was exchanged 16 h after transfection and virus supernatants were harvested 48 h after transfection. Donor cells for cell-to-cell infections were produced by nucleofection of 107 Jurkat cells with 4.5 μg of Gag-iCre plasmid and 120 μl Cell Line Nucleofector Solution V with supplement (Lonza) using program S-18 on the Amaxa Nucleofector II. Cells were incubated in RPMI 10% FBS for 16 h and viable cells recovered by Ficol hypaque centrifugation at 1200g followed by incubation in RPMI 10% FBS for 3 h before experiments.
p24 ELISA
The p24 ELISA was performed by a modified version of a previously published protocol (Moore et al., 1990). Costar 3922 flat-bottomed, high binding plates were incubated at room temperature with anti-p24 capture antibody overnight (Aalto D7320; 1:200 in 0.1 M NaHCO3). Plate was washed twice with 1 × TBST and blocked with 2% nonfat dry milk (Lab Scientific) for 1 h then washed in TBST. HIV supernatants treated with 1% Empigen (1:100 and 1:1000 in DMEM) along with titration of p24 standard are added to wells and incubated at room temperature for 2 h, then washed 4 × with TBST. Alkaline phosphatase conjugated mouse anti-HIV p24 (CLINIQA) was added (1:8000 in TBST 20% sheep serum) and incubated for 1 h followed by 6 TBST washes. 50 μl of Sapphire Substrate (Tropix) was added to each well and incubated for 20 min. Luminescence was quantitated on Fluo Star Optima plate reader and sample values calculated based on nonlinear regression of standard curve using Prism software (Graphpad software inc.).
Western blot analysis
293T cells were transfected using Polyjet Transfection reagent (Signagen) with 0.5 μg of plasmid DNA per well of 6 well plate. Cells were lysed with 1% Triton in TBS and protease inhibitor cocktail (Sigma). Lysate equivalent of approximately 2 × 105 cells per well were run on NuPage 4–12% Bis-Tris Gel (Novex) and transferred to Amersham Hybond-P PVDF membranes (GE Healthcare). Membranes were blocked with 2% nonfat dry milk (Lab Scientific), then probed with rabbit anti-cre recombinase (Novagen, 1:3000), human anti-HIV IgG (NIH AIDS Reagent Program, 1:10,000) primary antibodies then anti-rabbit (Jackson Immunoresearch) or anti-human horseradish peroxidase (Jackson Immunoresearch) conjugated secondary antibody.
Vpr Blam
Donor cells were created by nucleofecting pMM310 (Blam–Vpr) and NL4-3 at a 1:3 ratio. After ficol purification donor cells were co-cultured with Jurkat floxRG targets at a 1:1 ratio for 6 h. Cells were washed and resuspended in CO2 independent media containing 1.5 μM CCF2-AM substrate with 2.5 mM probenecid and incubated for 1.5 h at 23 °C. Cells were washed and resuspended in CO2 independent media with probenecid and incubated for 16 h to allow substrate cleavage. Cells were fixed in 4% PFA and and analyzed by FACS using an LSR II Fortessa at 425 nm and 525 nm wavelengths to quantitate CCF2-AM cleavage
High throughput screening
High throughput screening was carried out at the Mount Sinai Integrated Screening Core, which also provided the 1998 compound library (Microsource Spectrum Collection). Both donor Jurkat and target Jurkat floxRG cells were diluted to 5 × 105 cells/ml and 20 μl plated in 384 well plates using a Multi-Drop Combi-Microplate Dispenser (Thermo). Library compounds were pin transferred (4.2 μM final concentration) and plates incubated at 37 °C for 48 h. For follow up screens containing cell free infection, plating was carried out as above except that Jurkat floxRG target cells were resuspended at 1 × 106 cells/ml and then 20 μl (1ng p24 equivalent) of viral supernatant was added to each well. Secondary compound titrations were plated using an HP D300 digital dispenser, which dispenses compounds in DMSO in the pl to μl range and normalizes for DMSO concentration. After incubation cells were fixed with a final concentration of 1% paraformaldehyde then using a BD Accuri flow cytometer with Hypercyt autosampler (Intellicyt) with Hypercyt controller software.
Data analysis/Z factor
All samples were normalized to controls and represented as a percentage inhibition with the DMSO control representing 0% inhibition and the uninfected sample representing 100% inhibition. Toxicity was normalized in a similar manner with the DMSO control representing 0% toxicity and the highest toxicity sample in library representing 100% toxicity. The Z factor for the Gag-iCre assay was calculated by comparing AMD3100 treated samples to an untreated control, using the formula Z=1–3(σp–σn)/|μp–μn| where σ is the standard deviation, μ is the mean, p is treated sample and n is untreated control. Inhibition and toxicity was plotted using Prism (Graphpad software inc.) and a best fit curve with non-linear regression provided IC50 and LC50 values which were used to calculate therapeutic indices (IC50/LC50).
TZM assay
TZM-bl cells (from NIH AIDS Reagent Program from John Kappes) were plated at 2 × 104 cells/well in 96 well plates and incubated at 37 °C for 4 h. Test compounds were titrated at indicated concentrations. HIV NL4-3 viral supernatants (containing 2ng p24) were added to each well and cells were incubated for 48 h at 37 °C. Media was aspirated followed by lysis in Luciferase Cell Culture Lysis Reagent (Promega). 20 μl of each sample was read on Fluo Star Optima plate reader with injection of 100 μl of Luciferase Assay Reagent (Promega).
Viability assay
RG jurkats were plated at 5 × 105 cells per ml in 384 well plates and incubated at 37 °C with titrations of follow up compounds for 48 h. CellTiter Glo luminescent cell viability reagent (Promega) was added at 1:1 dilution according to manufacturers protocol and incubated for 10 min then read on Envision Multilabel Reader (Perkin Elmer). For TZM viability, the same protocol as above was followed except TZM cells were plated at 1 × 104 cells per well in 96 well plates.
Supplementary Material
Acknowledgements
This research was supported by grants NIH/NIAID AI112423 and NIH/NIGMS GM113885 to BKC, and NIH/NIAID grant AI109664 to CFB. We would like to thank the Icahn School of Medicine at Mount Sinai Dean’s flow Cytometry CORE.
Footnotes
Appendix A. Supplementary material
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.virol.2015.10.013.
References
- Alonso MA, Carrasco L. Relationship between membrane permeability and the translation capacity of human HeLa cells studied by means of the ionophore nigericin. Eur. J. Biochem. 1981;118:289–294. doi: 10.1111/j.1432-1033.1981.tb06399.x. [DOI] [PubMed] [Google Scholar]
- Briggs JA, Simon MN, Gross I, Krausslich HG, Fuller SD, Vogt VM, Johnson MC. The stoichiometry of Gag protein in HIV-1. Nat. Struct. Mol. Biol. 2004;11:672–675. doi: 10.1038/nsmb785. [DOI] [PubMed] [Google Scholar]
- Cavrois M, De Noronha C, Greene WC. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat. Biotechnol. 2002;20:1151–1154. doi: 10.1038/nbt745. [DOI] [PubMed] [Google Scholar]
- del Real G, Jimenez-Baranda S, Mira E, Lacalle RA, Lucas P, Gomez-Mouton C, Alegret M, Pena JM, Rodriguez-Zapata M, Alvarez-Mon M, Martinez AC, Manes S. Statins inhibit HIV-1 infection by down-regulating Rho activity. J. Exp. Med. 2004;200:541–547. doi: 10.1084/jem.20040061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O’Brien WA, Ratner L, Kappes JC, Shaw GM, Hunter E. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J. Virol. 2000;74:8358–8367. doi: 10.1128/jvi.74.18.8358-8367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov DS, Willey RL, Sato H, Chang LJ, Blumenthal R, Martin MA. Quantitation of human immunodeficiency virus type 1 infection kinetics. J. Virol. 1993;67:2182–2190. doi: 10.1128/jvi.67.4.2182-2190.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham ND, Yewdall AW, Chen P, Lee R, Zony C, Robinson JE, Chen BK. Neutralization resistance of virological synapse-mediated HIV-1 infection is regulated by the gp41 cytoplasmic tail. J. Virol. 2012;86:7484–7495. doi: 10.1128/JVI.00230-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez E, Grandjean J, Laszlo P. Ion transport by lasalocid A across red-blood-cell membranes. A multinuclear NMR study. Eur. J. Biochem. 1987;167:353–359. doi: 10.1111/j.1432-1033.1987.tb13344.x. [DOI] [PubMed] [Google Scholar]
- Herschhorn A, Finzi A, Jones DM, Courter JR, Sugawara A, Smith AB, 3rd, Sodroski JG. An inducible cell–cell fusion system with integrated ability to measure the efficiency and specificity of HIV-1 entry inhibitors. PLoS One. 2011;6:e26731. doi: 10.1371/journal.pone.0026731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner W, Chen P, Del Portillo A, Liu Y, Gordon RE, Chen BK. Sequence of human immunodeficiency virus type 1 (HIV-1) Gag localization and oligomerization monitored with live confocal imaging of a replication-competent, fluorescently tagged HIV-1. J. Virol. 2007;81:12596–12607. doi: 10.1128/JVI.01088-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta L, Lamoyi E, Baez-Saldana A, Larralde C. Human immunodeficiency virus envelope-dependent cell–cell fusion: a quantitative fluorescence cytometric assay. Cytometry. 2002;47:100–106. doi: 10.1002/cyto.10051. [DOI] [PubMed] [Google Scholar]
- Jolly C, Mitar I, Sattentau QJ. Requirement for an intact T-cell actin and tubulin cytoskeleton for efficient assembly and spread of human immunodeficiency virus type 1. J. Virol. 2007;81:5547–5560. doi: 10.1128/JVI.01469-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo BK, Stange DE, Sato T, Karthaus W, Farin HF, Huch M, van Es JH, Clevers H. Controlled gene expression in primary Lgr5 organoid cultures. Nat. Methods. 2011;9:81–83. doi: 10.1038/nmeth.1802. [DOI] [PubMed] [Google Scholar]
- Lai AL, Freed JH. HIV gp41 fusion peptide increases membrane ordering in a cholesterol-dependent fashion. Biophys. J. 2014;106:172–181. doi: 10.1016/j.bpj.2013.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowy RJ, Sarkar DP, Chen Y, Blumenthal R. Observation of single influenza virus–cell fusion and measurement by fluorescence video microscopy. Proc. Natl. Acad. Sci. USA. 1990;87:1850–1854. doi: 10.1073/pnas.87.5.1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncunill G, Negredo E, Bosch L, Vilarrasa J, Witvrouw M, Llano A, Clotet B, Este JA. Evaluation of the anti-HIV activity of statins. AIDS. 2005;19:1697–1700. doi: 10.1097/01.aids.0000183517.60384.db. [DOI] [PubMed] [Google Scholar]
- Moore JP, McKeating JA, Weiss RA, Sattentau QJ. Dissociation of gp120 from HIV-1 virions induced by soluble CD4. Science. 1990;250:1139–1142. doi: 10.1126/science.2251501. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Kunimoto S, Takahashi Y, Naganawa H, Sakaue M, Inoue S, Ohno T, Takeuchi T. Inhibitory effects of polyethers on human immunodeficiency virus replication. Antimicrob. Agents Chemother. 1992;36:492–494. doi: 10.1128/aac.36.2.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raviv Y, Viard M, Bess J, Jr., Blumenthal R. Quantitative measurement of fusion of HIV-1 and SIV with cultured cells using photosensitized labeling. Virology. 2002;293:243–251. doi: 10.1006/viro.2001.1237. [DOI] [PubMed] [Google Scholar]
- Sakamoto T, Ushijima H, Okitsu S, Suzuki E, Sakai K, Morikawa S, Muller WE. Establishment of an HIV cell–cell fusion assay by using two genetically modified HeLa cell lines and reporter gene. J. Virol. Methods. 2003;114:159–166. doi: 10.1016/j.jviromet.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Sigal A, Kim JT, Balazs AB, Dekel E, Mayo A, Milo R, Baltimore D. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature. 2011;477:95–98. doi: 10.1038/nature10347. [DOI] [PubMed] [Google Scholar]
- Sourisseau M, Sol-Foulon N, Porrot F, Blanchet F, Schwartz O. Inefficient human immunodeficiency virus replication in mobile lymphocytes. J. Virol. 2007;81:1000–1012. doi: 10.1128/JVI.01629-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Tomisato W, Hoshino T, Ishihara T, Namba T, Aburaya M, Katsu T, Suzuki K, Tsutsumi S, Mizushima T. Involvement of intracellular Ca2+ levels in nonsteroidal anti-inflammatory drug-induced apoptosis. J. Biol. Chem. 2005;280:31059–31067. doi: 10.1074/jbc.M502956200. [DOI] [PubMed] [Google Scholar]
- Wong RW, Balachandran A, Ostrowski MA, Cochrane A. Digoxin suppresses HIV-1 replication by altering viral RNA processing. PLoS Pathog. 2013;9:e1003241. doi: 10.1371/journal.ppat.1003241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyma DJ, Jiang J, Shi J, Zhou J, Lineberger JE, Miller MD, Aiken C. Coupling of human immunodeficiency virus type 1 fusion to virion maturation: a novel role of the gp41 cytoplasmic tail. J. Virol. 2004;78:3429–3435. doi: 10.1128/JVI.78.7.3429-3435.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A Simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.