

Abstract
Background
Fanconi anemia (FA) is a chromosomal instability syndrome characterized by increased frequency of chromosomal breakages, chromosomal radial figures and accelerated telomere shortening. In this work we performed detailed molecular-cytogenetic characterization of breakpoints in primary lymphocytes of FA-D2 patients in different stages of the disease using fluorescent in situ hybridization.
Results
We found that chromosomal breakpoints co-localize on the molecular level with common fragile sites, whereas their distribution pattern depends on the severity of the disease. Telomere quantitative fluorescent in situ hybridization revealed that telomere fusions and radial figures, especially radials which involve telomere sequences are the consequence of critically shortened telomeres that increase with the disease progression and could be considered as a predictive parameter during the course of the disease. Sex chromosomes in FA cells are also involved in radial formation indicating that specific X chromosome regions share homology with autosomes and also could serve as repair templates in resolving DNA damage.
Conclusions
FA-D2 chromosomal breakpoints co-localize with common fragile sites, but their distribution pattern depends on the disease stage. Telomere fusions and radials figures which involve telomere sequences are the consequence of shortened telomeres, increase with disease progression and could be of predictive value.
Keywords: Fanconi anemia, Fragile sites, Telomere fusions, Radial figures, X chromosome
Background
Fanconi anemia (FA) is a rare, inherited, genetically heterogeneous chromosomal instability syndrome. To date, 19 different complementation groups, which correspond to distinct DNA repair genes have been identified, FA-A, FA-B, FA-C, FA-D1 (BRCA2), FA-D2, FA-E, FA-F, FA-G, FA-I, FA-J (BRIP1), FA-M, FA-N (PALB2), FA-O (RAD51C), FA-P (SLX4), FA-Q (ERCC4), FA-S (BRCA1), FA-R (RAD51) and FA-T (UBE2T) [1]. The genes that correspond to different FA complementation groups are involved in the FA/BRCA DNA damage repair pathway having an essential function in the cellular response to stress induced by DNA alkylating agents [2]. The most frequent complementation group in Serbia is FA-D2 [3], which is in contrast to the FA-D2 frequency in the general population where it accounts for approximately 4 % of all complementation groups [4]. FA-D2 patients are characterized by a broad spectrum of congenital abnormalities and, unlike most of the other FA complementation groups, an early onset of hematological manifestations including acute myelogenous leukemia and bone marrow failure [5]. One of the most significant FA cellular characteristics is an increase of chromosomal aberrations induced by interstrand cross-linking (ICL) agents [6]. When exposed to these agents, FA cells show an increased frequency of chromosomal breakages and chromosomal radial figures, prolonged cell-cycle arrest in G2/M phase, reduced cellular survival and accelerated telomere shortening [7]. With respect to chromosomal breakages, Schoder and coworkers [8] reported that chromosomal breakpoints in cells derived from FA-A and FA-C patients co-localize with the chromosomal regions that are constitutionally prone to breakage in each individual, known as common fragile sites (CFSs). In addition to standard FS classification, it was shown that cells exposed to replication stress also display breakages in telomeric regions; therefore they were attributed to CFSs [9]. Recent studies of telomere maintenance in FA cells showed that telomere fusions arise either as a consequence of their critical shortening or altered capping function. High percentage of dysfunctional telomeres and marked telomere fragility was found to be typical for FA-D2 cellular phenotype [10]. However, the role of telomeres in formation of induced chromosomal aberrations such as radial figures has not been determined, yet.
The aim of this study was to provide the first molecular-cytogenetic characterization of breakpoint distribution and co-localization with FSs, formation of radial figures and involvement of telomeres in formation of chromosomal aberrations in FA-D2 primary lymphocytes originated from patients in different stages of the disease, severe and mild bone marrow failure stage.
Methods
Patients and sampling
A total of six patients (five females, one male) previously diagnosed with FA-D2 [3, 10] were included in our study. Mean age of children at the time of study was 8 ± 5 years. Routine control of those children was undertaken at the Mother and Child Health Care Institute of Serbia, Hematooncology department. Disease stage was determined based on complete blood count (CBC) and bone marrow examination using standard criteria. Children were divided into two groups accordingly: severe (group A) or mild (group B) bone marrow failure (BMF). Informed consents from the families were obtained and peripheral blood was collected in Li-heparin vaccutainers for additional testing. The study was approved by The Ethical Committee of Mother and Child Health Care Institute of Serbia.
Whole blood cultures
Aliquots of heparinized whole blood (0.5 ml) were set up in cultures containing PBmax-karyotyping medium (Invitrogen-Gibco, Paisley, UK) and treated with diepoxibuthan (DEB, Sigma Chemicals Co., Germany) (final concentration 0.1 μg/ml) 48 h after culture initiation and further harvested 72 h after initiation. Colchicine (Sigma-Aldrich, Munich, Germany) was added during the last 3 h (final concentration 2.5 μg/ml). Cells were collected by centrifugation and treated with hypotonic solution (0.56 % KCl). Cell suspensions were fixed in methanol/acetic acid (3:1), washed three times with fixative and dropped onto clean slides.
DAPI banding
Slides were incubated overnight at room temperature, dehydrated in series of ethanol (70, 95 and 100 %, respectively), 5 min each, and counterstained with 4′,6′-diamidino-2-phenylindole (DAPI)-containing Vectashield solution (Vector Laboratories Ltd, Peterborough, UK). DAPI selectively binds to heterochromatin regions, which produces a banding pattern known as inverse DAPI banding. According to the unique pattern of differentially stained regions each chromosome can be identified. Analysis was performed with a Zeiss-Axioimager A1 microscope and the ISIS imaging software package (MetaSystems, Altlussheim, Germany).
Locus-specific fluorescent in situ hybridization (Locus-specific FISH, BAC-FISH)
After determination of the most frequent chromosomal breakpoints by inverse DAPI staining, molecular characterization was performed by hybridization with appropriate bacterial artificial chromosome probes (BAC-probes) as described by Mrasek et al. [11]. In short, after removing DAPI, slides were incubated in pepsin solution for 5 min, washed in 1xPBS and fixed in 1 % formalin buffer solution. Prior to applying the BAC-probe, slides were dehydrated in series of ethanol (70, 90, 100 %, 5 min each), denatured in 70 % formamide solution for 2–3 min on a hot plate at 75 °C and immediately placed in 70 % ethanol (4 °C) to conserve target DNA single-stranded. Dehydration in 90 and 100 % ethanol followed. The probe mixture (the probe in hybridization buffer and COT1 Human DNA) was denatured in a thermocycler at 75 °C for 5 min, pre-annealed at 37 °C for 30 min and cooled down to 4 °C. Prepared denatured probes were applied to the slides and hybridized overnight in a dark humid chamber at 37 °C. After hybridization, the slides were washed in 4xSSC/0.2 % Tween20 at the room temperature and 1xSSC-solution (62–65 °C), dehydrated in series of ethanol, counterstained with DAPI solution and observed under the fluorescence microscope. The results were analyzed using ISIS software (MetaSystems, Germany). The chromosomal regions and associated BAC-probes used for hybridization are shown in Table 1.
Table 1.
BAC-probes used for fluorescence in situ hybridization for the most breakpoint-affected chromosomal regions
| Chromosomal region | Fragile site (FS) | FS typea | BAC-probe | Co-localization of FA breakpoints and FS |
|---|---|---|---|---|
| 1p13.3 | FRA1N | Aphidicolinb | RP11-242D10 | + |
| 1q21.2 | FRA1F | Aphidicolin | RP11-301 M17 | + |
| 1q42.2 | Near FRA1H | Azacytidin | RP11-109G24 | − |
| 2q35 | FRA2U | Aphidicolin | RP11-316O14 | + |
| 3p14.2 | FRA3B | Aphidicolin | RP11-129 K20 | + |
| 3p21.31 | FRA3H | New nomenclaturec | RP11-787O14 RP11-159A17 |
+ |
| 3q27.1 | FRA3C | Aphidicolin | RP11-110C15 | + |
| 5q13.2 | FRA5K | New nomenclatured | RP11-497H16 | + |
| 5q33.2 | FRA5O | New nomenclatured | RP11-265I24 RP11-494C5 |
+ |
| 7q22.3 | FRA7F | Aphidicolin | CTB-20D2 | + |
| 7q32.3 | FRA7H | Aphidicolin | RP11-138A9 RP11-36B6 |
+ |
| 11q13 | FRA11H | Aphidicolin | RP11-449G14 | + |
| 14q24.3 | FRA14G | Aphidicolinb | RP3-414A15 | + |
| 16q22.1 | FRA16C, FRA16B | Aphidicolin | RP11-106 J23 | + |
| 16q23 | FRA16D | Aphidicolin | RP11-358 L22 | + |
| 18q21.3 | FRA18B | Aphidicolin | RP11-15C15 | + |
BAC-probes for the appropriate FSs were selected according to the literature data:
aNomenclature according to Lukusa et al. [33]
bNew extended nomenclature according to Mrasek et al. [34]
cCommon according to the nomenclature by Borgaonkar [35]
dCommon according to the nomenclature by Simonic and Gericke [36]
Telomere quantitative fluorescent in situ hybridization (Q-FISH)
After metaphase analysis DAPI was removed, slides hybridized with telomere cPNA oligonucleotide probe (TTAGGG) as described by Slijepcevic [12]. Briefly, hybridization was performed with the Cy-3 labeled telomeric PNA probe (CCCTAA) 3′ supplemented with PNA centromeric probe for chromosome 2 (DAKO, Glostrup, Denmark) in final concentration 2 ng/ml. The PNA probe for centromere 2 was added in ready to use telomere probe. After hybridization slides were left in a dark humidified chamber for 2 h. The slides were then washed in 70 % formamide and stained with 4′,6′-diamidino-2-phenylindole (DAPI)-containing mounting medium (Vector Laboratories, UK). Analysis was performed using the ISIS software, MetaSystems (Altlussheim, Germany). Measurements were reported as arbitrary relative telomere length units (RTLU), which are defined as the ratio of signal intensity between telomeres and a centromere chromosome 2 reference signals.
Metaphase analysis
Metaphase spreads were analyzed according to the International System for Human Cytogenetic Nomenclature (ISCN, 2013). For each sample at least 1000 metaphases were analyzed for the breakpoint characterization, whereas at least 500 metaphases were scored for fusions and radial formation with regard to the chromosomes involved in their formation, and involvement of telomeres in arising of those aberrations.
Statistical analysis
FA breakpoints were expressed as the percentage of total number of breaks per patient. Telomere fusions and radial figures were presented as percentage of total number of metaphases analyzed and were statistically analyzed using nonparametric Mann-Whitney U test in the program SPSS 10 for Windows. Differences at p < 0.05 were considered as significant.
Results
FA breakpoints and assignment to FSs
Results of molecular cytogenetic characterization concerning chromosomal breakages, telomere fusions and radial figures in DEB treated peripheral blood derived from three FA-D2 patients in the severe stage of diseases (group A), and three patients in the mild stage of the disease (group B) are presented in Table 2.
Table 2.
FA-D2 patients’ karyotypes and molecular cytogenetic findings
| FA-D2 patient | Age (years) | Karyotype | Most frequent breakpoints | Fragile site (FS) | Breakpoint frequency (%) | Telomere fusion frequency (%) | Average telomere length (RTLUa ± SD) | Radial figures frequency (%) | |
|---|---|---|---|---|---|---|---|---|---|
| Group A | 1 | 8 | 46,XX | 1q42.2 | distal to FRA1H | 1.72 | 1.95 | 14.76 ± 3.23 | 7.06 |
| 14q24.3 | FRA14G | 3.09 | |||||||
| 18q21.3 | FRA18B | 1.72 | |||||||
| 2 | 6 | 46,XX | 2q35 | FRA2U | 2.4 | 1.54 | 23.02 ± 12.73 | 1.78 | |
| 3p14.2 | FRA3B | 2.4 | |||||||
| 5q33.2 | FRA5O | 2 | |||||||
| 7q32.3 | FRA7H | 1.6 | |||||||
| 3 | 17 | 46,XX | 3q27.1 | FRA3C | 2.75 | 1.62 | 35.72 ± 7.00 | 1.88 | |
| 5q13.2 | FRA5K | 2.75 | |||||||
| 5q33.2 | FRA5O | 3.29 | |||||||
| Group B | 4 | 3 | 46,XX | 3p14.2 | FRA3B | 11.07 | 0.08 | 20.48 ± 12.24 | 0.78 |
| 3p21.31 | FRA3H | 3.95 | |||||||
| 7q32.3 | FRA7H | 1.19 | |||||||
| 5 | 3 | 46,XX,der(21)t(15;21)(q15;p11.1)[5]/46,XX [15] | 1p13 | FRA1N | 1.76 | 0.72 | 21.01 ± 10.33 | 0.715 | |
| 1q21 | FRA1F | 1.32 | |||||||
| 3p21.31 | FRA3H | 1.32 | |||||||
| 11q13 | FRA11H | 0.88 | |||||||
| 6 | 11 | 46, XY, der(16)t(1;16)(q42;p11.2)del(1) (q42)[1]/46,XY[31] | 3p14.2 | FRA3B | 1.88 | 0.23 | 23.91 ± 6.92 | 0.23 | |
| 3p21.31 | FRA3H | 0.63 | |||||||
| 16q22.1 | FRA16C | 1.88 | |||||||
| 16q23 | FRA16D | 6.88 |
aRTLU–relative telomere length units
The FA breakpoint analysis showed that frequency of breakages, and breakpoint distribution pattern depend on the stage of the disease: most of the breakpoints in both groups of patients corresponded to CFSs, except for patient 1 in group A where one of the most frequent breaks was 1q42.2 (Fig. 1). In the group A patients, breakpoints were dispersed throughout the genome with a much lower frequency of breakage at the particular chromosomal locus, i.e. the most frequent breakpoints within this group did not exceed 3.297 % of the total number of breaks.
Fig. 1.
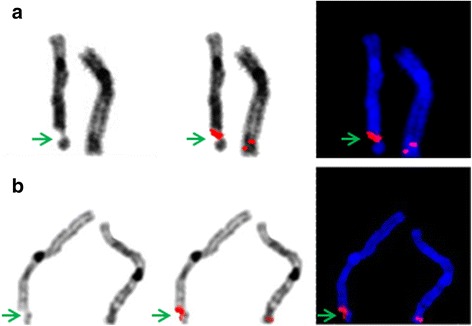
Representative images of BAC-FISH in FA-D2 metaphases, inverted DAPI and DAPI staining. Arrows indicate breakpoints and hybridized BAC-probes; RP11-265I24 located within FRA5O (a), and RP11-109G24 located within 1q42.2, near FRA1H (b)
In contrast, chromosomes in patients of B group tended to break with the higher frequency in the chromosomal regions where the most CFSs are reported (Table 2). Interindividual variability in frequency of the most common breakages was significant in this group, but the breakage pattern in two patients from this group was similar: in patients 4 and 6 the highest frequency of breakage was observed in regions that correspond to FRA3B (11.07 %) and FRA16D (6.87 %), respectively. However, patient 5 had a different breakage pattern compared to other two patients within this group, where low frequencies of the common breakages were found. However, this patient also shows an unbalanced karyotype, mos 46,XX,der(21)t(15;21)(q15;p11.1)[5]/46,XX[15].
Telomeres and radial figures
Frequency of telomere fusions and radial figures, with special regard to their structure and telomere involvement in radial figures formation, are presented in Table 2 and Figs. 2, 3 and 6.
Fig. 2.
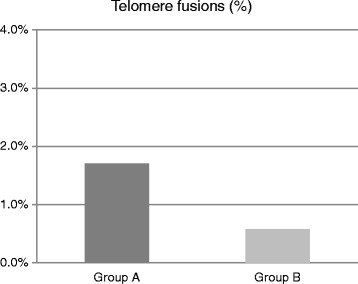
Percentage of telomere fusions in group A and B patients. Group A display more fusions than group B
Fig. 3.
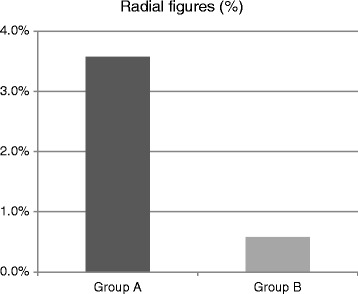
Percentage of radial figures in group A and B patients. In Group A incidence of radials was six fold higher compared to group B
Fig. 6.
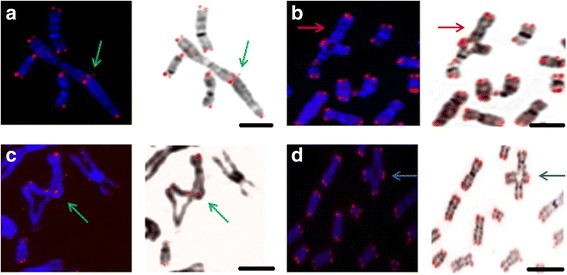
Representative images of telomere FISH in FA-D2 metaphases. Telomere signals at the fusion points are indicated by green arrow: telomere end-to-end fusion, three signals are present between two chromosomes (a), radial figure between chromosomes X and 13, red arrow indicates X chromosome in radial figure (b), green arrow indicates radial figure between telomere of one and interstitial region of another chromosome (c), blue arrow indicates simple radial figure (d). Scale bars = 10 μm
The results showed significantly higher frequency of telomere fusions in patients from group A versus group B (p = 0.05, Fig. 2). Within A group of patients, telomere fusions occurred most frequently between chromosomes 2p, 2q, 5q and 18q, whereas in group B fusions mostly occurred between 6q, 7q, 9p, 10q, 18q (Fig. 4). Relative quantification of telomere length revealed that the average telomere length (RTLU) was not significantly different between two groups (p = 0.442). However, quantification of telomere length at single chromosomes showed that p/q ratio ranged from 0.41 to 2.18 and that chromosomes with the shortest telomeres constituted telomere end-to-end fusions.
Fig. 4.
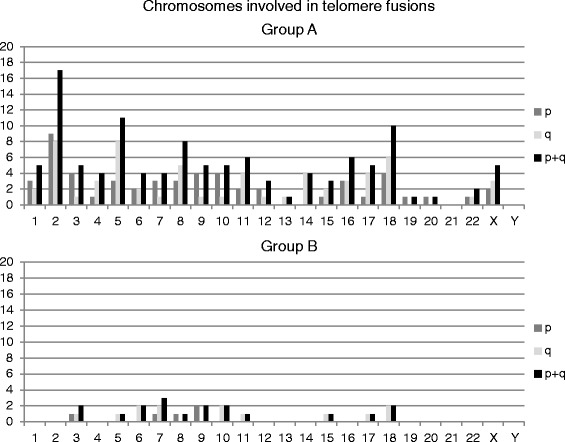
Chromosomes involved in telomere fusions in group A and B
The percentage of radial figures was low in group B (below 1 %), whereas in group A the percentage was on average six times higher than in group B (p = 0.05, Fig. 3). Regarding radial figure structure, great percentage of radials found in group A was a consequence of exchange between telomeres of one and non-telomeric, interstitial regions of other chromosomes, 25.49 % (50 % of all radials in patient 1, and 22.7 % for patients 2 and 3, each). Only two radials between telomeres and interstitial chromosomal sequences were observed in patient 4 and 5, and neither one was observed in patient 6. Although radials always arose between non-homologous chromosomes, distribution of involved chromosomes was heterogeneous–chromosomes 1, 2, 9, 10 and 14 were frequently present in radial figures in group A patients, and chromosome 1 was most frequent in group B. Chromosome X was found to be involved in radial figures in all group A patients and one group B patient (Fig. 5). Representative images of telomere fusions, radials and X chromosome in radial formation are presented in the Fig. 6.
Fig. 5.
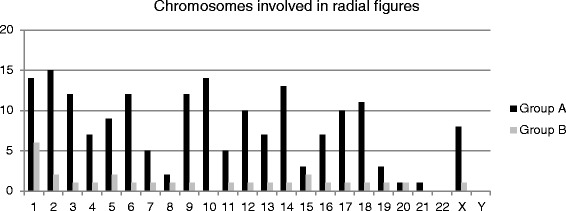
Chromosomes involved in radial figures in both patient groups
Discussion
The results of our study revealed that chromosomes are not equally prone to breakages in FA-D2 cells exposed to DEB. Matching the data of DAPI banding and BAC-FISH pointed out that regions with increased frequency of breakages co-localize with the CFSs, indicating that frequency of breakpoints as well as their distribution pattern is different among patients and is related to the disease stage.
Assignment of breakpoints to FS in FA-D2 cells is in accordance to previously reported data for FA-A and FA-C cell lines, although the disease stage for the patients in this study was not determined [8]. In FA-D2 patients in the severe stage of the disease (group A) the most frequent breakpoints were present within the CFSs regions (FRA14G, FRA5O, FRA5K and FRA3C, Table 2) except for patient 1 in group A where one of the most frequent breaks was 1q42.2 which is near the FRA1H [13]. However, previously it was not attributed as FS. Their distribution pattern was similar to that found in FA-A and FA-C patients, although different breakpoints were observed in these groups (FRA1A, FRA1D, FRA1E and FRA1J) [8]. The average incidence of chromosomal breaks didn’t increase with the disease progression, but the breakpoint distribution pattern was quite different. Mild stage patients (group B) displayed higher frequencies of breakpoints within the most common FSs, FRA3B and FRA16D except for the patient with unbalanced karyotype (Table 2).
FA pathway plays an essential role in regulation of maintenance and stability of CFSs [14]. Howlett et al. [15], was the first who establishes that disrupted FA pathway leads to increased chromosomal instability at CFSs underlying that their stability depends on the ATR-FANCD2 signaling pathway, i.e. following DNA replication stress, ATR kinase phosphorylates FANCD2 promoting its monoubiqutination, which activates homologous recombination (HR). Replication stress leads to accumulation of FANCD2 protein in the chromosomal regions prone to breakage, whether they are broken or not [16]; however altered FANCD2 protein in FA-D2 cells couldn’t maintain stability of existing FSs which in turn activates alternative mechanisms of repair. Apart from HR the CFSs are maintained by non-homologous end-joining (NHEJ) mechanisms, as well as with specialized polymerases that perform by-pass function in DNA synthesis [17, 18].
On the other hand, CFSs are AT-rich sequences prone to form DNA secondary structures which can lead to the replication fork collapse [19]. In addition, replication timing is delayed along CFSs, whereas exposure to DNA polymerase inhibitors intensifies the deceleration of the fork progression, so the CFSs enter G2 phase unreplicated [20]. Both give rise for chromosomal breaks.
Increasing body of evidence now show that large genes constitute the pool of CFSs [21] indicating relationship between instability and transcription of these genes. Since more than one cell cycle is required for their transcription, transcription and replication machineries collide and CFSs expression may occur [22]. Although different FS distribution pattern with the disease progression remains unclear, it is possible that with the progression of the disease different genes could be expressed as a cellular attempt to compensate progressive genomic instability, leading to different CFSs expression.
Up to now, 19 CFSs have been molecularly characterized [23]. Among them are the most common FSs (FRA3B and FRA16D, Table 3) which both contain very large genes (>1 Mbp) important for tumor suppression. These FSs are, along with FRA7H, most frequently expressed in mild stage. In severe stage of the disease different FSs appear as the most common. These chromosomal regions are also very large, extending from 2 to 6 Mbp, but are comprised by a great number of genes (Table 3); many of those are included in control of cell cycle and tumor suppression. For example, BCL2 gene (spans approximately 200 kbp), which is the first discovered anti-apoptotic gene, is located within FRA18B, whereas it is known that breakpoints and translocations within this region disrupt its function and lead to myeloproliferative diseases [24]. Similarly, protooncogene C-FOS, located within FRA14G, also has important role in regulation of cell proliferation and differentiation [25].
Table 3.
Genes located within common fragile sites according to the literature dataa, b, c, d
| Fragile site | Chromosomal region | Start | End | Gene | Gene length |
|---|---|---|---|---|---|
| Molecularly characterized fragile sites | |||||
| FRA3B | 3p14.2 | 58 600 001 | 63 800 000 | FHITa | >1.5 Mbp |
| FRA7H | 7q32.3 | 130 800 001 | 132 900 000 | /b | / |
| FRA16D | 16q23.2 | 79 200 001 | 81 600 000 | WWOXc | 1.1 Mbp |
| Not-characterized fragile sitesd | |||||
| FRA1F | 1q21.2 | 147 500 001 | 150 600 000 | 33 genes | / |
| FRA1N | 1p13.3 | 106 700 001 | 111 200 000 | 52 genes | / |
| FRA2U | 2q35 | 214 500 001 | 220 700 000 | 70 genes | / |
| FRA3C | 3q27.1 | 183 000 001 | 184 800 000 | 20 genes | / |
| FRA3H | 3p21.31 | 44 200 001 | 50 600 000 | 78 genes | / |
| FRA5K | 5q13.2 | 69 100 001 | 74 000 000 | 25 genes | / |
| FRA5O | 5q33.2 | 153 300 001 | 156 300 000 | 8 genes | / |
| FRA7F | 7q22.3 | 104 900 001 | 107 800 000 | 14 genes | / |
| FRA11H | 11q13.4 | 70 500 001 | 75 500 000 | 48 genes | / |
| FRA14G | 14q24.3 | 73 300 001 | 78 800 000 | 69 genes | / |
| FRA16C | 16q22.1 | 66 600 001 | 70 800 000 | 95 genes | / |
| FRA18B | 18q21.3 | 61 300 001 | 63 900 000 | 10 genes | / |
Patients in the severe stage of the disease have significantly higher percentage of telomere fusions compared to the patients in the mild stage. Interestingly, there was no significant difference in average telomere length between two groups of patients, but measurement of telomere length at each individual chromosome revealed that the chromosomes with the shortest telomeres were most frequently involved in telomere fusions. Our previous research showed that lymphocyte telomeres in FA-D2 patients are shortened when compared to the age-matched control [10]. Taking into account that FA cells are breakage prone, increased breakages at telomeres and complexity of their function could be the cause of their shortening. Recent review of Holohan et al. [26] pointed out that impaired telomere maintenance and attrition is a hallmark of FA-D2 group, not FA in general. Our previous study showed that FA-D2 cells displayed heterogeneous telomere length and high frequency of double-strand breaks in telomere regions (telomere dysfunction–induced foci - TIFs) which lead to telomere fragility [10]. Altered FANCD2 protein is not capable to maintain telomeres, leaving the telomeres unprotected and prone to increased fragility and attrition.
FA-D2 patients in the severe stage had significantly more radials than the patients in the mild phase of the disease, particularly radials formed between telomeres of one and interstitial chromosomal regions of other chromosomes, which was rarely observed in group B. This is unreported finding. Since shortened and fragile telomeres act as double strand breaks (DSB) interchromosomal recombination with other impaired chromosomal regions in attempt to repair the damage is not surprising. Additionally, two patients in severe stage developed bone marrow failure several months after cytogenetic examination and became candidates for bone marrow transplantation.
In both groups of patients, radials were composed of non-homologous chromosomes, which is consistent with the previously reported results [27, 28]. Distribution of involved chromosomes was heterogeneous and not specific for the disease stage. However, X chromosome was found in radials in both groups of patients, which is opposite to the report of Newell and colleagues [27], who found that sex chromosomes are not involved in FA-A and FA-G cells and suggested that alternative mechanisms of ICL repair, which avoid recombination between sex and autosome chromosomes, play the main repair role in FA cells. Involvement of Y chromosome was not observed, but only one male patient in the mild stage and with very low number of radials was part of the study. The presence of sex chromosomes in radial figures in FA patients was not previously reported.
Although radials are described in many genomic instability syndromes, only a few studies clarified the mechanism of their formation. Kuhn and Therman [29] and Scully et al. [30], suggested that they arise as an attempt of cellular repair machinery to resolve DSBs, especially when cells with hampered repair machinery are exposed to ICL-s employing homologous recombination [31, 32]. Radials formation in FA cells between non-homologous chromosomes, as well as between autosomes and sex chromosomes indicate that short dispersed chromosomal regions that share homology could act as repair templates in resolving the DNA damage. In FA-D2 cells the X chromosome is not spared from such recombination.
Conclusions
Overall, our results indicate that regions with increased frequency of breakage co-localize with CFSs, whereas their distribution pattern relates to the severity of the disease.
Telomere fusions, as well as interchromosomal recombination and radials formation which involve telomere sequences are the consequence of critically shortened telomeres, increase with disease progression and could be of predictive value.
Acknowledgement
The authors are thankful to patient, parents and medical staff of the Institute of mother and Child Health Care of Serbia. This work is financially supported by Ministry of Education, Science and Technological Development of the Republic of Serbia ON173046 and DAAD project No 57127754.
Funding
This study was funded by Ministry of Education, Science and Technological Development of the Republic of Serbia ON173046 and DAAD project No 57127754.
Availability of data and materials
The data sets supporting the conclusions of this article are included within the article. More details are available upon request.
Authors’ contributions
JF and IJ performed experiments and analytical procedures, and drafted the manuscript. GJ, DV, KM, AW and TL supervised the experiments, edited and approved the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national). Informed consent was obtained from all patients for being included in the study. Ethics committee of Mother and Child Health Care Institute of Serbia “Dr Vukan Cupic” approved the study and the approval is available upon request.
Contributor Information
Jelena Filipović, Email: filipovicj@vinca.rs.
Gordana Joksić, Phone: +381113408565, Email: gjoksic@vinca.rs.
Dragana Vujić, Email: vujicd@ptt.rs.
Ivana Joksić, Email: ivanajoksic@yahoo.com.
Kristin Mrasek, Email: Kristin.Mrasek@med.uni-jena.de.
Anja Weise, Email: anja.weise@med.uni-jena.de.
Thomas Liehr, Email: Thomas.Liehr@med.uni-jena.de.
References
- 1.Dong H, Nebert DW, Bruford EA, Thompson DC, Joenje H, Vasiliou V. Update of the human and mouse Fanconi anemia genes. Hum Genomics. 2015;9:32. doi: 10.1186/s40246-015-0054-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kee Y, D’Andrea AD. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Gene Dev. 2010;24:1680–1694. doi: 10.1101/gad.1955310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vujic D, Petrovic S, Lazic E, Kuzmanovic M, Leskovac A, Joksic I, Micic D, Jovanovic A, Zecevic Z, Guc-Scekic M, Cirkovic S, Joksic G. Prevalence of FA-D2 Rare Complementation Group of Fanconi Anemia in Serbia. Indian J Pediatr. 2014;81:260–265. doi: 10.1007/s12098-013-1284-4. [DOI] [PubMed] [Google Scholar]
- 4.Whitney M, Thayer M, Reifsteck C, Olson S, Smith L, Jakobs P, Leach R, Naylor S, Joenje H, Grompe M. Microcell mediated chromosome transfer maps the Fanconi anaemia group D gene to chromosome 3p. Nat Genet. 1995;11:341–343. doi: 10.1038/ng1195-341. [DOI] [PubMed] [Google Scholar]
- 5.Kalb R, Neveling K, Hoehn H, Schneider H, Linka Y, Batish SD, Hunt C, Berwick M, Callen E, Surralles J, Casado JA, Bueren J, Dasi A, Soulier J, Gluckman E, Zwaan CM, van Spaendonk R, Pals G, de Winter JP, Joenje H, Grompe M, Auerbach AD, Hanenberg H, Schindler D. Hypomorphic mutations in the gene encoding a key Fanconi anemia protein, FANCD2, sustain a significant group of FA-D2 patients with severe phenotype. Am J Hum Genet. 2007;80:895–910. doi: 10.1086/517616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res. 2009;668:4–10. doi: 10.1016/j.mrfmmm.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adelfalk C, Lorenz M, Serra V, von Zglinicki T, Hirsch-Kauffmann M, Schweiger M. Accelerated telomere shortening in Fanconi anemia fibroblasts–a longitudinal study. FEBS Lett. 2001;506:22–26. doi: 10.1016/S0014-5793(01)02869-1. [DOI] [PubMed] [Google Scholar]
- 8.Schoder C, Liehr T, Velleuer E, Wilhelm K, Blaurock N, Weise A, Mrasek K. New aspects on chromosomal instability: Chromosomal break-points in Fanconi anemia patients co-localize on the molecular level with fragile sites. Int J Oncol. 2010;36:307–312. [PubMed] [Google Scholar]
- 9.Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joksic I, Vujic D, Guc-Scekic M, Leskovac A, Petrovic S, Ojani M, Trujillo JP, Surralles J, Zivkovic M, Stankovic A, Slijepcevic P, Joksic G. Dysfunctional telomeres in primary cells from Fanconi anemia FANCD2 patients. Genome Integr. 2012;3:6. doi: 10.1186/2041-9414-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mrasek K, Wilhelm K, Quintana LG, Theuss L, Liehr T, Leskovac A, Filipovic J, Joksic G, Joksic I, Weise A. BAC-probes applied for characterization of fragile sites (FS) Methods Mol Biol. 2015;1227:289–298. doi: 10.1007/978-1-4939-1652-8_15. [DOI] [PubMed] [Google Scholar]
- 12.Slijepcevic P. Telomere length measurement by Q-FISH. Methods Cell Sci. 2001;23:17–22. doi: 10.1023/A:1013177128297. [DOI] [PubMed] [Google Scholar]
- 13.Curatolo A, Limongi ZM, Pelliccia F, Rocchi A. Molecular Characterization of the Human Common Fragile Site FRA1H. Genes Chromosomes Cancer. 2007;46:487–493. doi: 10.1002/gcc.20432. [DOI] [PubMed] [Google Scholar]
- 14.Durkin SG, Glover TW. Chromosome fragile sites. Annu Rev Genet. 2007;41:169–192. doi: 10.1146/annurev.genet.41.042007.165900. [DOI] [PubMed] [Google Scholar]
- 15.Howlett NG, Taniguchi T, Durkin SG, D’Andrea AD, Glover TW. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum Mol Genet. 2005;14:693–701. doi: 10.1093/hmg/ddi065. [DOI] [PubMed] [Google Scholar]
- 16.Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol. 2009;11:753–760. doi: 10.1038/ncb1882. [DOI] [PubMed] [Google Scholar]
- 17.Schwartz M, Zlotorynski E, Goldberg M, Ozeri E, Rahat A, Sage C, Chen BP, Chen DJ, Agami R, Kerem B. Homologous recombination and nonhomologous end-joining repair pathways regulate fragile site stability. Gene Dev. 2005;19:2715–2726. doi: 10.1101/gad.340905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergoglio V, Boyer AS, Walsh E, Naim V, Legube G, Lee MY, Rey L, Rosselli F, Cazaux C, Eckert KA, Hoffmann JS. DNA synthesis by Pol eta promotes fragile site stability by preventing under-replicated DNA in mitosis. J Cell Biol. 2013;201:395–408. doi: 10.1083/jcb.201207066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shah SN, Opresko PL, Meng X, Lee MY, Eckert KA. DNA structure and the Werner protein modulate human DNA polymerase δ-dependent replication dynamics within the common fragile site FRA16D. Nucleic Acids Res. 2010;38:1149–1162. doi: 10.1093/nar/gkp1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zlotorynski E, Rahat A, Skaug J, Ben-Porat N, Ozeri E, Hershberg R, Levi A, Scherer SW, Margalit H, Kerem B. Molecular basis for expression of common and rare fragile sites. Mol Cell Biol. 2003;23:7143–7151. doi: 10.1128/MCB.23.20.7143-7151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Le Tallec B, Koundrioukoff S, Wilhelm T, Letessier A, Brison O, Debatisse M. Updating the mechanisms of common fragile site instability: how to reconcile the different views? Cell Mol Life Sci. 2014;71:4489–4494. doi: 10.1007/s00018-014-1720-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helmrich A, Ballarino M, Tora L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell. 2011;44:966–977. doi: 10.1016/j.molcel.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 23.Ma K, Qiu L, Mrasek K, Zhang J, Liehr T, Quintana LG, Li Z. Common fragile sites: genomic hotspots of DNA damage and carcinogenesis. Int J Mol Sci. 2012;13:11974–11999. doi: 10.3390/ijms130911974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene. 2008;27:6398–6406. doi: 10.1038/onc.2008.307. [DOI] [PubMed] [Google Scholar]
- 25.Milde-Langosch K. The Fos family of transcription factors and their role in tumourigenesis. Eur J Cancer. 2005;41:2449–2461. doi: 10.1016/j.ejca.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 26.Holohan B, Wright WE, Shay JW. Telomeropathies: An emerging spectrum disorder. J Cell Biol. 2014;205:289–299. doi: 10.1083/jcb.201401012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Newell AE, Akkari YM, Torimaru Y, Rosenthal A, Reifsteck CA, Cox B, Grompe M, Olson SB. Interstrand crosslink-induced radials form between non-homologous chromosomes, but are absent in sex chromosomes. DNA Repair (Amst) 2004;3:535–542. doi: 10.1016/j.dnarep.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 28.Owen N, Hejna J, Rennie S, Mitchell A, Hanlon Newell A, Ziaie N, Moses RE, Olson SB. Bloom syndrome radials are predominantly non-homologous and are suppressed by phosphorylated BLM. Cytogenet Genome Res. 2014;144:255–263. doi: 10.1159/000375247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuhn EM, Therman E. Origin of symmetrical triradial chromosomes in human cells. Chromosoma. 1982;86:673–681. doi: 10.1007/BF00285610. [DOI] [PubMed] [Google Scholar]
- 30.Scully R, Puget N, Vlasakova K. DNA polymerase stalling, sister chromatid recombination and the BRCA genes. Oncogene. 2000;19:6176–6183. doi: 10.1038/sj.onc.1203971. [DOI] [PubMed] [Google Scholar]
- 31.Joksic I, Petrovic S, Leskovac A, Filipovic J, Guc-Scekic M, Vujic D, Joksic G. Enhanced frequency of sister chromatid exchanges induced by diepoxybutane is specific characteristic of Fanconi anemia cellular phenotype. Genetika. 2013;45:393–403. doi: 10.2298/GENSR1302393J. [DOI] [Google Scholar]
- 32.Thyagarajan B, Campbell C. Elevated homologous recombination activity in Fanconi anemia fibroblasts. J Biol Chem. 1997;272:23328–23333. doi: 10.1074/jbc.272.37.23328. [DOI] [PubMed] [Google Scholar]
- 33.Lukusa T, Fryns JP. Human chromosome fragility. Biochim Biophys Acta. 2008;1779:3–16. doi: 10.1016/j.bbagrm.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 34.Mrasek K, Schoder C, Teichmann AC, Behr K, Franze B, Wilhelm K, Blaurock N, Claussen U, Liehr T, Weise A. Global screening and extended nomenclature for 230 aphidicolin-inducible fragile sites, including 61 yet unreported ones. Int J Oncol. 2010;36:929–940. doi: 10.3892/ijo_00000572. [DOI] [PubMed] [Google Scholar]
- 35.Borgaonkar DS. Chromosomal Variation in Man. 7. New York: Wiley-Liss; 1994. [Google Scholar]
- 36.Simonic I, Gericke GS. The enigma of common fragile sites. Hum Genet. 1996;97:524–531. doi: 10.1007/BF02267080. [DOI] [PubMed] [Google Scholar]
- 37.Zimonjic DB, Druck T, Ohta M, Kastury K, Croce CM, Popescu NC, Huebner K. Positions of Chromosome 3p14.2 Fragile Sites (FRA3B) within the FHIT Gene. Cancer Res. 1997;57:1166–1170. [PubMed] [Google Scholar]
- 38.Mishmar D, Rahat A, Scherer SW, Nyakatura G, Hinzmann B, Kohwi Y, Mandel-Gutfroind Y, Lee JR, Drescher B, Sas DE, Margalit H, Platzer M, Weiss A, Tsui LC, Rosenthal A, Kerem B. Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site. Proc Natl Acad Sci U S A. 1998;95:8141–8146. doi: 10.1073/pnas.95.14.8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ried K, Finnis M, Hobson L, Mangelsdorf M, Dayan S, Nancarrow JK, Woollatt E, Kremmidiotis G, Gardner A, Venter D, Baker E, Richards RI. Common chromosomal fragile site FRA16D sequence: identification of the FOR gene spanning FRA16D and homozygous deletions and translocation breakpoints in cancer cells. Hum Mol Genet. 2000;9:1651–1663. doi: 10.1093/hmg/9.11.1651. [DOI] [PubMed] [Google Scholar]
- 40.National Center for Biotechnology Information. http://www.ncbi.nlm.nih.gov. Accessed 10 Jun 2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data sets supporting the conclusions of this article are included within the article. More details are available upon request.