

Abstract
CX3CR1-deficient mice develop very severe experimental autoimmune encephalomyelitis (EAE), associated with impaired NK cell recruitment into the CNS. Yet, the precise implications of NK cells in autoimmune neuroinflammation remain elusive. Here we investigated the pattern of NK cell mobilization and the contribution of CX3CR1 to NK cell dynamics in the EAE.
We show that in both wild-type (WT) and CX3CR1-deficient EAE mice, NK cells are mobilized from the periphery and accumulate in the inflamed CNS. However, in CX3CR1-deficient mice, the infiltrated NK cells displayed an immature phenotype contrasting with the mature infiltrates in WT mice. This shift in the immature/mature CNS ratio contributes to EAE exacerbation in CX3CR1-deficient mice, since transfer of mature WT NK cells prior to immunization exerted a protective effect and normalized the CNS NK cell ratio. Moreover, mature CD11b+ NK cells show higher degranulation in the presence of autoreactive 2D2 transgenic CD4+ T cells and kill these autoreactive cells more efficiently than the immature CD11b− fraction.
Together, these data suggest a protective role of mature NK cells in EAE, possibly through direct modulation of T cells inside the CNS, and demonstrate that mature and immature NK cells are recruited into the CNS by distinct chemotactic signals.
Keywords: NK cells, neuroinflammation, EAE, Chemokines, CX3CR1
Introduction
The role of natural killer (NK) cells in neuroinflammation remains controversial. We and others have presented evidence for deficient NK cell activity in patients with multiple sclerosis (MS), suggesting that NK cells may have a protective, disease-limiting role in neuroinflammation [1–6]. However, the results in the animal model of MS, experimental autoimmune encephalomyelitis (EAE), are contradictory; both a protective [7–9] as well as a pathogenic [10] role for NK cells in disease development has been described.
NK cells are highly heterogeneous and versatile cells with respect to their phenotype, function and organ distribution [11–15]. Thus, one could speculate that while a certain subtype may contribute to pathology, another subset may be protective in the appropriate environment [16].
Along this line, we reported reduced gene expression of CX3CR1 – the receptor for the chemokine CX3CL1 (fractalkine) – in MS patients compared with healthy individuals, and a correlation of the frequency of circulating CX3CR1-expressing NK cells with disease activity [3]. We also showed that the expression of CX3CR1 serves to discriminate fully mature from immature human NK cells [17]. Thus, diminished levels of CX3CR1-expressing NK cells may be associated with a diminished frequency of mature NK cells in MS. Consistent with this, mice deficient for CX3CR1 (CX3CR1GFP/GFP mice), experienced more severe EAE, corresponding with a selective reduction of NK cell infiltrates in the inflamed CNS compared to control animals [18]. This suggested that CX3CR1-expression may be related to the migration or effector function of “protective” NK cells. However, it still remains unclear whether a particular NK cell subtype confers protection, how and where protective NK cells exert their function and how they are mobilized during the course of neuroinflammation.
In this study, we investigated NK cell dynamics during the course of EAE, and the implications of the chemokine receptor CX3CR1 on NK cell migration and effector function using the MOG33–35-induced EAE model. We found that CX3CR1 mediates recruitment and migration of mature NK cells into the CNS, and that this process contributes to control CNS autoimmunity.
Results
NK cell frequency in peripheral blood decreases during EAE
To investigate how NK cells are distributed in vivo during neuroinflammation, we induced EAE in C57BL/6 WT mice and longitudinally monitored NK cell frequencies in peripheral blood during the course of disease (Fig. 1A). We found that NK cell frequencies in blood decreased directly after the peak of disease (day 16), precisely 20 days after immunization, from 5.01 % ± 1,43 % (day 0) to 2.67 % ± 0,95% (day 20) (p = 0,0136). This may point to a neuroinflammation-related altered fate of NK cells. To investigate the distribution of NK cells in the CNS and immune tissues at disease onset, and at time of the observed decrease of NK cells in blood, C57BL/6 WT EAE mice were sacrificed at day 10 and 20 post-immunization (p.i.) and NK cells numbers were assessed in blood, LN, spleen and CNS (Fig. 1B). We observed that already at day 10 p.i., NK cells accumulated into the CNS and decreased in lymph nodes and spleen. At day 20, elevated numbers of NK cells were found in the CNS, which corresponded with a dramatic reduction of the number of circulating NK cells in blood. Thus, at day 20 p.i. not only the frequency of NK cells (Fig. 1A) but also the absolute number of cells in the circulation were diminished.
Figure 1.
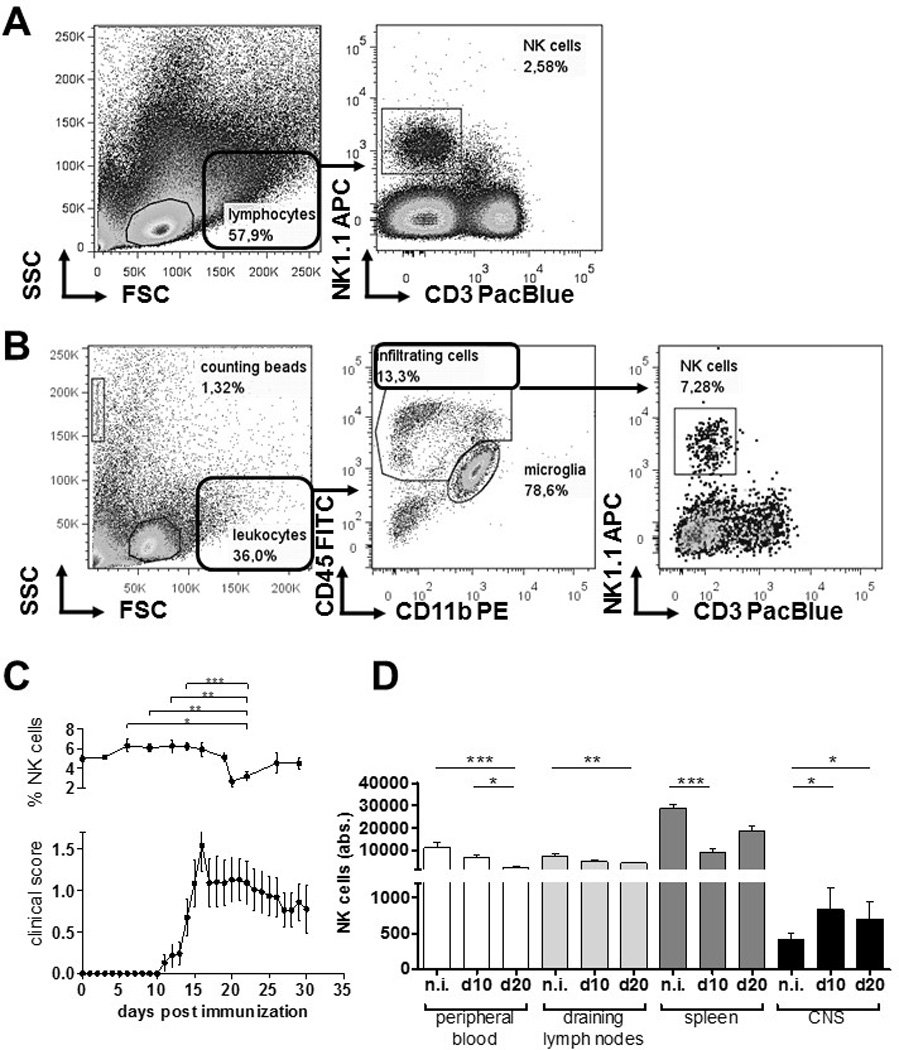
CX3CR1-deficient and WT NK cells show comparable migration behaviour during EAE
It was previously reported that CX3CR1-deficient animals experience a more severe EAE course characterized by a reduced migration of NK cells into the CNS [18]. Thus, we investigated how CX3CR1-deficiency might affect NK cell distribution during neuroinflammation. We confirmed that CX3CR1-deficient mice showed a significant increase in disease severity, and earlier disease onset, as well as an increased disease incidence (Table 2). The analysis of NK cell numbers in blood, spleen, draining lymph nodes and the CNS at day 20 p.i. also revealed an increase of the NK cell fraction in the CNS of CX3CR1-deficient mice from day 0 to day 20 after immunization. However, no differences were observed in the peripheral blood (Fig. 2A). Additionally, NK cell numbers were diminished in the spleen at day 10 after immunization, whereas no changes were observed in the draining lymph nodes (Fig. 2A).
Table 2.
Clinical EAE data of CX3CR1-deficient and WT mice.
| Group | N | Incidence | Mean disease onset ± SD |
Mean maximum clinical score ± SD |
|---|---|---|---|---|
| CX3CR1GFP/GFP | 10 | 8 / 10 | 16.0 ± 2.7 | 1.9 ± 0.6 |
| WT | 10 | 6 / 10 | 18.4 ± 2.6 | 1.4 ± 0.7 |
Figure 2.
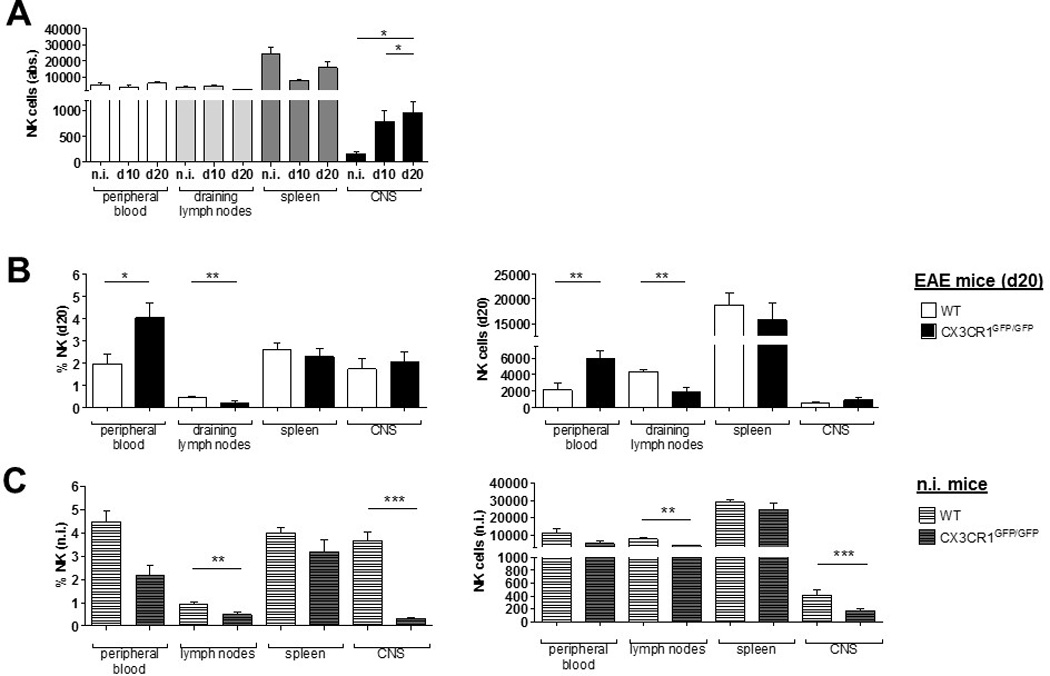
At day 20 p.i., the frequencies and absolute numbers of NK cells in the spleen and CNS did not differ between WT and CX3CR1-deficient mice (Fig. 2B shows the NK cell frequencies (left) and absolute numbers (right)). In blood, NK cell numbers and frequencies at day 20 p.i. were significantly higher in CX3CR1-deficient compared to WT mice (Fig. 2B). Absolute numbers of NK cells were significantly lower in the draining lymph nodes of CX3CR1-deficient mice compared to WT at this time point (Fig. 2B). Intriguingly, both frequency and absolute numbers of NK cells in non-immunized CX3CR1-deficient mice were extremely reduced in the CNS compared to WT animals (Fig. 2C).
Peripheral and not CNS CX3CR1-deficiency leads to a more severe EAE
We next investigated whether, in our hands, the increased EAE severity observed in CX3CR1GFP/GFP mice is due to the deficient expression of the receptor in peripheral immune cells. In bone-marrow (BM) chimera experiments complementary to those shown by Garcia et al. [19], we demonstrated that immune reconstitution of irradiated WT CX3CR1+/+ animals with CX3CR1-deficient bone marrow cells (CX3CR1GFP/GFP → WT) led to an aggravated EAE course (Supplemental Fig. 1A), and higher disease activity (Supplemental Fig. 1B) compared to control mice reconstituted with non-compromised CX3CR1+/+ BM cells (CX3CR1+/+→WT). Thus, deficient CX3CR1 expression in peripheral immune cells leads to EAE aggravation.
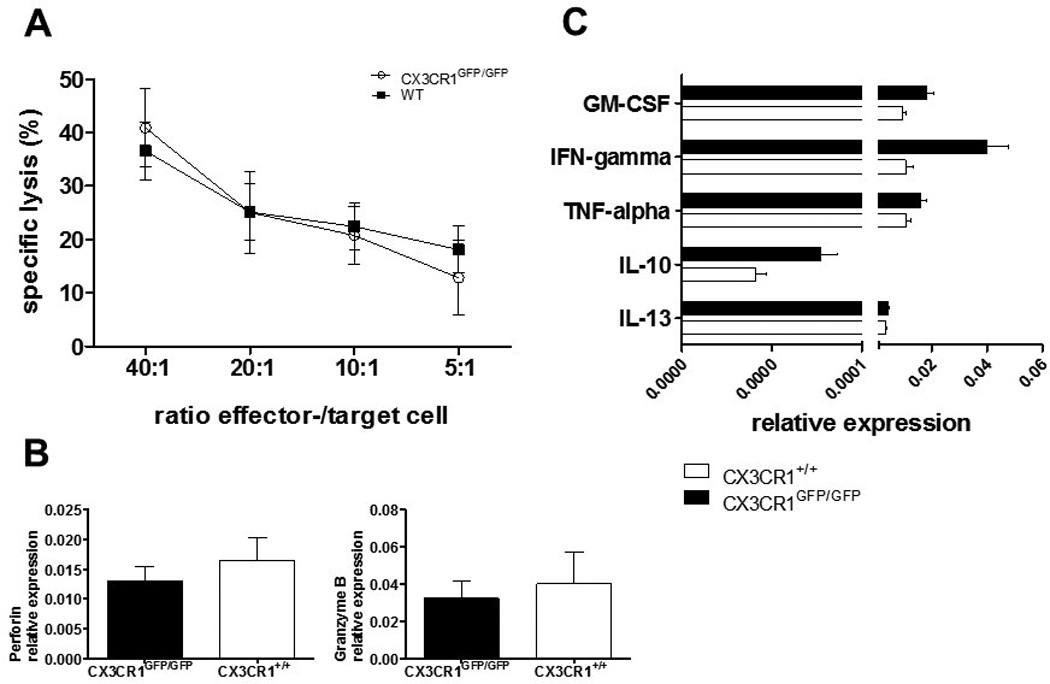
NK cells from CX3CR1 deficient mice show normal functional properties
We previously reported that CX3CR1 expression influenced human NK cell cytokine expression, activation status, maturation, cytotoxic activity, and proliferative responses [17]. Therefore, to rule out that CX3CR1-deficient NK cells may have intrinsic functional deficits that could influence our results, we examined the general cytotoxic activity, cytolytic protein and cytokine profiles from spleen-derived NK cells of unmanipulated WT and CX3CR1-deficient mice, by calcein-acetyoxymethyl release assay and quantitative real-time rtPCR, respectively. We observed the same NK-cell-effector-/YAC-1-target-cell ratio-dependent lysis capacity of CX3CR1-deficient NK cells compared to WT NK cells (Fig. 3A). Additionally, we observed that CX3CR1+/+ and CX3CR1-deficient murine NK cells expressed similar levels of the cytotoxicity mediators perforin and granzyme b (Fig. 3B) and the effector cytokines IFN-gamma, GM-CSF, TNF-alpha, IL-10 and IL-13 (Fig. 3C).
Figure 3.
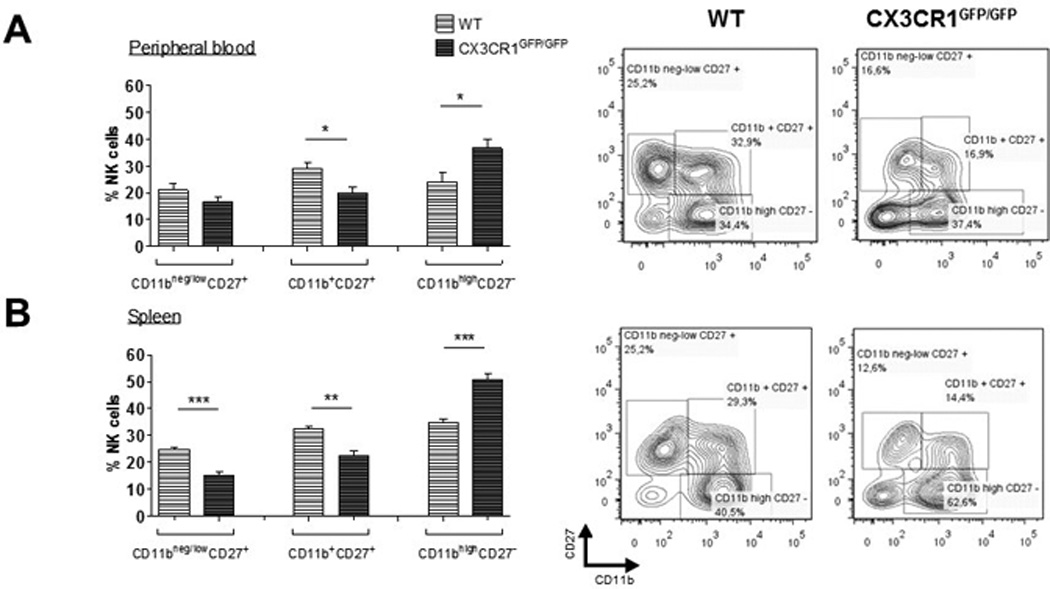
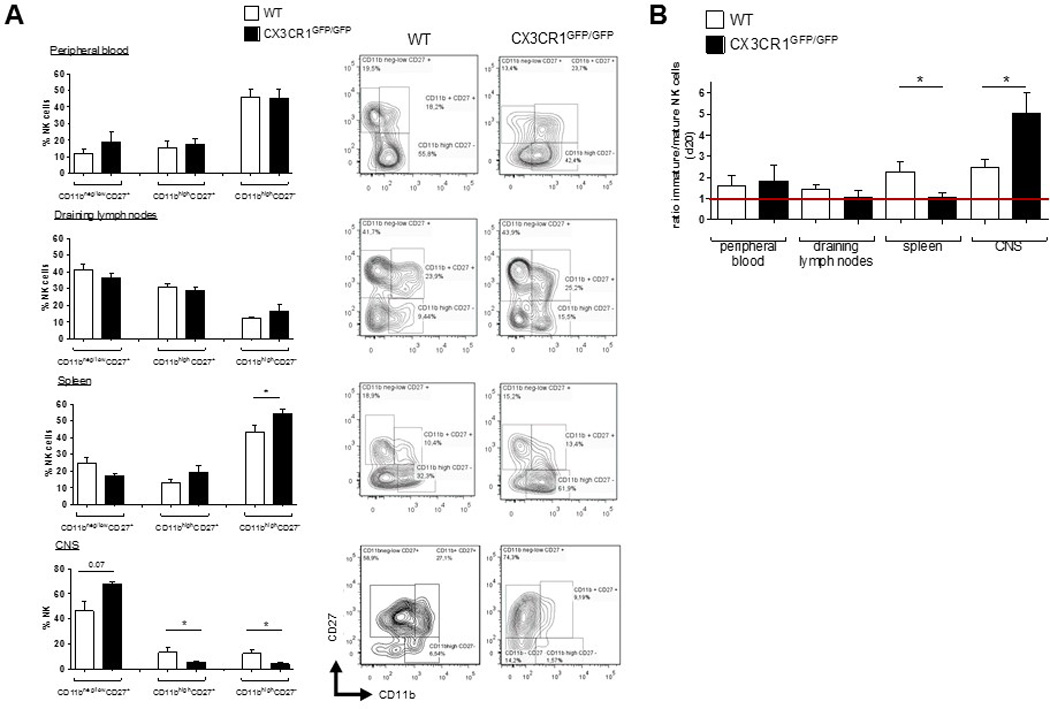
Moreover, lack of CX3CR1 did not affect NK cell maturation, according to the expression of CD11b and CD27 [20, 21]. All different CD3−NK1.1+ NK cell fractions of the immature (CD11bneg/lowCD27+), mature (CD11b+CD27+) and fully differentiated NK cells (CD11bhighCD27−) (Figure 4, right panels) were present in peripheral blood (Fig. 4A) and spleen (Fig. 4B) of CX3CR1-deficient mice. However, in CX3CR1-deficient mice the fraction of fully differentiated cells was even larger than in WT, while the fractions of immature and early mature cells were smaller.
Figure 4.
CX3CR1-deficient EAE mice show deficient migration of mature NK cells into the CNS
We next investigated whether CX3CR1 may be involved in the migration of a particular NK cell subtype into the CNS, as has been postulated for human NK cells [16, 22].
In WT animals, CX3CR1 is predominantly expressed on pre-mature/tolerant CD11b−CD27− (NK cells with predominantly inhibitory signals) [23, 24] and fully differentiated mature CD11b+CD27− (data not shown). Thus, during EAE, CX3CR1-deficiency may affect circulation of these distinct fractions.
At day 20 p.i., analysis of NK cells isolated from the CNS, LN, spleen and blood of WT and CX3CR1 deficient mice with EAE showed that the proportions of mature CD11bhigh CD27+ and fully differentiated CD11bhigh CD27− NK cells were significantly decreased in the CNS of CX3CR1-deficient, but were elevated in spleen, compared to WT (Fig. 5A). Altered distribution of mature vs. immature NK cell fractions in the CNS of CX3CR1-deficient EAE mice was reflected in the elevated immature/mature ratio observed under CX3CR1-deficiency (Fig. 5B).
Figure 5.
Transfer of splenic NK cells prior to EAE induction ameliorates EAE severity, delays disease onset and restores the NK cell phenotypic ratio inside the CNS
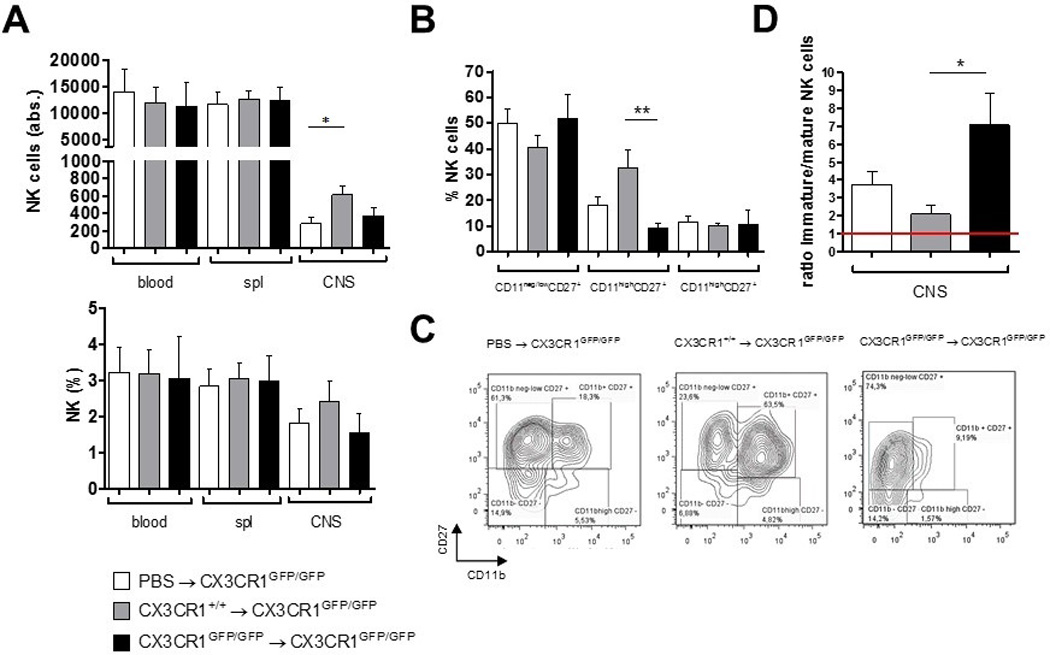
We showed that the frequency of mature NK cells migrating into the CNS during EAE was reduced in CX3CR1-deficient mice compared to WT mice, although the total amounts of NK cells in the CNS were comparable. Deficient NK cell activity inside the CNS may contribute to the increased EAE severity observed in CX3CR1 deficient animals. To examine whether functional splenic NK cells from WT mice may restrict EAE development, splenic NK cells from WT and CX3CR1-deficient mice (or PBS as control) were transferred into CX3CR1-deficient recipient mice one day before the induction of EAE. Transfer of WT NK cells reduced EAE incidence (Table 3), ameliorated clinical severity (Fig. 6A) and cumulative disease activity (Fig. 6B), and delayed disease onset (Fig. 6C). Mice transferred with NK cells from CX3CR1-deficient donors also appeared to benefit from the transfer. Next, we investigated in 9 and 5 of the mice receiving WT or CX3CR1-deficient NK cells, respectively, whether this single NK cell administration prior to immunization might affect the NK cell distribution inside the CNS compared to the PBS control mice. At day 20 p.i., frequencies and number of CNS NK cells were assessed, along with maturation phenotype defined by the expression of CD27 and CD11b. Mice that received a transfer of WT, but not of CX3CR1-deficient NK cells, showed higher numbers of NK cells in the CNS compared to the PBS-injected control group (p = 0.054) (Fig. 7A). Interestingly, the NK cell composition inside the CNS of CX3CR1GFP7GFP recipient mice resembled that of PBS-injected control mice, with high proportions of CD11bneg/lowCD27+ NK cells and only low levels of the potent CD11bhighCD27+ NK cells. However, in contrast, in mice transferred with WT-NK cells, the proportion of CD11bhighCD27+ cells was highly increased in the CNS (Fig. 7B and C). Accordingly, the ratio of immature to mature NK cells was significantly higher in these mice as compared to WT NK cell recipient mice (Fig. 7D). Frequencies and numbers of CD4 or CD8 T cells remained unaltered with respect to the control mice injected with PBS (data not shown).
Table 3.
Clinical data of CX3CR1-deficient mice transferred with NK cells or control PBS injection prior immunization.
| Genotype of transferred NK cells |
N | Incidence | Mean disease onset ± SD |
Mean maximum clinical score ± SD |
|---|---|---|---|---|
| CX3CR1GFP/GFP | 21 | 15 / 21 | 13.7 ± 1.6 | 1.7 ± 1.4 |
| CX3CR1+/+ | 20 | 13 / 20 | 13.9 ± 1.9 | 1.2 ± 1.3 |
| PBS | 18 | 17 / 18 | 11.6 ± 1.7 | 2.3 ± 1.1 |
Figure 6.
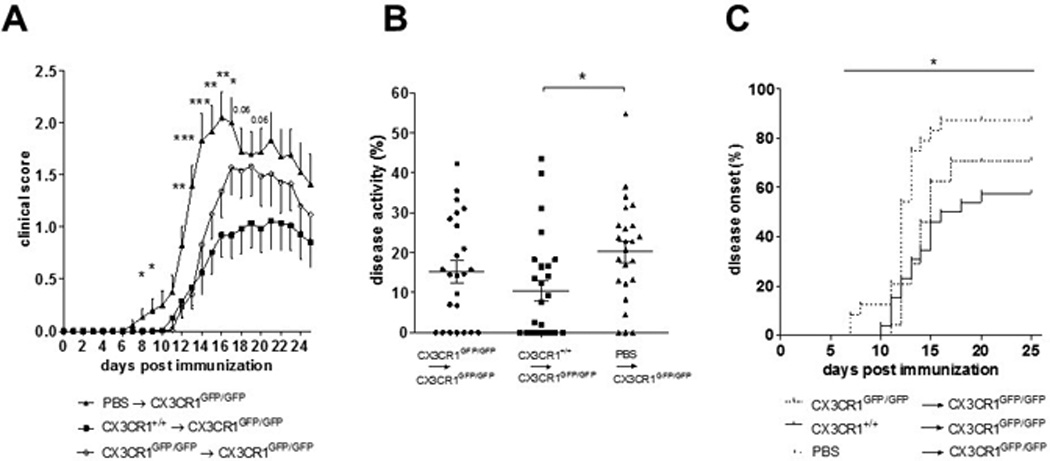
Figure 7.
Mature CD11b+ NK cells are more cytotoxic to autoreactive encephalitogenic CD4+ T cells than CD11b− NK cells
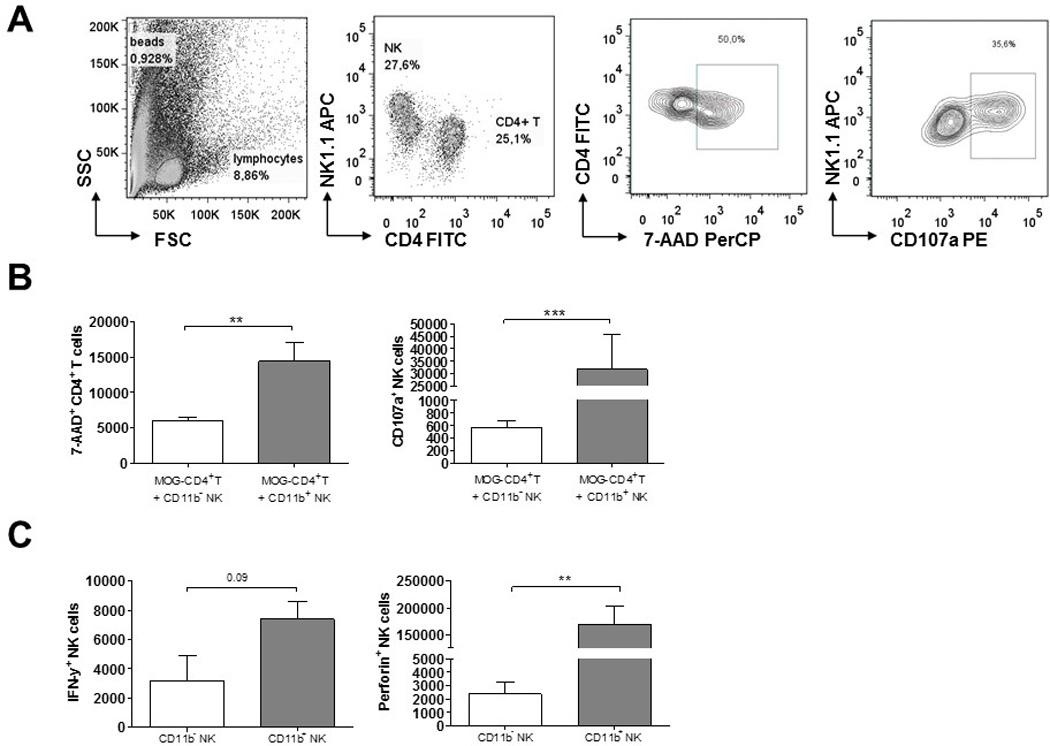
To investigate the mechanisms by which mature CD11b+ NK cells may protect from EAE, we investigated the cytotoxic capacities of CD11b+ and CD11b− NK cells towards encephalitogenic MOG-specific CD4+ T cells derived from the 2D2 mouse. For this, short-term CD4 cell lines were generated with the specific MOG antigen in vitro, as described in the Methods. At day 7, the cells were re-stimulated unspecifically with anti-CD3/anti-CD28 in the presence of CD11b+ or CD11b− NK cells that were previously activated with/ without IL-2. After 24 hours of co-culture, T cell viability and apoptosis was analysed by DNA-staining with 7-AAD. Similarly, NK cell degranulation was analysed by the surface expression of CD107a using flow cytometry. We found a higher numbers of CD107a positive cells within the CD11b+ NK cell population compared to the CD11b− NK cell subset. Correspondingly, we found that the CD11b+ NK cells killed autoreactive CD4+T cells more efficiently in vitro as indicated by the elevated numbers of T cells that were stained with 7-AAD (Fig. 8B). In addition, FACS sorted CD11b+ and CD11b− NK cells were analysed for their IFN-gamma and perforin expression by flow cytometry. Here, we found that after IL-2 stimulation, CD11b+ NK cells produced higher levels of IFN-gamma and, specially, much more perforin compared with the CD11b− subset (Fig. 8C).
Figure 8.
Discussion
In this study, we show that NK cells are recruited into the CNS during inflammation and that CX3CR1-mediated migration of mature CD11bhigh NK cells contributes to limit the severity of EAE.
We found that during EAE the frequency and absolute numbers of NK cells were diminished in peripheral blood and spleen, but increased inside the CNS, pointing to a selective NK cell migration from the periphery into the CNS during inflammation. NK cell mobilization to the target organs has been reported in the course of autoimmune [25, 26] and CNS pathology [16], suggesting that targeted migration of NK cells may either contribute to, or restrict the pathologic process. Our previous reports on NK cells in MS suggested that abnormal NK cell differentiation and activity may support chronic inflammation [3, 22, 27, 28]. Thus, we speculated that NK cell migration into the CNS after peak of disease could contribute to EAE amelioration. We tested this hypothesis using the well-described CX3CR1-deficient (CX3CR1GFP/GFP) mouse, which was shown to develop severe EAE, with deficient NK cell migration into the CNS [18].
In fact, we confirmed that CX3CR1-deficient mice are more susceptible to EAE than WT mice. However, in contrast to the previous report [18], we also observed a mobilization of NK cells from the periphery into the inflamed CNS in the CX3CR1-deficient EAE mice. This discrepancy on migration of CX3CR1-NK cells into the inflamed CNS may result from differences in the experimental approaches used in the two studies. In the current study, CNS infiltrating NK cells were assessed during the earlier EAE phases, i.e. until disease peak, whereas Huang et al. [18] analysed NK cells at the time of remission. Similarly, transient alterations in NK cell frequencies and cytotoxic activity have been described in MS patients during periods of disease activity [4, 29–32]. Thus in EAE, the dynamics of NK cells and disease activity may also be interrelated.
Our ex vivo experiments did not show evidence of intrinsic alteration in NK cell phenotype or function in CX3CR1-deficient mice. The comparison of CX3CR1-deficient and WT revealed similar NK cytotoxic capacity and production of cytolytic proteins and effector cytokines. Our data on the cytolytic capacity of CX3CR1-deficient and WT cells are in disagreement with the report by Ponzetta el al [33], which showed an enhanced cytotoxicity of splenic NK cells from CX3CR1-deficient mice. While Ponzetta at al. used whole splenocytes activated with IL-15 for their cytotoxic assay [33], we utilized sorted NK cells, to avoid contamination of cytotoxic CD8 T cells. Moreover, we did not pre-activate the cells with IL-15. These experimental differences may explain the divergent results. We further show that peripheral CX3CR1-deficient NK cells display the same distinct maturation stages as WT cells. Altogether, these data indicate that CX3CR1-signalling is not essential for NK cell activation or differentiation. As shown for human NK cells [17], we demonstrated that CX3CR1 is predominantly expressed on fully differentiated CD11bhighCD27− NK cells, confirming a previous report on CX3CR1 expression within the late differentiated fraction of CD11b+KLRG1+ NK cells [33]. Fractalkine (CX3CL1), the ligand of CX3CR1 is secreted by neurons, among other cells, during brain inflammation [34] and appears to direct the activity of CX3CR1-expressing microglia in various pathological scenarios in the brain [34–40]. Thus, during EAE, neuron-derived fractalkine may contribute to the recruitment of mature NK cells to inflamed sites. We show that at day 20 p.i., the CX3CR1-deficient mice display a diminished frequency of mature, and an elevated frequency of immature NK cells in the CNS, compared to WT animals. Interestingly, in non-immunized CX3CR1-deficient mice the proportion of mature NK cells was generally higher in peripheral blood and spleen compared to WT mice. We also confirmed the results of Poli et al. which showed a predominant presence of immature NK cells in the brains of healthy mice [16] (data not shown). Thus, our data point to a specific involvement of CX3CL1/fractalkine in the recruitment of mature NK cells into CNS during inflammation.
Finally, we demonstrated that transfer of CX3CR1+/+ NK cells, but not of CX3CR1-deficient NK cells, led to disease amelioration, delayed disease onset and reduced EAE incidence in CX3CR1-deficient mice. Moreover, transfer of WT NK cells appeared to restore the balance of immature and mature NK cell in the CNS during EAE, pointing to a protective role of mature NK cells into the EAE. It remains unclear why mature NK cells limit EAE while in an experimental stroke model they seem to contribute to exacerbation of brain infarction [41]. Deficient CX3CR1 expression on NK cells might further influence NK cell interactions with other immune cells, such as dendritic cells (DCs) or monocytes [42]. Although we cannot rule out that the beneficial effects of transferred NK cells were exerted in the periphery, our in vitro experiments using bone marrow derived DCs co-cultured with either CX3CR1-deficient or WT NK cells did not show evidence of different effects of these NK cells on DC differentiation and activation in vitro (data not shown). Thus, these findings point rather to modulatory effects inside the CNS, as postulated by Hao et al. [9]. We previously showed that neither CX3CR1neg/low nor CX3CR1high NK cells influenced the phenotype of monocytes, but rather differentially modulated their co-stimulatory capacities by increasing the expression of CD40 [17]. In this line, we are currently investigating the effects of the different NK cell subsets on infiltrating and resident myeloid cells.
Recently, Garcia et al. showed that CX3CR1 deficiency was associated with enhanced recruitment of CD115+Ly6C−CD11c+ dendritic cells into the inflamed EAE brain, correlating with increased severity of CNS pathology [19]. Here, we showed that NK cells also contribute to disease aggravation in the CX3CR1-deficient mice. We speculate that in CX3CR1-deficent mice, the presence of CX3CR1-negative immature NK cells may lead to altered immunomodulatory interactions between NK cells and pathogenic T cell and/ or NK cells and myeloid cells, such as DCs [19] and/or microglia [9, 43], contributing to an enhanced pathogenic T cell response, and hence increased EAE disease severity in CX3CR1-deficient mice. Here we found that CD11b+ NK cells produce high levels of perforin and IFN-gamma. Consistent with this result, we show that the CD11b+ subset degranulates more in the presence of encephalitogenic T cells, and kills these autoreactive CD4+ T cells more efficiently than the CD11b− counterpart, thus offering a possible explanation of the mechanism by which CD11b+ NK cells may exert neuroprotective effects. This presumption should be however confirmed in follow-up experiments using in our set-up NK cells from IFN-gamma- and perforin-deficient mice.
In summary, our data indicate that CX3CR1 deficient mice show impaired migration of mature NK cells into the CNS during EAE, suggesting that recruitment of fully matured NK cells may contribute to limit neuroinflammation. In human, we and others have presented data indicating that the enrichment of mature NK cells may contribute to the beneficial effects of certain MS therapies, including mitoxantrone in patients with secondary-progressive MS [28], natalizumab [44] and fingolimod [45]. For other therapies, such as daclizumab or beta-interferon, a treatment-induced enrichment of immature CD56bright cells has been postulated to be beneficial for patients [1, 46, 47]. Thus far, it remains uncertain which particular subsets may be beneficial in human disease [27], in part because data on human NK cells is available for blood and CSF but not for CNS tissue. We previously described that NK cells in the CSF of patients with neuroinflammation show a CD56bright, CX3CR1- immature phenotype, in contrast to the NK cells in the blood of the same patients [22]. Immature NK cells displayed similar chemotactic tools as the well-described central memory T cells (Tcm) observed in the CSF in patients with MS [48–50], which are only able to migrate into the CNS parenchyma after differentiating into effector memory cells [51]. Accordingly, immature CD56bright NK cells may become mature and upregulate CX3CR1 inside the CSF [22], allowing mature NK cells to penetrate into the CNS, where they might exert neuroprotective effects as we observed in mouse. Thus, the CSF may represent a separate compartment between blood and CNS tissue, suggesting that CSF NK cells might have a different phenotype than NK cells within the CNS tissue. Nevertheless, it remains to be established how the different NK cells subsets infiltrate the CNS, and how the migration route might affect their activity and maturation. Extended research is needed to obtain a more complete picture of NK cell migration into the CNS in mouse and human, and to identify which cell populations may be beneficial in various contexts.
Materials and methods
Mice
WT C57BL/6 mice were obtained from the Research Institute for Experimental Medicine (FEM) of the Charité (Berlin, Germany). Homozygote and heterozygote breeding of CX3CR1 deficient (CX3CR1GFP/GFP) mice was conducted at the FEM under specific pathogen-free conditions. T cell receptor (TCR)-transgenic 2D2 mice were kindly provided by Prof. Dr. Dirnagl (Center for Neurology, Neurosurgery and Psychiatry, Charité Berlin, Germany). All animal experiments were approved by the regional animal study committee of Berlin (LAGeSo) and performed in accordance to national and international guidelines.
Generation of bone marrow chimeric mice
Recipient C57BL/6 (WT) mice (4–5 week old) were irradiated with a dose of 900 rad and reconstituted with 25 × 106 cells isolated from CX3CR1-deficient (CX3CR1GFP/GFP) or WT donor mice as previously described [52]. Six weeks after reconstitution, the CX3CR1 genotype was determined using polymerase chain reaction (PCR) based genomic DNA analyses and flow cytometric analyses of expression of the inserted GFP gene [53]. The primers X, 5′-TTC ACG TTC GGT CTG GTG GG-3′ and Y, 5′-GGT TCC TAG TGG ACG TAG GG-3′ (annealing temperature: 60°C), were used for the amplification of the wild-type allele, while the primers Y and Z, 5′-GAT CAC TCT CGG CAT GGA CG-3′ (annealing: 60°C), were used for the amplification of the mutant allele. Mice were allowed to reconstitute for 6–7 weeks after bone marrow transfer before EAE induction.
Induction and assessment of EAE
Active EAE was induced in C57/BL5 as described previously [54, 55]. In brief, inbred mice were immunized subcutaneously with 250 µg myelin oligodendrocyte glycoprotein peptide 35–55 (MOG33–35) (Pepceuticals, Leicester, UK) emulsified in complete Freund's adjuvant (Difco Laboratories, USA) containing 800 µg Mycobacterium tuberculosis H37Ra (Difco Microbiology, USA). Pertussis toxin (PT, 200 ng) (Sigma Aldrich, Germany) was injected intraperitoneally the day of immunization and repeated 48 hours later. Mice were weighed and examined daily for EAE symptoms and scored as follows: 0, no signs of neurologic disease; 1, lack of tail tone; 2, abnormal gait, hind-limb weakness; 2.5, partial hind-limb paralysis; 3, complete hind-limb paralysis; 3.5, ascending paralysis; 4, tetraplegia; and 5, moribund or death. Mice were sacrificed when they reached a score of > 3.0. To determine the cumulative disease activity, the area under the curve (AUC) from the clinical score plot for each individual mouse was calculated and analysed with the Mann-Whitney test using GraphPad Prism 5.01.
NK cell Transfer
WT and CX3CR1GFP/GFP mice were killed by cervical dislocation, and the spleens were collected. Splenocytes were isolated by macerating the tissue through a 100 µm mesh (Becton Dickinson, Germany) and purified NK cells were obtained using negative cell separation (MACS NK Cell Isolation Kit, Miltenyi Biotech, Germany). 0.5–1 × 106 WT or CX3CR1GFP/GFP NK cells were injected intravenously into CX3CR1GFP/GFP recipient mice one day before EAE immunization. Intravenous injection with phosphate buffered saline (PBS) was performed as control. Active EAE was induced as described above.
Preparation of PBMCs and NK cell staining for flow cytometry
Mice were lethally anaesthetized with a mixture of ketamine (415 mg/kg) (Actavis, Germany) and xylazine (9,7 mg/kg) (Bayer Health Care, Germany) and perfused with PBS. Cells from blood, spleen, lymph nodes and the CNS were isolated and prepared as described previously [8, 56–58]. Single cell suspensions were washed with FACS buffer [PBS; 1% BSA (SERVA Electrophoresis GmbH, Germany), 0.5% NaN3 (Sigma Aldrich, Germany)] and incubated with mouse Fc Block™ (BD Pharmingen™, Germany) for 15 min to block nonspecific Ig-binding to the Fc receptor. Thereafter, NK cells were stained with a panel of conjugated antibodies diluted in FACS buffer for 20 min at 4°C. After washing cell pellets, samples were analysed by flow cytometry on a FACS Canto™ or a LSR Fortessa™ (BD Bioscience, Germany). The following anti-mouse antibodies were used: CD3 PacBlue (BD Pharmingen™, Germany, clone: 500A2), CD11b PE (BD Pharmingen™, Germany, clone M1/70), NK1.1 APC (Miltenyi Biotec, Germany, clone: PK136), CD27 PECy7 (eBioscience, San Diego, CA, clone: LG.7F9), CD45 FITC (eBioscience, San Diego, CA, clone: 30-F11), CX3CR1 FITC (R&D Systems, Minneapolis, USA, polyclonal) and CX3CR1 PerCPCy5 (Biolegend, San Diego, CA, clone: SA011F11). For intracellular stainings, cells were incubated for 20 min at 4°C in 0.5% saponin (in PBS) (Sigma Aldrich, Germany) after the surface staining, followed by a 20 min antibody incubation for intracellular markers (anti-mouse IFN-gamma and perforin, eBioscience, San Diego, CA, clone: XMG1.2 and eBio0MAK-D, respectively). Absolute cell numbers were determined using TrueCount™ Tubes as indicated by the manufacturer’s instructions (BD Bioscience, Germany). Lymphocytes were identified by their characteristic size and granularity in the sideward to forward scatter graph. NK cells were distinguished from other lymphocytes by their lack of CD3 and expression of NK1.1. NK cells were further sub-classified according to the expression of the maturation markers CD11b and CD27 in pre-mature (CD11b−CD27−), immature (CD11bneg/lowCD27+), mature (CD11b+CD27+) and fully differentiated NK cells (CD11bhighCD27−).
NK cytotoxicity measurements
Effector NK cells were negatively selected from splenocytes of unmanipulated WT and CX3CR1GFP/GFP mice using the NK Isolation kit (Miltenyi Biotech, Germany) according to the manufacturer’s instructions. The purity of enriched NK cells was consistently >90%. NK cells were diluted to 1.6 × 106 cells/ ml and used as effector cells in a cytotoxicity assay. Cytotoxicity was determined using the calcein-acetyoxymethyl release assay, as described previously [3, 59]. In brief, target YAC-1 lymphoma cells were long-term cultured in complete medium [RPMI 1640 (Gibco, Germany) supplemented with 2 mM L-glutamine (Gibco, Germany), 100 U/ ml penicillin (Seromed, Austria), 100 µg/ ml streptomycin (Seromed, Austria), 10% FCS, Sigma Aldrich Germany, and 1% HEPES (Gibco, Germany)] and showed a viability of >80%. After harvesting, YAC-1 cells were washed 3 times with PBS, incubated with 15 µM calcein-AM (MoBiTec GmbH, Germany) for 30 min at 37°C in 5% CO2, washed again 3 times with complete medium, and adjusted to 4 × 105 cells/ ml for the cytotoxicity assay. 100 µl of target cells and 100 µl of various effector cell dilutions were placed into a flat bottom 96-well microtiter plate (Corning/Costar, USA) (effector: target cell ratios ranging from 40:1 to 5:1). After incubation for 4 h at 37°C in 5% CO2, 40 µl of each supernatant were harvested and added to 60 µl of complete medium, then placed into a fresh flat-bottom plate. Samples were analyzed in quadruplicate and measured using a Wallac Victor2 1420 Multilabel spectrofluorimeter (Perkin Elmer, USA) (excitation filter: 485 nm; emission filter: 535 nm). Specific lysis (percentage of calcein-AM release) was calculated according to the following formula: [(test release − spontaneous release) / (maximum release − spontaneous release)] × 100. Spontaneous release refers to calcein-AM released from target cells in complete medium alone, while maximum release refers to calcein-AM released from target cells lysed in complete medium containing 1,8% Triton X-100 (Sigma Aldrich, Germany).
Quantitative real-time rtPCR
Lymphocyte RNA was isolated using the RNeasy® Mini Kit (QIAGEN, Germany) or the E.N.Z.A.® Total RNA Kit 1 (Omega, Bio-Tek, USA). Total RNA was then reversely transcribed to cDNA with random hexamers using the TaqMan Reverse Transcription Reagents (Perkin Elmer, Foster City, CA), according to the manufacturer’s instructions. Quantitative real-time rtPCR was performed on an ABI Prism 7000 Sequence Detection System (Applied Biosystems, Germany) [3, 17, 60]. Primers and probes are listed in Table 1. 18S rRNA was expressed at a constant level across all of the samples analysed (data not shown), and was used as the endogenous reference. All samples were run in duplicate. Comparative threshold method (ΔΔCt) was used to quantify the results obtained by real-time rtPCR. Target expression was normalized to the endogenous housekeeping gene (18S rRNA) for each sample.
Table 1.
Primers and probes for real RT-PCR.
| Name | Primer | Sequence 5' → 3' |
|---|---|---|
| m-18S | Forward | TTC GAA CGT CTG CCC TAT CAA |
| Reverse | TCC CCG TCA CCC ATG GT | |
| Probe | CGA TGG TAG TCG CCG TGC CTA CCA | |
| m-IL10 | Forward | TCG GCC AGA GCC ACA TG |
| Reverse | AGG TAA AAC TGG ATC ATT TCC GAT A | |
| Probe | TGC AGG ACT TTA AGG GTT ACT TGG GTT GC | |
| m-IL13 | Forward | AGC CTG TGG CCT GGT CC |
| Reverse | TCA AGA AGA AAT GTG CTC AAG CTG | |
| Probe | CAC AGG GCA ACT GAG GCA GGC A | |
| m-GM-CSF | Forward | GCC ATC AAA GAA GCC CTG AA |
| Reverse | GCG GGT CTG CAC ACA TGT TA | |
| Probe | ACA TGC CTG TCA CAT TGA ATG AAG AAG TAG AAG | |
| m-IFN-γ | Forward | CAG CAA CAG CAA GGC GAA A |
| Reverse | CTG GAC CTG TGG GTT GTT GAC | |
| Probe | AGG ATG CAT TCA TGA GTA TTG CCA AGT TTG A | |
| m-TNF-α | Forward | CCA AAT GGC CTC CCT CTC AT |
| Reverse | TCC TCC ACT TGG TGG TTT GC | |
| Probe* | CTC ACA CTC AGA TCA T | |
| m-granzyme B | TaqMan® Gene Expression Assay Applied Biosystems (Germany) | |
| m-perforin | TaqMan® Gene Expression Assay, Applied Biosystems (Germany) | |
All primers and probes were bought from Eurofins MWG GmbH, Germany.
Probe for m-TNF-α is labelled with MGB; all other probes are FAM-TAMRA labelled
In vitro co-culturing of CD4+ T cells and NK cells
CD4+ T cells specific for the myelin peptide MOG35–55 were isolated from lymph nodes of TCR-transgenic 2D2 mice [61], using the CD4+ T cell Isolation kit (Miltenyi Biotech, Germany). Splenocyte suspensions from the same donor mice were prepared as described above, irradiated (30 Gray) and incubated for 1h with 12,5µg/ml MOG35–35 peptide (Pepceuticals, UK). CD4+ T cells and irradiated splenocytes were co-cultured 1:1 in complete medium [RPMI 1640 (Gibco, Germany) supplemented with 2 mM L-glutamine (Gibco, Germany), 100 U/ml penicillin (Seromed, Austria), 100 µg/ml streptomycin (Seromed, Austria), 10% FCS, Sigma Aldrich Germany, and 1% HEPES (Gibco, Germany)]. Cultures were split if necessary and stimulated with IL-2 (100 U/ml) 48 hours later. After 5 days in culture, CD4+ T cells were reactivated with anti-CD3/anti-CD28 (3 µg/ml) overnight and cultured 1:1 with FACS-sorted splenic CD11b+ or CD11b− NK cell from wild-type mice, previously activated with IL-2 (100 U/ml) for 24 hours in complete medium. Cells were harvested after 24 hours and CD4+ T cells were stained for viability (7-AAD staining, Miltenyi Biotech, Germany), NK cells were stained for the degranulation marker CD107a (BD Pharmingen™, Germany, clone 1D4B) for FACS analysis as described above.
Statistical analyses
GraphPad Prism 5.01 was used for statistical analysis. The t-test was used for two-group comparisons. EAE clinical scores were analysed using the non-parametric Mann-Whitney test, or the Kruskal-Wallis test (with Dunn’s post-hoc test), for two- or three-group comparisons, respectively. EAE onset and survival were analysed using the log-rank (Mantel-Cox) test, and incidence analysed with Fisher’s exact test. P-values < 0.05 were considered significant (* p < 0.05, ** p < 0.01, *** p < 0.001). Figures show bars or dots indicating mean ± SEM.
Supplementary Material
{kind=link}
Acknowledgments
We thank Prof. Dr. Hans-Dieter Volk, head of the Institute for Medical Immunology, Charité – University Medicine Berlin, Germany, for helpful discussions, and N. Asselborn and T. Hohnstein for technical support. We thank Desirée Klunkel, Toralf Kaiser and Jenny Kirsch from the flow cytometry and cell sorting core facilities of the Berlin-Brandenburg Centrum für Regenerative Therapien (BCRT) and the Deutsches Rheuma-Forschungszentrum (DRFZ), repectively, for cell sorting. This study was supported by the Charité – Universitätsmedizin Berlin (Stipend to L. Hertwig. Rahel Hirsch Stipend to Dr. C. Infante Duarte) and the DFG (SFB 650, TP13). Dr. A. Cardona is funded by a grant of the National Institutes of Health (SC1GM095426).
Abbreviations
- CNS
central nervous system
- CX3CL1
fractalkine
- CX3CR1
fractalkine receptor
- EAE
experimental autoimmune encephalomyelitis
- MOG
myelin oligodendrocyte glycoprotein
- NK cell
natural killer cell
- WT
wild-type
Footnotes
Author contributions
Conceived and designed the experiments: L. Hertwig, I. Hamann, A. E. Cardona, R. M. Ransohoff, C. Infante-Duarte. Performed the experiments: L. Hertwig, I. Hamann, A. E. Cardona, J. M. Millward, S. Romero Suarez, R. Pietrek, K. Pollok, H. Stuis, Analyzed the data: L. Hertwig, J. M. Millward, I. Hamann. Wrote and edited the paper: L. Hertwig, C. Infante-Duarte.
Conflict of interest disclosure
The authors declare no conflict of interests
References
- 1.Bielekova B, Catalfamo M, Reichert-Scrivner S, Packer A, Cerna M, Waldmann TA, McFarland H, Henkart PA, Martin R. Regulatory CD56(bright) natural killer cells mediate immunomodulatory effects of IL-2Ralpha-targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci U S A. 2006;103:5941–5946. doi: 10.1073/pnas.0601335103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Airas L, Saraste M, Rinta S, Elovaara I, Huang YH, Wiendl H, Finnish Multiple S Pregnancy Study, G. Immunoregulatory factors in multiple sclerosis patients during and after pregnancy: relevance of natural killer cells. Clin Exp Immunol. 2008;151:235–243. doi: 10.1111/j.1365-2249.2007.03555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Infante-Duarte C, Weber A, Kratzschmar J, Prozorovski T, Pikol S, Hamann I, Bellmann-Strobl J, Aktas O, Dorr J, Wuerfel J, Sturzebecher CS, Zipp F. Frequency of blood CX3CR1-positive natural killer cells correlates with disease activity in multiple sclerosis patients. FASEB J. 2005;19:1902–1904. doi: 10.1096/fj.05-3832fje. [DOI] [PubMed] [Google Scholar]
- 4.Benczur M, Petranyl GG, Palffy G, Varga M, Talas M, Kotsy B, Foldes I, Hollan SR. Dysfunction of natural killer cells in multiple sclerosis: a possible pathogenetic factor. Clin Exp Immunol. 1980;39:657–662. [PMC free article] [PubMed] [Google Scholar]
- 5.Hirsch RL, Johnson KP. Natural killer cell activity in multiple sclerosis patients treated with recombinant interferon-alpha 2. Clin Immunol Immunopathol. 1985;37:236–244. doi: 10.1016/0090-1229(85)90155-2. [DOI] [PubMed] [Google Scholar]
- 6.Uchida A, Maida EM, Lenzhofer R, Micksche M. Natural killer cell activity in patients with multiple sclerosis: interferon and plasmapheresis. Immunobiology. 1982;160:392–402. doi: 10.1016/S0171-2985(82)80003-X. [DOI] [PubMed] [Google Scholar]
- 7.Xu W, Fazekas G, Hara H, Tabira T. Mechanism of natural killer (NK) cell regulatory role in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;163:24–30. doi: 10.1016/j.jneuroim.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 8.Zhang B, Yamamura T, Kondo T, Fujiwara M, Tabira T. Regulation of experimental autoimmune encephalomyelitis by natural killer (NK) cells. J Exp Med. 1997;186:1677–1687. doi: 10.1084/jem.186.10.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hao J, Liu R, Piao W, Zhou Q, Vollmer TL, Campagnolo DI, Xiang R, La Cava A, Van Kaer L, Shi FD. Central nervous system (CNS)-resident natural killer cells suppress Th17 responses and CNS autoimmune pathology. J Exp Med. 2010;207:1907–1921. doi: 10.1084/jem.20092749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Winkler-Pickett R, Young HA, Cherry JM, Diehl J, Wine J, Back T, Bere WE, Mason AT, Ortaldo JR. In vivo regulation of experimental autoimmune encephalomyelitis by NK cells: alteration of primary adaptive responses. J Immunol. 2008;180:4495–4506. doi: 10.4049/jimmunol.180.7.4495. [DOI] [PubMed] [Google Scholar]
- 11.Shi FD, Ljunggren HG, Sarvetnick N. Innate immunity and autoimmunity: from self-protection to self-destruction. Trends Immunol. 2001;22:97–101. doi: 10.1016/s1471-4906(00)01821-4. [DOI] [PubMed] [Google Scholar]
- 12.Lunemann A, Lunemann JD, Munz C. Regulatory NK-cell functions in inflammation and autoimmunity. Mol Med. 2009;15:352–358. doi: 10.2119/molmed.2009.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi FD, Ljunggren HG, La Cava A, Van Kaer L. Organ-specific features of natural killer cells. Nat Rev Immunol. 2011;11:658–671. doi: 10.1038/nri3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huntington ND, Vosshenrich CA, Di Santo JP. Developmental pathways that generate natural-killer-cell diversity in mice and humans. Nat Rev Immunol. 2007;7:703–714. doi: 10.1038/nri2154. [DOI] [PubMed] [Google Scholar]
- 15.Hayakawa Y, Huntington ND, Nutt SL, Smyth MJ. Functional subsets of mouse natural killer cells. Immunol Rev. 2006;214:47–55. doi: 10.1111/j.1600-065X.2006.00454.x. [DOI] [PubMed] [Google Scholar]
- 16.Poli A, Kmiecik J, Domingues O, Hentges F, Blery M, Chekenya M, Boucraut J, Zimmer J. NK cells in central nervous system disorders. J Immunol. 2013;190:5355–5362. doi: 10.4049/jimmunol.1203401. [DOI] [PubMed] [Google Scholar]
- 17.Hamann I, Unterwalder N, Cardona AE, Meisel C, Zipp F, Ransohoff RM, Infante-Duarte C. Analyses of phenotypic and functional characteristics of CX3CR1-expressing natural killer cells. Immunology. 2011;133:62–73. doi: 10.1111/j.1365-2567.2011.03409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang D, Shi FD, Jung S, Pien GC, Wang J, Salazar-Mather TP, He TT, Weaver JT, Ljunggren HG, Biron CA, Littman DR, Ransohoff RM. The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J. 2006;20:896–905. doi: 10.1096/fj.05-5465com. [DOI] [PubMed] [Google Scholar]
- 19.Garcia JA, Pino PA, Mizutani M, Cardona SM, Charo IF, Ransohoff RM, Forsthuber TG, Cardona AE. Regulation of adaptive immunity by the fractalkine receptor during autoimmune inflammation. J Immunol. 2013;191:1063–1072. doi: 10.4049/jimmunol.1300040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gregoire C, Chasson L, Luci C, Tomasello E, Geissmann F, Vivier E, Walzer T. The trafficking of natural killer cells. Immunol Rev. 2007;220:169–182. doi: 10.1111/j.1600-065X.2007.00563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, Walzer T. Maturation of mouse NK cells is a 4-stage developmental program. Blood. 2009;113:5488–5496. doi: 10.1182/blood-2008-10-187179. [DOI] [PubMed] [Google Scholar]
- 22.Hamann I, Dorr J, Glumm R, Chanvillard C, Janssen A, Millward JM, Paul F, Ransohoff RM, Infante-Duarte C. Characterization of natural killer cells in paired CSF and blood samples during neuroinflammation. J Neuroimmunol. 2013;254:165–169. doi: 10.1016/j.jneuroim.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 23.Fu B, Tian Z, Wei H. Subsets of human natural killer cells and their regulatory effects. Immunology. 2014;141:483–489. doi: 10.1111/imm.12224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu B, Wang F, Sun R, Ling B, Tian Z, Wei H. CD11b and CD27 reflect distinct population and functional specialization in human natural killer cells. Immunology. 2011;133:350–359. doi: 10.1111/j.1365-2567.2011.03446.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flodstrom-Tullberg M, Bryceson YT, Shi FD, Hoglund P, Ljunggren HG. Natural killer cells in human autoimmunity. Curr Opin Immunol. 2009;21:634–640. doi: 10.1016/j.coi.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 26.Mandal A, Viswanathan C. Natural killer cells: In health and disease. Hematol Oncol Stem Cell Ther. 2014 doi: 10.1016/j.hemonc.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 27.Chanvillard C, Jacolik RF, Infante-Duarte C, Nayak RC. The role of natural killer cells in multiple sclerosis and their therapeutic implications. Front Immunol. 2013;4:63. doi: 10.3389/fimmu.2013.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chanvillard C, Millward JM, Lozano M, Hamann I, Paul F, Zipp F, Dorr J, Infante-Duarte C. Mitoxantrone induces natural killer cell maturation in patients with secondary progressive multiple sclerosis. PLoS One. 2012;7:e39625. doi: 10.1371/journal.pone.0039625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kastrukoff LF, Lau A, Wee R, Zecchini D, White R, Paty DW. Clinical relapses of multiple sclerosis are associated with 'novel' valleys in natural killer cell functional activity. J Neuroimmunol. 2003;145:103–114. doi: 10.1016/j.jneuroim.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 30.Kastrukoff LF, Morgan NG, Zecchini D, White R, Petkau AJ, Satoh J, Paty DW. A role for natural killer cells in the immunopathogenesis of multiple sclerosis. J Neuroimmunol. 1998;86:123–133. doi: 10.1016/s0165-5728(98)00014-9. [DOI] [PubMed] [Google Scholar]
- 31.Merrill J, Jondal M, Seeley J, Ullberg M, Siden A. Decreased NK killing in patients with multiple sclerosis: an analysis on the level of the single effector cell in peripheral blood and cerebrospinal fluid in relation to the activity in the disease. Clin Exp Immunol. 1982;47:419–430. [PMC free article] [PubMed] [Google Scholar]
- 32.Hauser SL, Ault KA, Levin MJ, Garovoy MR, Weiner HL. Natural killer cell activity in multiple sclerosis. J Immunol. 1981;127:1114–1117. [PubMed] [Google Scholar]
- 33.Ponzetta A, Sciume G, Benigni G, Antonangeli F, Morrone S, Santoni A, Bernardini G. CX3CR1 regulates the maintenance of KLRG1+ NK cells into the bone marrow by promoting their entry into circulation. J Immunol. 2013;191:5684–5694. doi: 10.4049/jimmunol.1300090. [DOI] [PubMed] [Google Scholar]
- 34.Limatola C, Ransohoff RM. Modulating neurotoxicity through CX3CL1/CX3CR1 signaling. Front Cell Neurosci. 2014;8:229. doi: 10.3389/fncel.2014.00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 36.Garcia JA, Cardona SM, Cardona AE. Analyses of microglia effector function using CX3CR1-GFP knock-in mice. Methods Mol Biol. 2013;1041:307–317. doi: 10.1007/978-1-62703-520-0_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pabon MM, Bachstetter AD, Hudson CE, Gemma C, Bickford PC. CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson's disease. J Neuroinflammation. 2011;8:9. doi: 10.1186/1742-2094-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zujovic V, Benavides J, Vige X, Carter C, Taupin V. Fractalkine modulates TNF-alpha secretion and neurotoxicity induced by microglial activation. Glia. 2000;29:305–315. [PubMed] [Google Scholar]
- 39.Nash KR, Lee DC, Hunt JB, Jr, Morganti JM, Selenica ML, Moran P, Reid P, Brownlow M, Guang-Yu Yang C, Savalia M, Gemma C, Bickford PC, Gordon MN, Morgan D. Fractalkine overexpression suppresses tau pathology in a mouse model of tauopathy. Neurobiol Aging. 2013;34:1540–1548. doi: 10.1016/j.neurobiolaging.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. Am J Pathol. 2010;177:2549–2562. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gan Y, Liu Q, Wu W, Yin JX, Bai XF, Shen R, Wang Y, Chen J, La Cava A, Poursine-Laurent J, Yokoyama W, Shi FD. Ischemic neurons recruit natural killer cells that accelerate brain infarction. Proc Natl Acad Sci U S A. 2014;111:2704–2709. doi: 10.1073/pnas.1315943111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pallandre JR, Krzewski K, Bedel R, Ryffel B, Caignard A, Rohrlich PS, Pivot X, Tiberghien P, Zitvogel L, Strominger JL, Borg C. Dendritic cell and natural killer cell cross-talk: a pivotal role of CX3CL1 in NK cytoskeleton organization and activation. Blood. 2008;112:4420–4424. doi: 10.1182/blood-2007-12-126888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lunemann A, Lunemann JD, Roberts S, Messmer B, Barreira da Silva R, Raine CS, Munz C. Human NK cells kill resting but not activated microglia via NKG2D- and NKp46-mediated recognition. J Immunol. 2008;181:6170–6177. doi: 10.4049/jimmunol.181.9.6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mellergard J, Edstrom M, Jenmalm MC, Dahle C, Vrethem M, Ernerudh J. Increased B cell and cytotoxic NK cell proportions and increased T cell responsiveness in blood of natalizumab-treated multiple sclerosis patients. PLoS One. 2013;8:e81685. doi: 10.1371/journal.pone.0081685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson TA, Evans BL, Durafourt BA, Blain M, Lapierre Y, Bar-Or A, Antel JP. Reduction of the peripheral blood CD56(bright) NK lymphocyte subset in FTY720-treated multiple sclerosis patients. J Immunol. 2011;187:570–579. doi: 10.4049/jimmunol.1003823. [DOI] [PubMed] [Google Scholar]
- 46.Lin YC, Winokur P, Blake A, Wu T, Romm E, Bielekova B. Daclizumab reverses intrathecal immune cell abnormalities in multiple sclerosis. Ann Clin Transl Neurol. 2015;2:445–455. doi: 10.1002/acn3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saraste M, Irjala H, Airas L. Expansion of CD56Bright natural killer cells in the peripheral blood of multiple sclerosis patients treated with interferon-beta. Neurol Sci. 2007;28:121–126. doi: 10.1007/s10072-007-0803-3. [DOI] [PubMed] [Google Scholar]
- 48.Kivisakk P, Mahad DJ, Callahan MK, Sikora K, Trebst C, Tucky B, Wujek J, Ravid R, Staugaitis SM, Lassmann H, Ransohoff RM. Expression of CCR7 in multiple sclerosis: implications for CNS immunity. Ann Neurol. 2004;55:627–638. doi: 10.1002/ana.20049. [DOI] [PubMed] [Google Scholar]
- 49.Berahovich RD, Lai NL, Wei Z, Lanier LL, Schall TJ. Evidence for NK cell subsets based on chemokine receptor expression. J Immunol. 2006;177:7833–7840. doi: 10.4049/jimmunol.177.11.7833. [DOI] [PubMed] [Google Scholar]
- 50.Vitale M, Della Chiesa M, Carlomagno S, Romagnani C, Thiel A, Moretta L, Moretta A. The small subset of CD56brightCD16- natural killer cells is selectively responsible for both cell proliferation and interferon-gamma production upon interaction with dendritic cells. Eur J Immunol. 2004;34:1715–1722. doi: 10.1002/eji.200425100. [DOI] [PubMed] [Google Scholar]
- 51.Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol. 2003;3:569–581. doi: 10.1038/nri1130. [DOI] [PubMed] [Google Scholar]
- 52.Cardona AE, Sasse ME, Liu L, Cardona SM, Mizutani M, Savarin C, Hu T, Ransohoff RM. Scavenging roles of chemokine receptors: chemokine receptor deficiency is associated with increased levels of ligand in circulation and tissues. Blood. 2008;112:256–263. doi: 10.1182/blood-2007-10-118497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fiebiger SM, Bros H, Grobosch T, Janssen A, Chanvillard C, Paul F, Dorr J, Millward JM, Infante-Duarte C. The antioxidant idebenone fails to prevent or attenuate chronic experimental autoimmune encephalomyelitis in the mouse. J Neuroimmunol. 2013;262:66–71. doi: 10.1016/j.jneuroim.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 55.Millward JM, Guo J, Berndt D, Braun J, Sack I, Infante-Duarte C. Tissue structure and inflammatory processes shape viscoelastic properties of the mouse brain. NMR Biomed. 2015 doi: 10.1002/nbm.3319. [DOI] [PubMed] [Google Scholar]
- 56.Kruisbeek AM. Isolation of mouse mononuclear cells. Curr Protoc Immunol. 2001;Chapter 3(Unit 3):1. doi: 10.1002/0471142735.im0301s39. [DOI] [PubMed] [Google Scholar]
- 57.Beeton C, Chandy KG. Isolation of mononuclear cells from the central nervous system of rats with EAE. J Vis Exp. 2007:527. doi: 10.3791/527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kouro T, Yokota T, Welner R, Kincade PW. Isolation of prolymphocytes from bone marrow and fetal liver. Curr Protoc Immunol. 2005;Chapter 22(Unit 22F):21. doi: 10.1002/0471142735.im22f01s66. [DOI] [PubMed] [Google Scholar]
- 59.Neri S, Mariani E, Meneghetti A, Cattini L, Facchini A. Calcein-acetyoxymethyl cytotoxicity assay: standardization of a method allowing additional analyses on recovered effector cells and supernatants. Clin Diagn Lab Immunol. 2001;8:1131–1135. doi: 10.1128/CDLI.8.6.1131-1135.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wandinger KP, Sturzebecher CS, Bielekova B, Detore G, Rosenwald A, Staudt LM, McFarland HF, Martin R. Complex immunomodulatory effects of interferon-beta in multiple sclerosis include the upregulation of T helper 1-associated marker genes. Ann Neurol. 2001;50:349–357. doi: 10.1002/ana.1096. [DOI] [PubMed] [Google Scholar]
- 61.Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med. 2003;197:1073–1081. doi: 10.1084/jem.20021603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.