

Abstract
Methionine sulfoxide reductase A (msrA) reduces methionine sulfoxide in proteins back to methionine. Its catalytic cysteine (Cys72-SH) has a low pKa that facilitates oxidation by methionine sulfoxide to cysteine sulfenic. If the catalytic cycle proceeds efficiently, the sulfenic acid is reduced back to cysteine at the expense of thioredoxin. However, the sulfenic acid is vulnerable to “irreversible” oxidation to cysteine sulfinic acid that inactivates msrA (hyperoxidation). We observed that human msrA is resistant to hyperoxidation while mouse msrA is readily hyperoxidized by micromolar concentrations of hydrogen peroxide. We investigated the basis of this difference in susceptibility to hyperoxidation and established that it is controlled by the presence or absence of a Met residue in the carboxyl terminal domain of the enzyme, Met229. This residue is Val in human msrA and when it was mutated to Met, human msrA became sensitive to hyperoxidation. Conversely, mouse msrA was rendered insensitive to hyperoxidation when the Met229 was mutated to Val or to one of 5 other residues. Positioning of the methionine at residue 229 is not critical, as hyperoxidation occurred so long at the methionine was located within the 14 carboxyl terminal residues. The carboxyl domain of msrA is known to be flexible and to have access to the active site, and Met residues are known to form stable, non-covalent bonds with aromatic residues through interaction of the sulfur atom with the aromatic ring. We propose that Met229 forms such a bond with Trp74 at the active site, preventing formation of a protective sulfenylamide with Cys72 sulfenic acid. As a consequence, the sulfenic acid is available for facile, irreversible oxidation to cysteine sulfinic acid.
Graphical Abstract
Reactive oxygen species (ROS) are inevitably produced by a variety of the cellular activities. Some processes generate hydrogen peroxide and activate specific cellular signaling.1–3 The sulfur containing residues of proteins, Cys and Met, are relatively readily oxidized and are common targets of reactive species in cells under oxidative stress. Oxidative modifications of Cys that function in cellular regulation have been widely studied while those of Met are only recently gaining attention. The most common oxidations are disulfide formation by Cys and methionine sulfoxide (MetO) formation from Met. Unlike oxidations of other amino acids, both disulfide formation and MetO formation are reversible. Disulfides may be reduced back to the thiol form by various reductases, often utilizing thioredoxin.4 MetO is reduced back to Met by the methionine sulfoxide reductases that are thioredoxin-dependent enzymes which are virtually universal among aerobic organisms.5, 6 Oxidation of Met to MetO introduces a chiral center at the sulfur atom so there are two epimers of MetO; R-MetO and S-MetO. The S and R epimers are reduced stereospecifically by msrA and msrB, respectively. Mammalian msrA is found both in the cytosol and mitochondria, encoded by a single gene through two initiation sites.7 The cytosolic form is myristoylated, a modification that is essential for msrA to protect the heart against ischemia-reperfusion injury.8 Deletion of msrA renders both prokaryotic and eukaryotic cells more susceptible to killing by oxidative stresss.9, 10
msrA from a variety of organisms has been studied in great detail, initially by Weissbach and colleagues11 and subsequently by many investigators.12–14 The active site is highly conserved among organisms and contains a low pKa Cys residue that is central to its catalytic function. The enzyme is bifunctional, being capable of acting as a methionine sulfoxide reductase or as a methionine peroxidase15 (Figure 1). The first step in the reductase cycle is reduction of the sulfoxide with concomitant oxidation of the cysteine thiol to cysteine sulfenic acid. Normally the sulfenic acid is rapidly reduced by two “resolving cysteines” in the carboxy domain that then form a disulfide. This disulfide is reduced by thioredoxin at the ultimate expense of cellular NADPH.16, 17
Figure 1.

Mouse msrA is both a reductase and peroxidase. The upper panel shows the catalytic cycle when acting as a reductase. The lower panel shows the catalytic cycle when acting as an oxidase. Please see the Introduction for details.
The reductase is converted to a peroxidase by depletion of reducing power or by blocking the resolving cysteines through action of a proposed regulatory protein (Figure 1). In the first step of the peroxidase catalytic cycle, MetO can again oxidize the active site cysteine thiol to its sulfenic acid. msrA also reacts with hydrogen peroxide to form a sulfenic acid, a reaction that may function in cellular signaling.15, 18. Because either thioredoxin is not available or the carboxy domain is blocked, the sulfenic acid is potentially susceptible to further oxidation to cysteine sulfinic acid. MetO is a relatively weak oxidizing agent19 and does not further oxidize the cysteine sulfenic acid. Hydrogen peroxide is a relatively strong oxidizing agent that readily oxidizes the cysteine sulfenic acid to cysteine sulfinic acid. Formation of the sulfinic acid is effectively an irreversible oxidation20 that has been termed “hyperoxidation”. Hyperoxidation can be prevented if the cysteine sulfenic acid can form a sulfenylamide with the amide nitrogen of a nearby residue21. When cellular reducing power again becomes available, the sulfenylamide is reduced back to the thiol. Sulfenylamide formation was first noted in a structural study of protein tyrosine phosphatase 1B whose catalytic cycle also depends on generation of a sulfenic acid at its active site.21 A similar protective mechanism has recently been proposed for glutathione peroxidase, which has an active site selenocysteine.22
The sulfenic acid form of the active site Cys72 of mouse msrA can also form a sulfenylamide, in this case with the close-by amide nitrogen of Trp74. This was demonstrated in studies in which the sulfenic acid was formed by reacting mouse msrA with methionine sulfoxide in the absence of reducing power.15 Human and mouse msrA are highly homologous, with 88% amino acid identity (Figure 2). In the course of studies on human and mouse msrA, we noted that both formed a sulfenylamide when the active site Cys was oxidized by MetO. However, when oxidized by hydrogen peroxide, the human msrA again formed a sulfenylamide while the mouse msrA was hyperoxidized and irreversibly inactivated. We investigated the basis of this difference in susceptibility to hyperoxidation and established that it is controlled by the presence or absence of a Met residue in the carboxyl terminal domain of the enzyme.
Figure 2.

Alignment of mouse and human msrA amino acid sequences without their mitochondrial leader sequences. The hexapeptide substrate’s sequence is that of the six underlined amino acids in the carboxyl terminus of mouse msrA. The active site consensus motif (-GCFWG-) is indicated in bold italic letters, and the two resolving cysteines, Cys218 and Cys227, are marked with asterisks. Met229 in mouse msrA and Val231 in human msrA are shown in bold letters.
MATERIALS AND METHODS
Materials
Met, MetO, and hydrogen peroxide (30%) were obtained from Sigma-Aldrich. Oxidized hexapeptide (Pro-MetO-Ala-Ile-Lys-Lys) and native hexapeptide (Pro-Met-Ala-Ile-Lys-Lys) were purchased from American Peptide (Sunnyvale, CA). Throughout the paper msrA refers to the mouse form unless the human is specified. Recombinant, wild-type, non-myristoylated7 human msrA, mouse msrA, and their site specific mutants msrA variants were prepared and purified as described previously except for the studies in Figure 5. 7, 23 Wild type, Met229Leu, C107S/C218S/C227S (C3S), C107S/C218S/C227/M229K (C3S-M229K), C107S/C218S/C227/M229Q (C3S-M229Q), C107S/C218S/C227S/Δ228-233 (C3S Δ228-233), and all other site-directed point mutants were cloned into the pET17b plasmid (Novagen) and expressed in E. coli. 7, 23 The protein concentration of solutions was determined from their absorbance at 280 nm using molar extinction coefficients calculated by GPMAW version 9 (Lighthouse data, Odense, Denmark). The purity of all preparations, including those used for Figure 5, was measured from the integrated areas of their reverse phase chromatograms monitored at 210 nm. The gradient was developed as described below for the mass spectrometric analysis of intact proteins. All preparations were >90% pure. Before use in any experiment, the fully reduced form of msrA was prepared by incubation with 10 mM DTT in 50 mM sodium phosphate, pH 7.5 with 1 mM diethylenetriamine-pentaacetic acid (DTPA) for 30 min at 37° C followed by dialysis against the same buffer without DTT.
Figure 5.

A carboxyl terminal methionine is required to induce sensitivity to hyperoxidation and inactivation by H2O2. Only methionine induces sensitivity, and the Met residue must be in the carboxyl terminus, at or after residue 220. Proteins were incubated at 37° C for 5 min with 1 mM H2O2 at which time DTT was added to 10 mM to scavenge remaining H2O2 and reduce any disulfide bonds or sulfenic acid. The solution was incubated another 5 min and then analyzed by reverse phase HPLC-mass spectrometry as described in Materials and Methods. Protein peak areas were obtained from the 210 nm chromatogram and used to calculate the percentage hyperoxidized.
For the studies reported in Figure 5, 10 ml growths of E. coli expressing each mutant were used, allowing use of a simpler batch DEAE purification. Expression was induced by 0.5 mM IPTG for 3 h at 37° C. Cells were disrupted by sonication, and the homogenate was centrifuged at 21,000 g for 5 min. The supernatant was made 1% in streptomycin sulfate, held at 4° C for 30 min, and again centrifuged for 5 min at 21,000 g. The supernatant was brought to 80% saturation with ammonium sulfate by addition of solid ammonium sulfate and held for another 30 min at 4° C. Following centrifugation, the pellet was dissolved in 300 μl buffer with 10 mM DTT, incubated for 30 min at 37° C, and then dialyzed to remove DTT. The protein solution was then mixed with 100 μl DEAE Sephacel® (50% slurry), to which neither mouse nor human msrA binds. The mixture was held at 4° C overnight, and then centrifuged. The supernatant containing msrA of >90% purity was stored at −20° C. The concentration of msrA was determined by quantitative infrared fluorescence of Coomassie Blue stained SDS gels. 24
Mass spectrometric analysis of intact proteins
The mass of intact proteins was determined by an HPLC-mass spectrometry system with an accuracy of better than ±1 Da. msrA was treated with 100 μM hydrogen peroxide in 50 mM phosphate/1 mM DTPA, pH 7.4. The reaction was stopped by making the solution 0.5% in acetic acid or 10 mM DTT. 118 ng of each sample was loaded onto a reverse phase column on an HPLC equipped with an autosampler set to 4°C (Agilent 1100 series HPLC, Agilent Technologies, Santa Clara, CA). The system contained a Zorbax 300Å StableBond C18 MicroBore column (865630–902, 1.0 × 50mm, 3.5μm). The initial solvent was 0.05 % trifluoroacetic acid with gradient elution by acetonitrile/0.05 % trifluoroacetic acid increasing at 2 %/min with a flow rate of 20 μL/min. The effluent from the spectrophotometric detector was mixed in a tee with acetic acid pumped at 20 μl/min by a separate Agilent 1100 series HPLC. Positive ion electrospray ionization (ESI) mass spectra were obtained with an Agilent 6520 mass spectrometer equipped with a time-of-flight detector. The capillary voltage was 3500 V, and data was collected in the mass range of 500–2500 m/z. Mass spectra were analyzed and deconvoluted using Agilent software, MassHunter version B.05. After identification of the peaks by their mass, the 210 nm UV chromatograms with and without DTT treatment were integrated and the areas used for quantitation. (DTT converts any sulfenylamide or sulfenic acid back to the cysteine thiol.) The sulfenylamide form (−2 Da from unmodified) was quantitated in the analysis without DTT and the hyperoxidized form (+32 Da from unmodified) in the analysis with DTT.
Peptide mapping and sequencing
85 μM C3S msrA was treated with 1 mM hydrogen peroxide for 15 min at 37°C. The solution was then incubated with 10 mM DTT for 10 min to stop the reaction and reduce any sulfenylamide present. It was then dialyzed against 50 mM Tris/1 mM DTPA, pH 7.4 at 4°C. Twenty μg protein in 21 μl was digested at 37°C overnight with 1 μg chymotrypsin (# 84975820, Boeringer Mannheim). The digestion was stopped by adding 0.2 μl of 50% acetic acid, giving a final concentration of 0.5%. 7.5 μg was analyzed by reverse phase HPLC- tandem mass spectrometry as described above for protein mass spectroscopy except that the acetonitrile gradient was developed at 1%/min from 0 to 45%. Spectra were deconvoluted with the same Agilent MassHunter software and MS/MS spectra were matched to those predicted with GPMAW and confirmed by de novo sequencing with PEAKS version 7.0 (Bioinformatics Solutions, Ontario, Canada).
msrA activity assay
Reductase activity was measured with MetO or the oxidized hexapeptide (Pro-MetO-Ala-Ile-Lys-Lys) and oxidase activity with the native hexapeptide (Pro-Met-Ala-Ile-Lys-Lys) at 37° C for 6 min as described.15 In the oxidase assay, the oxidized and reduced peptides were separated by HPLC-mass spectrometry as described above. The oxidized peptide eluted 1 min before the reduced peptide, and their peaks were baseline separated. The integrated area of each peptide was obtained from the UV chromatogram at 210 nm. Km and Vmax were obtained by fitting the curve of substrate concentration versus product to a hyperbola with Prism version 6 (GraphPad Software, La Jolla, CA).
RESULTS
Mouse msrA is readily hyperoxidized by hydrogen peroxide while human msrA is not
The sequences of mouse and human msrA are 88% identical, and 20 residues on both the amino and carboxy sites of the active site Cys72 are identical (Figure 2). Moreover, the active sites of mammalian msrA are superimposable in their three dimensional folded forms25, 26 and those from other organisms including bacteria are either superimposable or extremely similar.12, 27 As detailed in the Introduction, When mouse msrA is incubated with MetO in the absence of reducing agents, the enzyme reduces 3 molecules of methionine sulfoxide causing formation of a disulfide bond, oxidation of Met229 to MetO, and formation of a sulfenylamide at the active site. The mass of the protein molecule is increased by 12 Da (−2 Da for the disulfide, −2 Da for the sulfenylamide, and +16 Da for the sulfoxide). Human msrA lacks Met229 so when incubated with methionine sulfoxide its mass decreases by 4 Da with disulfide and sulfenylamide formation (Figure 3a–d). As with mouse msrA, human msrA does not undergo further oxidation with prolonged incubation with methionine sulfoxide.
Figure 3.

Comparison of mouse and human msrA incubated with MetO or H2O2. (AD), Protein Mass. The enzymes were incubated for 5 min with 10 mM MetO and then injected into the HPLC-mass spectrometer. The major form of the mouse msrA in (B) is +12 Da which, as explained in the main text, is the net change from oxidation of Met229 to its sulfoxide (+16 Da), oxidation of the resolving cysteines to their disulfide (−2 Da), and oxidation of the active site cys to its sulfenamide (−2 Da). The minor form is ~−4 Da because Met229 was not oxidized before formation of the disulfide and sulfenamide terminated the oxidation reactions. (D) shows that human msrA forms the disulfide and sulfenamide for a net change of −4 Da. (E) Mouse msrA is inactivated by H2O2 exposure while human msrA is relatively resistant. This panel figure shows the time courses of loss of reductase activity of 2 μM mouse (●) and human (○) msrA upon exposure to 1 mM H2O2. The oxidized hexapeptide (Pro-MetO-Ala-Ile-Lys-Lys) was the substrate and DTT provided reducing equivalents. Each data point is the average of analyses run on two different days.
Incubation of human msrA with hydrogen peroxide also generated the −4 Da form whose active site Cys72 was resistant to hyperoxidation to the sulfinic acid (Figure 3e). The native form with full activity was regenerated by incubation with the reducing agent DTT, consistent with the expected facile reduction of the disulfide and sulfenylamide. In contrast, exposure of mouse msrA caused rapid loss of activity and a gain in mass of 30 Da. Subsequent incubation with DTT generated a form that was 32 Da greater than the unmodified, native form. This +32 Da form is catalytically inactive. The +32 Da mass change is consistent with addition of 2 oxygen atoms, most likely on the active site Cys72 or Met229. Peptide mapping with sequencing by MS/MS established that Cys72 was hyperoxidized to the sulfinic acid while Met229 was not oxidized (Figure 4). This result implies that hyperoxidation of the Cys72 sulfenic acid by hydrogen peroxide is much faster than oxidation of Met229 by the sulfenic acid.
Figure 4.

Incubation of mouse msrA with H2O2 oxidizes the active site Cys72 to its sulfinic acid. To avoid complications from oxidation or disulfide formation with other cysteine residues, the 3 non-active site cys were mutated to ser [C107S/C218S/C227S].15 The enzyme was incubated for 15 min with or without 1 mM H2O2, then cleaved with chymotrypsin and mapped by HPLC-MS/MS as described.15 The active site Cys72 is contained in the peptide of residues 69–74. The parent peptide’s mass was measured to be 732.2485 Da, and its calculated mass was 732.2480, an error of 0.7 ppm. The expected mass of each b and y ion was calculated by GPMAW and compared to the observed mass, demonstrating that Cys72 was oxidized to the sulfinic acid (represented by a lower case c in the figure). The error in mass measurement for this mass spectrometer in MS-MS mode is ≤ 10 ppm. ND, not detected.
Met229 in mouse msrA renders Cys72 susceptible to hyperoxidation by hydrogen peroxide
Given the impressive similarity of the primary, secondary, and tertiary structures of the mouse and human msrA, we wanted to identify the difference(s) that controlled susceptibility to hyperoxidation. Recognizing that the mouse Met229 is a Val in the human, we focused our initial investigations on the carboxyl terminal region. Mutating the Val to Met in human msrA rendered it susceptible to hyperoxidation.
We also prepared various mutants of mouse msrA, confirmed that all were catalytically active, and tested them for susceptibility to hyperoxidation (Figure 5). Increasing deletions from the carboxyl terminus of the C3S form of msrA revealed that deletion of the last 3 amino acids did not significantly alter hyperoxidizability. Deletion of 4 residues from carboxyl terminus left Met229 as the carboxyl terminus, with some decrease in hyperoxidizability. However, when Met229, was deleted, the truncated form was rendered substantially resistant to hyperoxidation. In a full length mouse msrA, mutating Met229 to any of 6 other residues also conferred resistance to hyperoxidation, including mutation to the hydrophobic Leu or Val. We then constructed mutants in which the Met was placed at residues other than 229 (Figure 5). A Met at residue 220 or closer to the carboxyl terminus rendered the enzyme susceptible to hyperoxidation while placement at residue 219 or earlier markedly decreased sensitivity.
Glutamine is an analogue of methionine so that site-specific mutation of methionine to glutamine provides a genetic method for testing the effect of a constitutive methionine sulfoxide.28 Substitution of Gln for Met229 converted the enzyme to a form resistant to hyperoxidation, indicating that only methionine and not its sulfoxide was capable of inducing susceptibility (Figure 5). We tested this proposal directly by preparing msrA with MetO229 by incubating the enzyme with MetO. After dialysis to remove MetO, it was exposed to varying concentrations of hydrogen peroxide along with the wild-type and Met229Leu forms (Figure 6). The MetO229 and Met229Leu forms were both resistant to hyperoxidation while the control Met229 form was very sensitive. Thus, the reduced sulfur of Met is important in controlling sensitivity.
Figure 6.

Mutation or oxidation of Met229 in mouse msrA prevents inactivation by H2O2. Enzyme activity was measured after 30 min exposure to the indicated concentration of H2O2 with 2 mM oxidized hexapeptide as substrate. Wild-type msrA (○); wild-type msrA with Met229 oxidized to MetO before incubation with H2O2 (□); Met229Leu (△).
Is oxidation from cysteine sulfenic to the sulfinic mediated by hydrogen peroxide or by intermolecular oxidation by msrA?
Following formation of the Cys72 sulfenic acid in msrA, further oxidation could be mediated either by another molecule of hydrogen peroxide or by a second molecule of the sulfenic acid form of msrA. The two mechanisms would differ in the effect of varying the concentration of msrA during the oxidation. If hydrogen peroxide is the oxidizing agent then the rate of sulfinic acid formation would be independent of the concentration of msrA. If msrA is the oxidizing species then the reaction is second order in msrA and the rate would be proportional to [msrA].2 We found that the rate of hyperoxidation was independent of the concentration of msrA, and we conclude that the oxidation is mediated by hydrogen peroxide and not by msrA acting as an oxidase (Figure 7).
Figure 7.

The rate of hyperoxidation is independent of the msrA concentration. The time course of hyperoxidative inactivation by 100 μM H2O2 was determined. Aliquots were taken at the plotted times, and the reaction was stopped by addition of DTT to 10 mM. The fraction inactivated was determined from the areas of the native and hyperoxidized peaks in the 210 nm chromatogram from the HPLC UV detector. Although graphed here as a linear plot to allow easier visualization, the regression line was fit to each series as a semi-logarithmic plot. The intercepts and slopes were averaged and used to generate the plotted fit line and to calculate the lag time and half-time for inactivation. The lag is presumably due to the first oxidation of the active site cysteine to the sulfenic form followed by regeneration of the thiol and formation of a disulfide bond by the carboxyl terminal cysteine residues.
Why evolve an msrA susceptible to hyperoxidation?
Sulfur containing amino acids are metabolically expensive and are in limited availability to mammals living in the wild, and hyperoxidation of cysteine to the sulfinic acid causes irreversible inactivation of msrA. Thus, there must be counterbalancing benefits to rendering the enzyme susceptibile to hyperoxidation. For example, when the active site of peroxiredoxin II is oxidized to the sulfonic acid, the protein oligomerizes to a decamer with a marked gain of function as a chaperone.29 We utilized dynamic light scattering to assess whether hyperoxidized msrA also undergoes oligomerization. The results shown in Figure 8 demonstrate that it remains monomeric when hyperoxidized. We then considered the possibility that introduction of a methionine residue in the carboxyl domain modulated the activity of msrA, either as a reductase or oxidase. We compared the kinetics of mouse wild type and Met229Leu msrAs and found that they were the same in both forms in both the oxidase and reductase directions, with the values matching those previously published18 (Table 1). The benefit of introducing a Met residue in the carboxyl domain thus remains to be elucidated
Figure 8.

Both wild-type and Met229Leu remain monomeric when incubated with H2O2. Dynamic light scattering was utilized to determine the distribution of the molecular radius or radii. Each measurement was performed 10 times on two different days, and the results were averaged. This procedure was repeated and each panel shows the average of two sets of 10 measurements. The mass-averaged hydrodynamic radius of the peak of the distribution is shown in each panel and corresponds to a monomer.
Table 1.
Kinetic Parameters of Mouse msrA
| msrA | Km (mM) | Vmax (μmol min−1 mg−1) | ||
|---|---|---|---|---|
| Reductase | Oxidase | Reductase | Oxidase | |
| Wild-Type | 3.1 | 4.0 | 6.2 | 0.20 |
| Met229Leu | 2.8 | 4.7 | 5.6 | 0.28 |
DISCUSSION
We have described a novel mechanism for modulating the susceptibility of a low pKa cysteine to hyperoxidation to the sulfinic acid. The presence of a single methionine residue in the carboxyl terminal domain of mouse msrA suffices to induce susceptibility. The carboxyl domain of msrA is known to be flexible and to have access to the active site.12, 25, 26 Further, Met residues are known to form stable, non-covalent bonds with aromatic residues through interaction of the sulfur atom with the aromatic ring.30–32 We propose that Met229 forms such a bond with Trp74 at the active site, preventing formation of a sulfenylamide with Cys72 sulfenic acid (Figure 9). As a consequence, the sulfenic acid is available for facile, irreversible oxidation to cysteine sulfinic acid. Consistent with this proposal is our finding that neither Met229 sulfoxide nor Met229Gln induces susceptibility because neither can form a bond with Trp74.
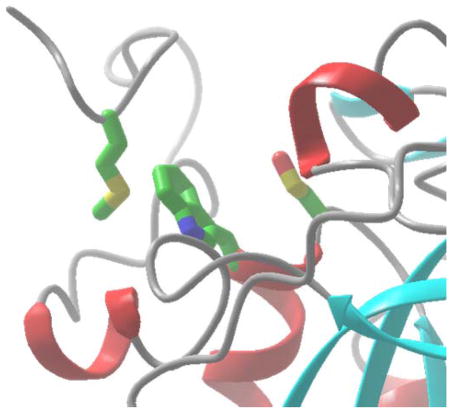
Figure 9.

Hypothesis: Methionine prevents sulfenylamide formation through bonding of Met229 to the aromatic ring of Trp74. The active site region and the carboxyl terminal domain are shown in this figure. H2O2 oxidizes the active site Cys72 to the sulfenic acid (red arrow in panel A) which can be further oxidized by peroxide because a Met229-Trp74 bond has formed (black solid line). In panel B, Leu229 cannot form such a bond, allowing the sulfenic acid to form a sulfenylamide with the amide of Trp74 (blue arrow). The figure was prepared by James Gruschus.
Of the mammalian msrA whose sequences are available, the mouse and the Rhesus monkey have such a carboxyl-domain methionine residue. As noted in the results, sulfur containing amino acids are often nutritionally limiting for animals living in the wild. The introduction of a methionine residue through evolution would be expected to have a functional advantage. The finding that the carboxyl terminal domain methionine in msrA renders its active site cysteine much more susceptible to hyperoxidation is therefore unexpected. While the added methionine residue may have an as-yet undiscovered function, the simplest explanation would be that hyperoxidation of the active site cysteine has a function or advantage. We considered two possible advantages, namely an increased catalytic efficiency or an induction of oligomerization. Our experimental data demonstrated that neither occurred. At present, neither the mouse nor the monkey has revealed to us the advantage of the carboxyl terminal methionine residue.
Acknowledgments
Funding
This research was supported by the Intramural Research Division of the National Heart, Lung, and Blood Institute (ZIA HL000225).
We thank Dr. James M. Gruschus for advice and for preparing Figure 9.
ABBREVIATIONS
- C3S
C107S/C218S/C227S
- C3S-M229K
C107S/C218S/C227/M229K
- C3S-M229Q
C107S/C218S/C227/M229Q
- C3S Δ228-233
C107S/C218S/C227S/Δ228-233
- DTPA
diethylenetriamine-pentaacetic acid
- MetO
methionine sulfoxide
- msr
methionine sulfoxide reductase
- msrA
methionine sulfoxide reductase A
Footnotes
The authors declare that they have no competing financial interests.
References
- 1.Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 2.Rhee SG. H2O2, a Necessary Evil for Cell Signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 3.Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, Rhee SG. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem. 1997;272:217–221. [PubMed] [Google Scholar]
- 4.Arner ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem. 2000;267:6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 5.Brot N, Weissbach H. Biochemistry and physiological role of methionine sulfoxide reductase in proteins. Arch Biochem Biophys. 1983;223:271–281. doi: 10.1016/0003-9861(83)90592-1. [DOI] [PubMed] [Google Scholar]
- 6.Zhang XH, Weissbach H. Origin and evolution of the protein-repairing enzymes methionine sulphoxide reductases. Biol Rev Camb Philos Soc. 2008;83:249–257. doi: 10.1111/j.1469-185X.2008.00042.x. [DOI] [PubMed] [Google Scholar]
- 7.Kim G, Cole NB, Lim JC, Zhao H, Levine RL. Dual sites of protein initiation control the localization and myristoylation of methionine sulfoxide reductase A. J Biol Chem. 2010;285:18085–18094. doi: 10.1074/jbc.M110.119701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao H, Sun J, Deschamps AM, Kim G, Liu C, Murphy E, Levine RL. Myristoylated Methionine Sulfoxide Reductase A Protects the Heart from Ischemia-Reperfusion Injury. Am J Physiol Heart Circ Physiol. 2011;301:H1513–H1518. doi: 10.1152/ajpheart.00441.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moskovitz J, Berlett BS, Poston JM, Stadtman ER. The yeast peptide-methionine sulfoxide reductase functions as an antioxidant in vivo. Proc Natl Acad Sci USA. 1997;94:9585–9589. doi: 10.1073/pnas.94.18.9585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moskovitz J, Rahman MA, Strassman J, Yancey SO, Kushner SR, Brot N, Weissbach H. Escherichia coli peptide methionine sulfoxide reductase gene: regulation of expression and role in protecting against oxidative damage. J Bacteriol. 177:502–507. doi: 10.1128/jb.177.3.502-507.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weissbach H, Resnick L, Brot N. Methionine sulfoxide reductases: history and cellular role in protecting against oxidative damage. Biochim Biophys Acta. 2005;1703:203–212. doi: 10.1016/j.bbapap.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 12.Boschi-Muller S, Olry A, Antoine M, Branlant G. The enzymology and biochemistry of methionine sulfoxide reductases. Biochim Biophys Acta. 2005;1703:231–238. doi: 10.1016/j.bbapap.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 13.Kim HY, Gladyshev VN. Methionine sulfoxide reductases: selenoprotein forms and roles in antioxidant protein repair in mammals. Biochem J. 2007;407:321–329. doi: 10.1042/BJ20070929. [DOI] [PubMed] [Google Scholar]
- 14.Levine RL, Moskovitz J, Stadtman ER. Oxidation of methionine in proteins: Roles in antioxidant defense and cellular regulation. IUBMB Life. 2000;50:301–307. doi: 10.1080/713803735. [DOI] [PubMed] [Google Scholar]
- 15.Lim JC, You Z, Kim G, Levine RL. Methionine sulfoxide reductase A is a stereospecific methionine oxidase. Proc Natl Acad Sci USA. 2011;108:10472–10477. doi: 10.1073/pnas.1101275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lowther WT, Brot N, Weissbach H, Honek JF, Matthews BW. Thiol-disulfide exchange is involved in the catalytic mechanism of peptide methionine sulfoxide reductase. Proc Natl Acad Sci USA. 2000;97:6463–6468. doi: 10.1073/pnas.97.12.6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boschi-Muller S, Azza S, Sanglier-Cianferani S, Talfournier F, Van Dorsselear A, Branlant G. A sulfenic acid enzyme intermediate is involved in the catalytic mechanism of peptide methionine sulfoxide reductase from Escherichia coli. J Biol Chem. 2000;275:35908–35913. doi: 10.1074/jbc.M006137200. [DOI] [PubMed] [Google Scholar]
- 18.Lim JC, Gruschus JM, Kim G, Berlett BS, Tjandra N, Levine RL. A Low pKa cysteine at the active site of mouse methionine sulfoxide reductase A. J Biol Chem. 2012;275:25596–25601. doi: 10.1074/jbc.M112.369116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wood PM. The redox potential for dimethyl sulphoxide reduction to dimethyl sulphide: evaluation and biochemical implications. FEBS Lett. 1981;124:11–14. doi: 10.1016/0014-5793(81)80042-7. [DOI] [PubMed] [Google Scholar]
- 20.Klomsiri C, Karplus PA, Poole LB. Cysteine-based redox switches in enzymes. Antioxid Redox Signal. 2011;14:1065–1077. doi: 10.1089/ars.2010.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 22.Orian L, Mauri P, Roveri A, Toppo S, Benazzi L, Bosello-Travain V, De Palma A, Maiorino M, Miotto G, Zaccarin M, Polimeno A, Flohé L, Ursini F. Selenocysteine oxidation in glutathione peroxidase catalysis: an MS-supported quantum mechanics study. Free Radic Biol Med. 2015;87:1–14. doi: 10.1016/j.freeradbiomed.2015.06.011. [DOI] [PubMed] [Google Scholar]
- 23.Kim G, Selengut J, Levine RL. Carbonic anhydrase III: the phosphatase activity is extrinsic. Arch Biochem Biophys. 2000;377:334–340. doi: 10.1006/abbi.2000.1793. [DOI] [PubMed] [Google Scholar]
- 24.Luo S, Wehr NB, Levine RL. Quantitation of protein on gels and blots by infrared fluorescence of Coomassie blue and Fast Green. Anal Biochem. 2006;350:233–238. doi: 10.1016/j.ab.2005.10.048. [DOI] [PubMed] [Google Scholar]
- 25.Lim JC, Gruschus JM, Ghesquiere B, Kim G, Piszczek G, Tjandra N, Levine RL. Characterization and solution structure of mouse myristoylated methionine sulfoxide reductase A. J Biol Chem. 2012;287:25589–25595. doi: 10.1074/jbc.M112.368936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lowther WT, Brot N, Weissbach H, Matthews BW. Structure and mechanism of peptide methionine sulfoxide reductase, an “anti-oxidation” enzyme. Biochemistry. 2000;39:13307–13312. doi: 10.1021/bi0020269. [DOI] [PubMed] [Google Scholar]
- 27.Taylor AB, Benglis DM, Jr, Dhandayuthapani S, Hart PJ. Structure of Mycobacterium tuberculosis methionine sulfoxide reductase A in complex with protein-bound methionine. J Bacteriol. 2003;185:4119–4126. doi: 10.1128/JB.185.14.4119-4126.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chin D, Means AR. Methionine to glutamine substitutions in the C-terminal domain of calmodulin impair the activation of three protein kinases. J Biol Chem. 1996;271:30465–30471. doi: 10.1074/jbc.271.48.30465. [DOI] [PubMed] [Google Scholar]
- 29.Lim JC, Choi HI, Park YS, Nam HW, Woo HA, Kwon KS, Kim YS, Rhee SG, Kim K, Chae HZ. Irreversible oxidation of the active-site cysteine of peroxiredoxin to cysteine sulfonic acid for enhanced molecular chaperone activity. J Biol Chem. 2008;283:28873–28880. doi: 10.1074/jbc.M804087200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morgan RS, Tatsch CE, Gushard RH, McAdon J, Warme PK. Chains of alternating sulfur and pi-bonded atoms in eight small proteins. Int J Pept Protein Res. 1978;11:209–217. doi: 10.1111/j.1399-3011.1978.tb02841.x. [DOI] [PubMed] [Google Scholar]
- 31.Zauhar RJ, Colbert CL, Morgan RS, Welsh WJ. Evidence for a strong sulfur-aromatic interaction derived from crystallographic data. Biopolymers. 2000;53:233–248. doi: 10.1002/(SICI)1097-0282(200003)53:3<233::AID-BIP3>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 32.Valley CC, Cembran A, Perlmutter JD, Lewis AK, Labello NP, Gao J, Sachs JN. The Methionine-aromatic Motif Plays a Unique Role in Stabilizing Protein Structure. J Biol Chem. 2012;287:34979–34991. doi: 10.1074/jbc.M112.374504. [DOI] [PMC free article] [PubMed] [Google Scholar]