

Abstract
A novel recombinant protein tetra-H2A (TH) derived from histone H2A has been developed to replace protamine as a conditionally reversible, nucleic acid condensing agent. The novel protein will address the insufficient release of nucleic acid therapeutics, which is captured by protamine for siRNA delivery. TH is composed of 4 tandem repeats of the histone H2A N-terminal sequence, intervened by the cathepsin D cleavage site. The repeating H2A sequence enhances the binding affinity to anionic nucleic acids, forming more stable condensates, as demonstrated by the binding affinity assay. The TH/siRNA condensates are formulated into a core-membrane structured liposomal nanoparticle (NP). The endosomes of cancer cells are rich in cathepsin D, allowing on-site degradation of TH and facilitating the intracellular release of siRNA. The NPs assembled with TH produced a higher silencing efficiency of target genes in vitro and in vivo than the NPs assembled with protamine as the nucleic acid condensing agent. The exploitation of TH in the NP formulation exhibited a biocompatibility profile similar to that of protamine, with minimal immunostimulating and systemic toxicity observed after repeated administration.
Keywords: RNAi, recombinant protein, liposome, nanoparticle, tumor targeted delivery
Graphical Abstract

1. Introduction
RNA interference (RNAi), discovered by Fire and Mello, had a strong impact on therapeutic drug development [1]. In particular, short-interference RNA (siRNA) can knockdown the target gene with high specificity and efficiency. Although the number of RNAi-related patent applications has skyrocketed, the systemic delivery of these vulnerable and negatively charged macromolecules to target cells remains an obstacle to clinical applications of RNAi therapy [2]. The advent of a versatile platform that can be easily tailored to target different cell populations and optimize the performance of various siRNA-based therapeutics is in high demand.
Nanoparticulate devices for drug delivery confer multiple advantages over free drugs, mainly through the improvement of their pharmacokinetics. Liposome-based formulations of anticancer drugs are examples of this strategy. The carriers are biodegradable, biologically inert, minimally immunogenic and readily modifiable for targeting purposes. Over the past few years, our group has been dedicated to the development of a series of PEGylated, liposome-based nanoparticles (NPs) that can efficiently deliver siRNA [3–6], pDNA [7], peptides [8], chemodrugs [9], and proteins [10] to solid tumors and metastatic lesions. In our Liposome-Protamine-Hyaluronic acid (LPH) formulation, the mix of anionic hyaluronic acid (HA) and siRNA is usually condensed into a nanometric complex using protamine-a naturally existing, arginine-rich polypeptide. The complex is then coated with lipids and extensively PEGylated [4]. The utilization of protamine as the condensing agent of nucleic acids enables the formation of a stable core that supports an oppositely charged lipid bilayer. However, the DNA condensing capacity of protamine is so strong that it does not readily release the nucleic acid cargo intracellularly, compromising the bioavailability of the delivered cargo.
In nature, protamines are replacements for histone proteins in the haploid phase of spermatogenesis [11], generating a more condensed packaging of genomic DNA and protecting the genetic message delivered by spermatozoa [12]. After fertilization, the highly packed nucleoprotamine complex is decondensed to allow the assembly of the nucleosome structure by histone proteins [13, 14]. The wisdom of nature inspired us to use a histone, a natural nucleic acid condensing reagent, with a weaker binding affinity, as a replacement for protamine in our formulation. The arginine rich nuclear proteins, histone proteins [15–18] and histone protein derivatives [19, 20], have been extensively investigated as transfection reagents in vitro and in vivo. However, histone-mediated gene transfer resulted in relatively low transgene expression possibly due to insufficient condensation and/or endosome escape [21]. We attempted to address such issues by constructing a histone-based recombinant protein with protein engineering technology based on rational design.
In this study, we investigated the possibility of replacing protamine with a recombinant protein that was composed of four tandem repeats of human histone H2A peptide [19, 22], intervened by cathepsin D cleavage sites [22–24]. A pH-responsive membrane-lytic peptide, namely GALA [25], was incorporated into the C-terminal of the recombinant protein so as to facilitate the endosome escape of the cargo. We hypothesized that the recombinant protein, tetra-H2A (TH), was able to condense HA and siRNA into a stable complex that could support a cationic lipid coating and a high degree of PEGylation, forming Lipid-tetra-H2A-Hyaluronic acid (LHH) NP, as illustrated in Fig 3a. The distal end of the PEG chain was conjugated with anisamide (AA) to target cancer cells that overexpress sigma receptor. Once the NP was taken up by the cells through receptor-mediated endocytosis, the cathepsin D cleavage sites in the TH were subject to enzymatic digestion in the endosome compartment as pH decreased. The degradation of the condensing agent was supposed to enhance the cytoplasmic release of nucleic acid cargos. The current study was designed to test these hypotheses. The delivery efficiency of the LHH NP was examined in NCI-H460, a human lung carcinoma cell line that overexpresses the sigma receptor [3, 5]. This cell line was also stably transduced with firefly luciferase gene, which allows convenient evaluation of the knockdown efficiency of the target gene through a luciferase assay.
Figure 3. Preparation and characterization of LHH NP.

a) Schematic illustration of LHH NP preparation. Anionic siRNA and HA is mixed with cationic TH and DOTAP/Cholesterol liposome to form the core-shell structure complex. The NP is then PEGylated through post-insertion by heating. b) Particle size and zeta potential characterization by Dynamic Light Scattering and Laser Doppler Velocimetry. c) TEM imaging of LHH NP after uranyl acetate staining. Scale bar: 100 nm.
2. Materials and Methods
2.1. Cell line and culture conditions
A human lung carcinoma cell line, NCI-H460 (ATCC, Manassas, VA) was cultured in RPMI-1640 supplemented with 10% fetal bovine serum (Life Technologies, Carlsbad, California), and penicillin/streptomycin (Life Technologies, Carlsbad, California). The H460 cells were stably transduced with GL3 firefly luciferase genes with a retroviral vector to create H460 cells (H460-luc) that express luciferase by Pilar Blancafort’s laboratory at the University of North Carolina at Chapel Hill.
2.2. Cloning, expression and purification of TH
Gene encoding TH and TH mutants were synthesized by Blue Heron Biotech (Bothell, WA). The gene was cloned into a pET28b vector (Merck KGaA, Germany). The fidelity of the gene was confirmed using DNA sequencing. pET28b vectors with corresponding TH genes were transformed into BL21 (DE3) E. Coli expression host cell line (Merck KGaA, Germany). The transformant colony was grown in Circlegrow® media (MP Biomedicals, Solon, OH). The protein expression was induced with 0.6 mM ITPG (Fermentas, Glen Burnie, MD) at 30°C for 4h. Culture was centrifuged and lysed with a lysis buffer (100 mM NaH2PO4, 10 mM Tris, 500 mM NaCl, 1% Triton and 1 mM β-mercaptoethanol). The target protein contained Histidine x6 tag at the N-terminal. The lysate was spun at 30,000 xg for 45 min and the supernatant was loaded onto a 1 mL Ni-NTA agarose column (Qiagen, Valencia, CA) in the presence of 5 mM imidazole. The column was then washed with 20 mL wash buffer (100 mM NaH2PO4, 10 mM Tris, 500 mM NaCl, 20 mM imidazole and 1mM β-mercaptoethanol). Protein was eluted with 5 mL elution buffer (100 mM NaH2PO4, 10 mM Tris, 500 mM NaCl, 200 mM imidazole and 1 mM β-mercaptoethanol). The protein was then desalted and buffer-exchanged using ultrafiltration with 3k Ultracel (Merck KGaA, Germany). The final protein was solubilized in 50 mM HEPES buffer (pH 7.0). The purity of the TH was examined by SDS-PAGE. The identity of the TH was also confirmed by western blot analysis by probing Histidine x6 tag.
2.3. Preparation of LHH NPs
1,2-Dioleoyl-3-trimethylammonium-propane (DOTAP) (Avanti Polar Lipids Inc., Alabaster, Alabama) and cholesterol (1:1, mol:mol) (Sigma Aldrich, St. Louis, MO) were dissolved in chloroform and the solvent was removed under reduced pressure. The lipid film was hydrated overnight with distilled water to form cationic liposomes (10 mM), which were sequentially extruded through 400, 200, and 100 nm polycarbonate membranes (Millipore, Billerica, MA). The core of LHH-NPs was prepared by mixing 28 µg of TH in 100 µL deionized (DI) water with 10 µg of a mixture of HA and siRNA (1:1, w:w) in 100 µL DI water so that the N/P ratio between TH and the HA/siRNA mixture was approximately 2.5:1, where N equals the positive charges carried by amine groups and P equals the negative charges carried by nucleic acid. To prepare the core of Lipid Protamine-HA (LPH)-NPs, 8 µg of protamine in 100 µL DI water was complexed with 10 µg of a mixture of hyaluronic acid and siRNA (1:1, w:w) so that the N/P ratio was 1:1. After incubation at room temperature for 10 min, the core was mixed with 9 µL cationic liposome (20 mM) and incubated for another 10 min for lipid coating. The lipid-coated NPs were PEGylated using a post-insertional approach by adding 7 µL micelles (3.5mM) composed of DSPE-PEG and DSPE-PEG-AA (1:1; mol:mol) and incubating the NPs at 50°C for 15 min. DSPE-PEG-AA was synthesized in Dr. Leaf Huang’s laboratory based on a previously established protocol [26]. The size and zeta potential of the NPs were characterized using a Malvern Zetasizer ZS90 (Worcestershire, UK).
2.4. SYBR Green exclusion assay
SYBR Green I nucleic acid stain (Life Technologies, Carlsbad, California) was incubated with plasmid DNA (2 µg/mL) in TAE buffer according to the manufacturer’s instructions. One microgram of plasmid was then complexed with protamine (Sigma Aldrich, St. Louis, MO), H2A peptide (peptide2.0, Chantilly, VA) or TH at various N/P ratios. The complexes were incubated at room temperature for 15 min before measurement. The fluorescence of SYBR Green was determined using a PerkinElmer fluorescence spectrometer LS55 (PerkinElmer Inc., Waltham, MA).
2.5. Cathepsin D cleavage digestion
Ten micrograms of the TH with or without cathepsin D cleavage sites were incubated with 2 units of human liver cathepsin D (EMD Millipore, Billerica, MA) in the presence of 50 mM acetate buffer (pH 3.8) at 37°C. The digestion products were then resolved by 4–12% SDS-PAGE (Life Technologies) and visualized using Coomassie staining.
To quantify the Cathepsin D mediated digestion of polycations and the resulting release of nucleic acids, complexes were made by mixing five micrograms SYBR Green treated plasmid DNA with TH (2.5: 1 N/P ratio), TH/ΔCathD (2.5: 1 N/P ratio), H2A peptide (2.5: 1 N/P ratio) or protamine (1:1 N/P ratio). These complexes were incubated with 2 units of cathepsin D in the presence of 50 mM acetate buffer (pH 3.8) at 37°C. Samples were assayed at different time points. The samples were neutralized by adding alkaline solution before the fluorescence intensity of SYBR Green was determined using a fluorescence spectrometer (PerkinElmer Inc.).
2.6. Hemolytic assay
The pH-responsive, membrane-lytic ability was determined using a hemolytic assay. Sheep red blood cells (RBC) (Innovative Research, Novi, MI) were washed thoroughly with DPBS and spun down at 1000 xg for 10 min. The RBCs were then reconstituted into 5×108 cells/ml at pH 5.0 or pH 7.4 with sodium phosphate/sodium acetate buffer (50 mM). Ten micrograms of TH or TH/ΔGALA were added to red blood cells with mild shaking (50 rpm) and incubated for 1 h at 37°C. The RBCs were then spun down at top speed with a tabletop centrifuge. The lysis of RBCs was determined by reading the absorbance of the supernatant at 541 nm using spectrometer (PerkinElmer Inc.).
2.7. In Vitro transfection and imaging
H460-luc cells stably transfected with luciferase genes were seeded in 96 wells with a seeding density of 5×103 cells/well. To demonstrate the targeted delivery of the NPs, siRNA against luciferase (target sequence 5′-CTT ACG CTG AGT ACT TCG A-3′) was encapsulated into LHH-NPs with or without AA, and added to the H460-luc cells at a final concentration of 50nM. To study the dose-response silencing effect, anti-luciferase siRNA was encapsulated into LHH-NPs prepared with TH, TH/ΔGALA, TH/ΔCathD and TH/ΔGALA/ΔCathD. LPH-NP prepared with protamine was also used as a control. The NPs were administered to H460-luc cells so that the final concentration of siRNA was 5, 10, 25, 50, 100, or 200 nM. After four hours of transfection, the NPs were removed and the medium was refreshed. The silencing of the luciferase genes was determined 24 h post-transfection using a luciferase assay kit (Promega, Madison, MI) following the manufacture’s instruction.
For in vitro imaging studies, 2×104 H460 cells were seeded in Lab-tek II 8-well chamber slides (Nalge Nunc International, Rochester, NY), Half a microgram of 6-carboxyfluorescein labeled double strand oligodeoxynucleotides (dsODN) (IDTDNA, Coralville, IA) was encapsulated into the LHH-NPs or LPH-NP and administered to the cells. The cells were then incubated with the NPs for 30 min before extensive washing with DPBS. To visualize the nuclei and lysosomes, the Hochest 33342 (Life Technologies) and LysoTracker (Life Technologies) were used to treat the cells for 30 min before observing according to the manufacturer’s instructions. The cells were imaged using an Olympus FV1000 multiphoton confocal microscope (Olympus America Inc) and images were analyzed with FV10-ASW Viewer3.1.
2.8. In Vivo biodistribution study of LHH-NPs
Female athymic nude mice, 6–8 weeks old, were subcutaneously inoculated with 5×106 H460 cells in the flank. LHH-NPs loaded with 10 µg of Texas Red labeled dsODN (IDTDNA, Coralville, IA) were prepared and injected intravenously (IV) into the tumor bearing mice. Four hours post-injection, animals were sacrificed and the organs were collected (heart, liver, spleen, lung, kidneys, tumor). Biodistribution of the labeled dsODN was qualitatively determined using a Kodak in vivo imaging system FX Pro (Rochester, NY). To quantify the in vivo biodistribution, ten micrograms of dsODN was labeled with 3H [27] and used to label LHH-NP. Major organs were collected 4 h post IV injection of 3H-labeled LHH-NP. One-hundred grams of each tissue were treated with 1 mL NCS-II tissue solubilizer (GE Healthcare Biosciences, Pittsburgh, PA) overnight and heated at 50°C. Three-hundred microliters of the samples were then mixed with 4 mL Ultra Gold LSC-Cocktail (Perkin Elmer Inc., Waltham, MA). The 3H reading was measured with a scintillation counter. All animal protocols were approved by the University of North Carolina at Chapel Hill’s Institutional Animal Care and Use Committee.
2.9. In Vivo target gene silencing mediated by LHH-NPs
In vivo transfection studies were conducted by formulating 10 µg anti-luciferase siRNA into LHH-NPs or LPH-NP and IV injecting the final particles into H460-luc xenograft bearing nude mice when the tumor size is about 200 mm3 (n=3). Mice were sacrificed 24 h post-injection and tumor tissues were collected and homogenized. The tissue lysates were subject to luciferase assay (Promega, Madison, MI) according to the manufacturer’s instruction.
2.10. AST, ALT, BUN assay and cytokine induction assay
CD-1 mice (n=5) were IV injected with LHH-NPs (equivalent to 10 µg of dsODN) for three consecutive days. On day 4, mice were sacrificed and sera were prepared for AST, ALT and BUN assays performed by the UNC core facility. To evaluate the acute immune-stimulating property of the LHH-NPs, CD-1 mice (n=3) were sacrificed 4 h after injection with LHH-NP or LPH-NP (equivalent to 10 µg of dsODN). Sera were collected from these mice for IL-6, IL-12, IFN-α and TNF-α level with ELISA assay kit (BD Biosciences, San Diego, CA) according to manufacturer’s manual.
3. Results
3.1. The recombinant protein TH was expressed and purified with high purity
Based on the repetition of histone H2A sequences in TH, the gene encoding TH was codon-optimized for higher expression in E. coli. The gene was then commercially synthesized, cloned into pET28b vectors and transformed into the expression host strain, BL21(DE3). Expression was optimized using various ITPG induction concentrations at different temperatures. Induction of the protein with 0.6 mM IPTG at 30°C for 4 h produced the highest protein yield with the fewest degradation products. TH was purified under native conditions using Ni-NTA affinity chromatography by capturing the Hisx6 tag of the N-terminal of TH. Purified TH was then buffer-exchanged with ultrafiltration. The final yield was approximately 5 mg per liter of culture. The purity of the TH was determined using a SDS-PAGE followed by coomassie staining. The results indicated an estimated purity greater than 95% (Fig. 1b). The identity of the purified TH was confirmed by western blot analysis conducted with a monoclonal anti-Hisx6 tag antibody (Fig. 1c). To validate the functionality of each motif, a series of mutant THs that lack either one of or both GALA and cathepsin D cleavage sites were expressed and purified as controls (termed TH/ΔGALA, TH/ΔCathD and TH/ΔGALA/ΔCathD, respectively).
Figure 1. Expression and purification of TH biopolymer.

a) Amino acid sequence of the TH. The recombinant, protein-based TH is composed of three functional motifs including condensation, dissociation and endosomal disruption motif, which are genetically engineered in one single molecule. b) SDS-PAGE analysis of purified TH followed by coomassie staining. The purity of the TH was estimated to be greater than 95%. c) Western blot analysis with mouse anti-Histidine x6 antibody to identify the TH.
3.2. The placement of motifs maintains their individual functionalities in the TH
The accessibility of cathepsin D cleavage sites in the TH to their corresponding enzymes was examined using a digestion assay at the optimal reaction conditions of human cathepsin D. The TH was incubated with cathepsin D for 1 h and the digestion products were analyzed using SDS-PAGE. The densitometry analysis of the bands stained with coomassie blue indicated that there was 46% degradation of TH (Fig. 2a) after one-hour exposure to cathepsin D. The digestion products were approximately 4 KDa and 13 KDa, which corresponded to an H2A monomer and trimer (or dimer with GALA). Although the cleavage sites were incorporated into the recombinant protein for enhanced degradation, the accessibility of the sites to the enzyme was different in a molecule due to steric hindrance. If no cleavage sites were available, 90% of TH/ΔCathD remained intact after cathepsin D digestion (Fig. 2a). The minimal degradation of TH/ΔCathD was most likely due to non-specific digestion after long-term incubation. Thus, the inclusion of cleavage sites responded to cathepsin D and facilated the degradation as expected.
Figure 2. The incorporation of multiple functional amino acid sequences in one molecule has preserved the functionality of each sequence.

a) SDS-PAGE of digested TH with cathepsin D protease. Lane 1: ladder marker, Lane 2: undigested TH, lane 3: TH digested by cathepsin D. Lane 4: TH/ΔCathD digested by cathepsin D. Bottom chart: Quantification of relative amount of protein based on densitometry analysis. The incorporation of cathepsin D substrate in the TH facilitated the degradation in the presence of corresponding enzyme. b) Hemolysis activity of TH at acidic (pH=5.0) or neutral (pH=7.4) pH. 1% TritonX-100 was used as a positive control (representing 100% hemolysis) and buffer alone was used as a negative control (n=3). The endosome disruptive sequence, GALA, in the TH induced lysis of the membrane exclusively at an acidic pH. Error bars show mean ± s.d. Student t-test. c) SYBR Green exclusion assay. Plasmid DNA with SYBR Green I stain was complexed with protamine, H2A peptide (37mer) or TH at various N/P ratios. (n=3). TH-mediated DNA condensation was equivalent to that of protamine, whereas H2A could only condense DNA into a less compact degree. Error bars show mean ± s.d. d) Cathepsin D induced decondensation. Plasmid DNA was complexed with TH, TH/ΔCathD, H2A peptide and protamine at optimal N/P ratio, followed by cathepsin D incubation. The incorporation of cathepsin cleavage enabled the TH to respond to the stimulus in a prompt way, facilitating the release of nucleic acids in an enzyme dependent manner.
A hemolysis assay was used to illustrate the pH-responsive, membrane lytic activity of the GALA peptide, which resides in the C-terminal region of TH. TH and TH/ΔGALA were incubated with sheep’s red blood cells at a neutral (pH 7.4) or acidic pH (pH 5.0). The hemolytic activities of the THs were quantified using the release of hemoglobin as determined by absorbance at 541nm. The results showed that the acidic pH activated the hemolytic activity of GALA in the C-terminal of the TH, which resulted in ~35% hemolysis. This activity, however, did not occur in neutral pH conditions (Fig. 2b). TH/ΔGALA, was a stringent control and did not induce hemolysis regardless of pH, ruling out the possibility that the lysis of the membrane was due to the multimerized H2A sequence. These results, combined with the results illustrating Cathepsin D digestion, demonstrate that both cathepsin D substrate and GALA peptide sequences have retained their designated functionality after incorporation into TH.
3.3. The incorporation of cathepsin D facilitates the release of nucleic acid cargo upon enzymatic digestion
It is observed that polycations with higher molecular weights, such as polyethyleneimine, polylysine and chitosan [28], have demonstrated higher binding affinity to the polyanionic nucleic acids. To enhance the binding affinity between the condensing agent and nucleic acid cargo, the histone H2A derived peptide was multimerized to increase the molecular weight. The DNA condensing capacity of the TH was assayed using a SYBR Green exclusion assay. The binding of SYBR Green to DNA strands increases the fluorescence intensity. This can be reversed through polycation-mediated DNA condensation because the process excludes the fluorescent probe. H2A peptide, protamine and TH were used to complex with plasmid DNA (pDNA) pretreated with SYBR Green at various N/P ratios. The fluorescence intensity emitted by the complexes was measured. The activity of inducing pDNA condensation by TH was higher than that of the protamine; complete condensation of pDNA occurred at a higher N/P ratio for TH than protamine (2.5:1 vs. 1.5:1) (Fig. 2c). In contrast, H2A peptide had induced only a partial condensation even at very high N/P ratios. Thus, multimerization has indeed increased the binding affinity of H2A.
In order to study enzyme-triggered release profiles of the nucleic acid, the condensed pDNA by H2A, protamine, TH and TH/ΔCathD at the optimal N/P ratios were incubated with human cathepsin D enzyme. Protamine was used to condense pDNA at a 1:1 N/P ratio, while H2A peptide, recombinant TH or TH derivatives condensed pDNA at a 2.5:1 N/P ratio. An increase of the fluorescence intensity was observed in the TH condensed pDNA complex when incubated with the enzyme, indicating a loosened structure due to the degradation of biopolymer by cathepsin D (Fig. 2d). The fluorescence intensities of TH/ΔCathD or protamine mediated condensation did not change in the presence of the enzyme at all the time points observed. Additionally, no significant change was observed in the TH/pDNA complexes incubated with BSA, which suggested that the disassembly was not caused by charge-related competitive interaction.
3.4. The TH replaces protamine and forms highly PEGylated LHH NPs that targets sigma receptors to promote siRNA delivery
The optimization of the NP formulation (Fig. 3a) was performed with a series of characterization studies to sequentially determine the optimal ratios for core complexes, liposome coating and the degree of PEGylation (data not shown). The core of the NP was formed by electrostatic interactions between cationic TH and anionic HA/siRNA mixture. The optimum N/P ratio for forming the core complexes were 2.5:1 for TH or TH derivatives, and 1:1 for protamine, so that the HA/siRNA (or dsODN) was condensed to the highest degree, and yet the core complex possessed a negative surface charge. The negatively charged core complex was then coated with cationic liposomes and then PEGylated through post-insertion. The LHH-NP or LPH-NP used in the following experiments were prepared using the same protocol as that described here. The LHH-NPs were characterized using dynamic light scattering and Laser Doppler Velocimetry, regarding the physicochemical properties. The final NP was determined to be ~50nm in diameter and with a surface charge of ~15mV (Fig. 3b). The size and uniformity of the LHH NPs were further confirmed using transmission electron microscopy (TEM) after uranyl acetate staining (Fig. 3c).
In order to validate the targeting ability of the AA that was conjugated to the distal end of the PEG chain, siRNA against the luciferase gene was encapsulated into LHH-NPs with or without AA, and added to H460 cells that were stably expressing luciferase. The silencing efficiency was assayed 24 h post-transfection using a luciferase assay. The results demonstrated that the LHH-NP with AA resulted in ~75% knockdown of luciferase gene (Fig. 4a). In contrast, siRNA encapsulated into LHH-NP without AA resulted in only ~36% knockdown efficiency. The cytotoxicity of LHH-NP was also evaluated in H460 cells using a MTS assay. The dose dependent cytotoxicity data, together with the results of the dose dependent silencing assay demonstrated that even at an extremely high dose, 100 times higher than the IC50 (4.3 nM), minimal cytotoxicity was observed in the cell cultures treated with LHH-NP (Fig. 4b).
Figure 4. The AA functionalized LHH-NP has demonstrated cell targeting ability and low cytotoxicity.

a) Ligand dependent knockdown of the target gene. LHH NPs with or without AA was loaded with siRNA against luciferase and administered to H460-luc cells. The knockdown efficiency was determined using a luciferase assay (n=3). The presence of AA on the surface of the NP significantly enhanced silencing efficiency. Error bars show mean ± s.d. Student t-test. b): Dose-dependent cytotoxicity versus dose-dependent knockdown activity (n=3). Error bars show mean ± s.d. The LHH NP demonstrated minimal cytotoxicity even at a concentration 100 times higher than the IC50.
3.5. The TH facilitated intracellular release of nucleic acid cargo, resulting in a higher efficiency of gene silencing
To study the influences of different condensing agents on the intracellular distribution of nucleic acids delivered by the NPs, a confocal microscopy study was conducted. The results demonstrated that FAM-labeled dsODNs (green) appeared in the cytoplasm 30 min after administration (Fig. 5a). Moreover, the dsODNs did not co-localize with LysoTracker (red) labeled lysosomes, which indicated an efficient endosome escape regardless of the condensing agent used. LHH-NP prepared with TH resulted in a homogenous intracellular distribution of FAM-labeled dsODNs. In contrast, dsODNs delivered by LHH/ΔCathD-NP (Fig. 5a-2) prepared with the TH/ΔCathD, or delivered by LPH-NP prepared with the protamine, showed a punctate distribution pattern (Fig. 5a-4). This suggested insufficient release of the cargo from the NPs, probably due to the lack of degradation mechanism of the condensing agent. Interestingly, the FAM-labeled dsODNs that were delivered by LHH/ΔGALA-NP, prepared with TH/GALA, showed a homogenous distribution as well as an efficient endosomal escape. Since all NPs were coated with cationic liposomes composed of DOTAP/Cholesterol, the results suggested that the cationic lipid was the major driving force to destabilize the endosome membrane due to the ion-pair mechanism [29]. The qualitative imaging assay revealed that the GALA peptide only played a minor role in facilitating endosomal escape.
Figure 5. The incorporation of TH into the LHH NP has demonstrated an enhanced intracellular release of the cargos and an increased siRNA silencing efficiency in vitro.

a) Confocal microscopic observation of cytoplasmic distribution of FAM labeled dsODNs delivered by LHH-NPs or LPH-NP. Nucleus of the cells was stained with Hoechst (blue) and lysosomes were labeled with LysoTracker (red). dsODNs delivered by NPs composed of TH (image 1) and TH/ΔGALA (image 3) have displayed more homogenously dispersed pattern. However, the dsODNs delivered by the NPs composed of protamine (image 4) or TH/ΔCathD (image 2) have demonstrated an aggregated pattern. b) Dose dependent luciferase knockdown activity of anti-luciferase siRNA when encapsulated in different NP formulations (n=3). The incorporation of TH in the NP formulation resulted in an enhanced target gene silencing efficiency compared with protamine. Error bars show mean ± s.d. c) IC50 of siRNA that is delivered by different NPs formulation, which was determined by the dose-response curve generated with Prism 5.0c software.
To evaluate the influences of various condensing agents on the knockdown efficiency of the NPs, anti-luciferase siRNA was formulated into the NPs and administered to H460-luc cells that were stably transfected with luciferase (Fig. 5b). Dose-response curves of different formulations were plotted using Prism software. The IC50, which indicates the dose required to achieve 50% knockdown of the target gene, was calculated. The IC50 for LHH-NP mediated transfection was about 4.3nM (Fig. 5c), which was 3 times lower than LPH-NP mediated transfection (13.1nM). Due to the lack of disassembly mechanism, the IC50 of siRNA delivered by LHH/ΔCathD was equivalent (13.2nM) to that of LPH. These data were in agreement with the results from confocal microscopic observations regarding to the release profiles.
3.6. Systemic administration of LHH-NP resulted in efficient target gene silencing
To study the in vivo biodistribution of the NPs after systemic administration, Texas-red labeled dsODN was encapsulated into LHH-NP and IV injected into H460-luc xenograft bearing nude mice. The tumor and major organs were harvested 4 h post-administration and the biodistribution of LHH-NP was determined using an in vivo imaging system. The result showed that the Texas-red labeled dsODN accumulated primarily in the tumors, with some deposits in the liver (Fig. 6a). Consistent with cell culture confocal microscopic data, there was a homogenous, intracellular distribution of Texas-red labeled dsODN in the cryosection of the tumor tissue. As was observed that fluorescence based imaging approach did not reflect the distribution quantitatively due to the quenching issues [30], 3H-labeled dsODNs were encapsulated into LHH-NP and IV injected to the xenograft bearing mice to accurately quantify the biodistribution of the therapeutics delivered by LHH-NP. Based on the activity detected from the organs and tumors collected 4 h post-injection, there was ~5% injected dose per gram of tissue accumulated in the tumor (Fig 6c). The overall tumor accumulation of LHH-NP was not significantly different from that of LPH-NP, and was independent of the presence of AA as a targeting ligand (data not shown).
Figure 6. The evaluation of biodistribution and transfection efficiency of the LHH-NP in vivo.

a) Biodistribution profiles of Tex-red labeled dsODN in major organs of H460 xenograft bearing nude mice 4 h post-administration of LHH-NP. The NPs predominately accumulated in the liver, kidneys and tumors after IV injection. b) Confocal microscopic images of tumor cryosections after systemic delivery of LHH-NP encapsulating Texas-red labeled dsODNs. c) Quantification of LHH biodistribution by tracking 3H labeled dsODNs 4 h post-injection (n=3). There was ~5% injected dose per gram tissue accumulated in the tumor. Error bars show mean ± s.d. d) In vivo luciferase gene silencing effects 24 h after IV injection of LHH and LPH NPs to the H460-luc bearing nude mice (0.25mg/kg) (n=3). siRNA delivered by LHH-NP resulted in significantly higher silencing effect than that of LPH-NP. Error bars show mean ± s.d. Student t-test.
The in vivo efficiency of gene silencing by tumor-targeted siRNA was evaluated in H460-luc xenograft-bearing nude mice. Anti-luciferase siRNA was encapsulated into LHH and LPH NPs and IV injected into the mice. Twenty-four hour post administration, the animals were sacrificed and the tumor lysate was assayed for luciferase activity. The data demonstrated that a single dose (0.25mg/kg), IV injection of LHH-NP loaded with anti-luciferase siRNA resulted in a ~66% silencing of the target gene, significantly higher than that of LPH-NP mediated knockdown (34%) (Fig. 6d). The increased knockdown is most likely a result of the enhanced release of siRNA. The LHH-NP without targeting ligand was able to cause a ~44% silencing, although there was no significant difference in the tumor accumulation of the targeted and non-targeted NPs. The results suggest that the presence of the targeting ligand probably facilitated the internalization of the NPs, which, in turn, promoted the siRNA delivery efficiency.
3.7. Systemic administration of LHH NP demonstrated minimal immune stimulating activity and negligible systemic toxicity
A panel of four cytokines, including IFN-α, TNF-α, IL-6 and IL-12, was assayed 4 h post-administration of the LHH-NP (Fig. 7a). The results indicated the immunestimulating effects of the formulation. There was no detectable induction of IFN-α or IL-6. A mild elevation of TNF-α (~135pg/ml for LHH-NP and 123pg/ml for LPH-NP) was observed and a relatively high level IL-12 induction (~1278pg/ml for LHH-NP and 1310pg/ml for LPH-NP) was detected. However, there was not a significant difference between the treatment with LPH and LHH NPs regarding to the cytokine induction levels. The immunogenicity of TH was examined after repeated injection for three weeks and there was no TH-specific antibody detected (data not shown).
Figure 7. The evaluation of immunostimulating effect and systemic toxicity of the LHH-NP.

a) Serum cytokine levels of the LHH-NP and the LPH-NP treated CD-1 mice examined by ELISA assay. A slight increase of TNF-α and relatively high level of IL12 was observed after injection. No significant difference was observed between LHH and LPH treatments. Error bars show mean ± s.d. b) AST, ALT and BUN levels of CD-1 mice after repeated injections (3 times × 0.5mg/kg) of LHH-NP. The repeated treatments of LHH-NP didn’t incur liver and kidney damage according to the blood biochemistry tests.
As was demonstrated by the biodistribution study, the liver and the kidneys were the two major organs that accumulated the most NPs in addition to the tumor. Therefore, it is necessary to check the liver and kidney infraction degree after the systemic administration of the LHH-NP. The serum alanine transaminase (ALT), aspartate transaminase (AST), liver damage markers, and blood urea nitrogen (BUN), a kidney infraction indicator, were examined after the mice received three injections of the NP (Fig. 7b). All parameters tested showed no significant difference between the treated group and the control group, which demonstrated that the NPs did not cause dysfunction of these two major organs.
4. Discussion
We have demonstrated that the use of a repetitive histone H2A peptide with intervening cathepsin D cleavage sites as a nucleic acid condensing agent to replace protamine in the LPH formulation, leads to an enhanced intracellular release of nucleic acid cargos after systemic administration. The enhanced release profile increased the intracellular bioavailability of the cargo and effectively boosted the silencing efficiency of targeted siRNA delivery. Compared to synthetic, cationic polymers, such as polyamidoamine dendrimer, poly-(L-lysine) and polyethyleneimine [31–33] that are usually generated by the polymerization of a single unit, the building blocks for protein-based “biopolymers” are composed of 20 different amino acids and infinite patterns of combination. The number of combinations available allows more diverse physicochemical or biological functions of the biopolymers than the charge alone.
Nature affords us many examples that merit attention regarding the patterns of building blocks. For example, cell penetrating [34, 35], nuclear localization [36, 37], and cell-targeting [38] sequences have been successfully incorporated into the design of many gene/siRNA delivery carriers to increase their delivery efficiency and specificity. From a technical perspective, the advancement of technology in the molecular biology field enables the DNA templates with repetitive sequences to be synthesized with high efficiency, encoding longer biopolymers with arranged placements of building blocks, defined size, and stereochemistry [39]. The development of these biopolymers offers an attractive alternative for nucleic acid delivery because they are non-toxic, biodegradable and biocompatible when compared to their chemically synthetic counterparts.
In the present study, we exploited the N-terminal sequence of human histone H2A, which was able to deliver genes to the cells in vitro according to the report by Balicki et al. [19]. Similar to the low molecular weight synthetic cationic polymers, the histone-derived peptide (~4 KDa) did not provide sufficient binding affinity to condense the nucleic acid (Fig. 2c). For the core-membrane structure NPs, a solid core complex is necessary to support the lipid coating and extensive PEGylation, which allows extended blood circulation and tumor targeting of the NPs [40]. To compensate for the low binding affinity, we have multimerized the H2A peptides to enable the relatively higher molecular weight polypeptide (~24 kDa) to condense the nucleic acids to a degree approaching to that of protamine (Fig. 2c). Although the charge density in the biopolymer was much lower than that in chemical polymers, with only 19% of positively charged amino acids in the molecule, the two short α-helices interrupted by a short loop in each H2A peptide possibly allowed the biopolymer to anchor the nucleic acid strands and rendered higher binding affinity [19]. The structure-based binding in addition to electrostatic binding significantly reduced the requirement of positive charges in a condensing agent for complete condensation. This was beneficial to the development of safe siRNA delivery vectors, as material related cell death was often observed in the cationic polymeric condensing agents mediated transfection, such as polyethyleneimine or poly-(L-lysine). The mechanistic studies suggested that the electrostatic interaction between the polymers and mitochondria was the central mechanistic role for cell necrosis or apoptosis induced by the highly charged, cationic polymers [41]. Contrarily, the use of the biopolymer as condensing agent abolished the toxicity as demonstrated by the wide therapeutic window determined by MTS cytotoxicity assay (Fig. 4b), possibly due to the relatively low charge-based reactivity and high degradability.
In order to facilitate the dissociation between the vectors and the nucleic acid cargos, cathepsin D cleavage sites were incorporated between H2A sequences as a switch for expedited degradation when exposed to the enzyme intracellularly. Cathepsin D is an aspartic protease that is abundant in the endosomes of many cancer cells. The drop of pH in the endosome protonates the critical aspartate residue and activates the enzyme for sequence-specific cleavage of the substrate [42]. The incubation of TH-condensed NPs with cathepsin D could reverse the condensation of the complex as demonstrated by SYBR Green exclusion assay (Fig. 2d). A condensing agent that is stimulus-dependent and that induces reversible effects is the critical component of the design; it resolves the dilemma of poor condensing or poor release efficiency mediated by the traditional condensing agents.
The liposomal NP based on the core-membrane structure is a representative of NPs that have been designed to disassemble in a multistage manner after systemic administration (Fig. 8). The PEG coating and targeting ligand functionalize the surface of the liposomal membrane, allowing the NPs to be long-circulating in the blood stream and to efficiently accumulate in the interstitial space of tumor tissues. The targeting ligand grafted onto the surface of the NPs recognizes the tumor-specific receptors and facilitates the cellular internalization of the NPs. After endocytosis, the abundance of lipases in the endosomal compartment starts to strip the lipid coating, leaving the core complexes exposed to the environment of the endosomal compartment. The cathepsin D is activated accompanying the drop of pH in the endosome. The protease-mediated degradation of the condensing agent weakens its binding affinity to the nucleic acids. Simultaneously, the remaining cationic lipid forms ion-pair with lipid of compartmental membrane, which destabilizes the endosome. The pH-responsive, membrane-lytic peptide in the C-terminal of the TH offers an additional mechanism for the escape of the cargo. Once the loosened core complex enters the cytoplasm, the nucleic acids dissociates from condensing agent by charge-based competitive displacement with cytoplasmic proteins.
Figure 8. Schematic illustration of proposed LHH NP intracellular trafficking after systemic delivery.

i. The pegylated LHH NP shows extended blood circulation by evading the capture of reticuloendothelial system and extravsates into the interstitial space of the tumor due to the EPR effect. ii. The targeting ligand at the distal end of the PEG facilitates the cellular internalization of the NPs. iii, the NPs are entrapped in the endosomal compartment where the pH starts to drop. The lipase will strip the lipid membrane and the core is exposed to pH-activated cathepsin D enzyme. iv. The cationic lipid starts to destabilize the membrane structure due to the ion-exchange mechanism. Meanwhile, the pH-activated GALA peptide inserts into the compartment membrane and forms the barrel structure in the membrane to release endosome. v. After endosome escape, the degraded TH by cathepsin D demonstrates weakened binding affinity to the nucleic acids, leaving the anionic cargos to be easily released by the charge-based competitive displacement.
The data acquired from gene knockdown assay and biodistribution assay allowed us to establish a correlation between the in vitro and in vivo delivery efficiency of the nanoparticles. According to the biodistribution data from four-hour post-injection (Fig. 6c), the tumor accumulation of siRNA was approximately 5% injected dose per gram of tissue when 0.5 mg/kg siRNA was IV injected to the mice. Assuming the distribution pattern is applicable to the scenario of 0.25 mg/kg siRNA dose and that the local NP concentration remained constant in the tumor tissue for 24 hours post injection, there was approximately 0.25 µg siRNA per gram of tissue. Given that the density of the tissue is 1.4 g/ml, the approximate concentration of siRNA was 26 nM on condition that the siRNA molecules were homogenously distributed over the whole tissue. However, NPs larger than 60 nm primarily accumulate in the interstitial space in the vicinity of the leaky vasculature, but are typically unable to penetrate deep into the tissue due to extracellular matrix and elevated interstitial pressure. Therefore, the effective dose of siRNA to the certain cell population under the influence was much higher than 26nM. Although our LHH formulation was able to achieve efficient siRNA delivery in vitro and in vivo, it was still difficult to directly predict in vivo delivery efficiency based on the in vitro data due to the complexity and heterogeneity of the tumor microenvironment.
This LHH NP was designed based on a solution-driven strategy. The knowledge of extracellular and intracellular NP trafficking routes as well as the understanding of the barriers to systemic delivery of siRNA leads to the recognition of problems, enabling us to devise novel NPs with appropriate solutions. The NPs were engineered in an attempt to mimic lipid-enveloped viruses in terms of structure and delivery process, which are acknowledged as the most efficient gene delivery vectors in vivo. However, these NPs abrogate the high risk of immunogenic and oncogenic concerns raised when using viral vectors. Moreover, with relatively easy tunability of both the lipid coating and the biopolymer based condensing agent, these artificial virus-like gene delivery vectors are likely to catch up the performance of the viral particles, regarding the transfection ability, with minimal toxicity and immunogenicity.
5. Conclusion
We have successfully designed a histone derived recombinant protein with a reversible activity of capturing nucleic acid. The core formed by the engineered protein and nucleic acid can be further stabilized by lipid membrane coating and PEGylation. The biochemical studies validated the functionality of each designated motif in the molecule. The in vitro and in vivo transfection studies showed the enhanced delivery efficiency, which was likely to be attributed to the proposed mechanisms. Generally, the study demonstrated a strategy regarding to the enhancement of systemic siRNA delivery efficiency by manipulating the sequences of recombinant protein based condensing agent. It can be envisaged that more of these biopolymer-based materials will come up in the future studies of delivery of nucleic acid therapeutics.
Supplementary Material
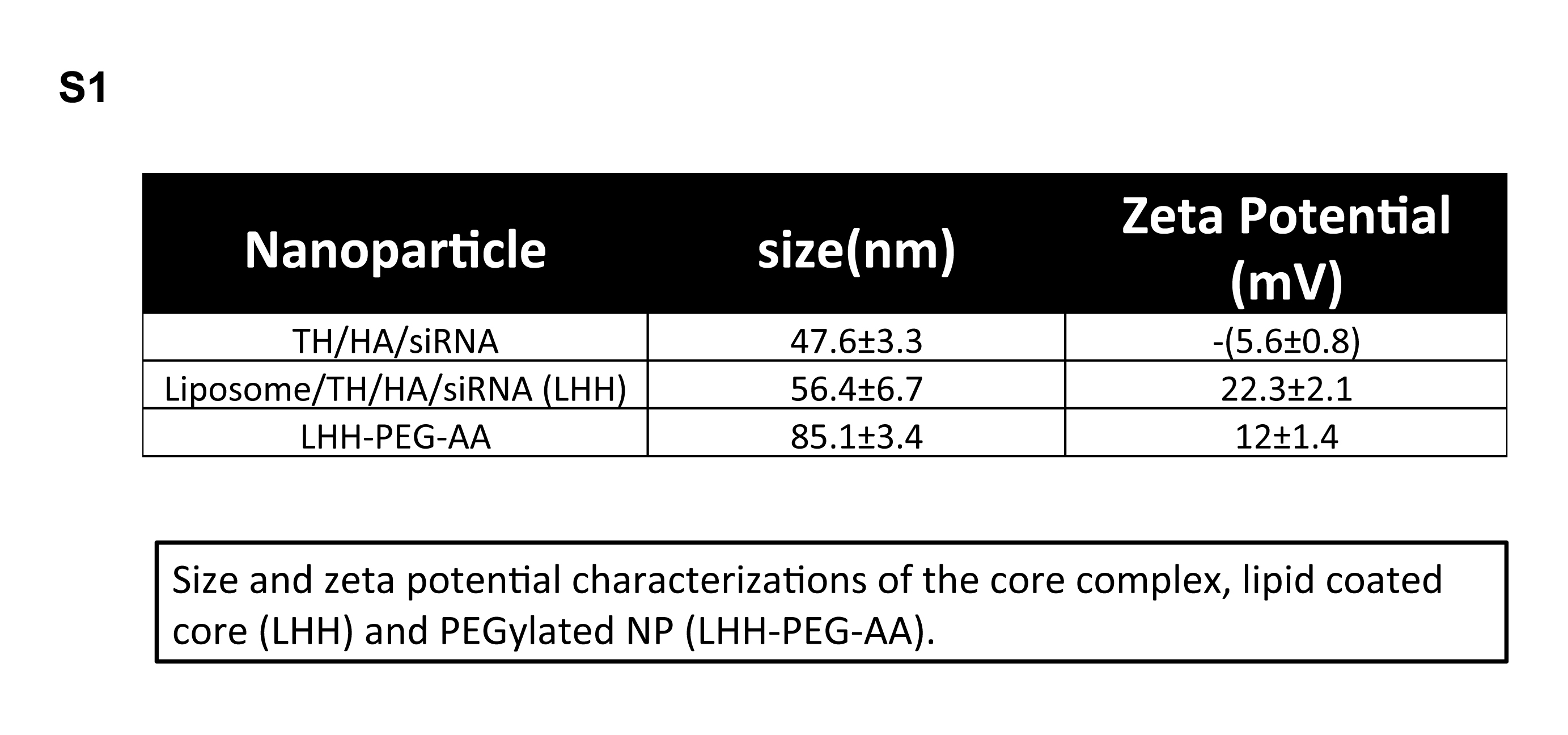{kind=link}
Acknowledgments
This work was supported by NIH grants CA129835, CA129421, CA151652, CA151455 and CA149363. This work was partly funded by the Department of Defense Prostate Cancer New Investigator Award to A. Hatefi (W81XWH-09-1-0303). We thank Dr. Pilar Blancafort for the H460-luc cells and Ms. Kelly Ann Racette for manuscript editing.
Abbreviation list
- TH
tetra-H2A polypeptide
- TH/ΔGALA
tetra-H2A polypeptide without GALA
- TH/ΔCathD
tetra-H2A polypeptide without cathepsin D cleavage site
- TH/ΔGALA/ΔCathD
tetra-H2A polypeptide without GALA or cathepsin D cleavage site
- DOTAP
1,2-dioleoyl-3-trimethylammonium-propane
- DSPE
1,2-dioctadecanoyl-sn-glycero-3-phosphoethanolamine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- 1.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 2.Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nature reviews Drug discovery. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Y, Sen J, Bathula SR, Yang Q, Fittipaldi R, Huang L. Novel cationic lipid that delivers siRNA and enhances therapeutic effect in lung cancer cells. Mol Pharm. 2009;6:696–705. doi: 10.1021/mp800136v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chono S, Li SD, Conwell CC, Huang L. An efficient and low immunostimulatory nanoparticle formulation for systemic siRNA delivery to the tumor. J Control Release. 2008;131:64–69. doi: 10.1016/j.jconrel.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li SD, Chen YC, Hackett MJ, Huang L. Tumor-targeted delivery of siRNA by self-assembled nanoparticles. Molecular therapy : the journal of the American Society of Gene Therapy. 2008;16:163–169. doi: 10.1038/sj.mt.6300323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li SD, Chono S, Huang L. Efficient gene silencing in metastatic tumor by siRNA formulated in surface-modified nanoparticles. J Control Release. 2008;126:77–84. doi: 10.1016/j.jconrel.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ueno NT, Bartholomeusz C, Xia W, Anklesaria P, Bruckheimer EM, Mebel E, et al. Systemic gene therapy in human xenograft tumor models by liposomal delivery of the E1A gene. Cancer Res. 2002;62:6712–6716. [PubMed] [Google Scholar]
- 8.Kim SK, Foote MB, Huang L. Targeted delivery of EV peptide to tumor cell cytoplasm using lipid coated calcium carbonate nanoparticles. Cancer letters. 2012 doi: 10.1016/j.canlet.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y, Bathula SR, Li J, Huang L. Multifunctional nanoparticles delivering small interfering RNA and doxorubicin overcome drug resistance in cancer. The Journal of biological chemistry. 2010;285:22639–22650. doi: 10.1074/jbc.M110.125906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim SK, Foote MB, Huang L. The targeted intracellular delivery of cytochrome C protein to tumors using lipid-apolipoprotein nanoparticles. Biomaterials. 2012;33:3959–3966. doi: 10.1016/j.biomaterials.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balhorn R. The protamine family of sperm nuclear proteins. Genome biology. 2007;8:227. doi: 10.1186/gb-2007-8-9-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliva R. Protamines and male infertility. Human reproduction update. 2006;12:417–435. doi: 10.1093/humupd/dml009. [DOI] [PubMed] [Google Scholar]
- 13.Romanato M, Cameo MS, Bertolesi G, Baldini C, Calvo JC, Calvo L. Heparan sulphate: a putative decondensing agent for human spermatozoa in vivo. Hum Reprod. 2003;18:1868–1873. doi: 10.1093/humrep/deg354. [DOI] [PubMed] [Google Scholar]
- 14.Romanato M, Regueira E, Cameo MS, Baldini C, Calvo L, Calvo JC. Further evidence on the role of heparan sulfate as protamine acceptor during the decondensation of human spermatozoa. Hum Reprod. 2005;20:2784–2789. doi: 10.1093/humrep/dei124. [DOI] [PubMed] [Google Scholar]
- 15.Kaouass M, Beaulieu R, Balicki D. Histonefection: Novel and potent non-viral gene delivery. J Control Release. 2006;113:245–254. doi: 10.1016/j.jconrel.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 16.Demirhan I, Hasselmayer O, Chandra A, Ehemann M, Chandra P. Histone-mediated transfer and expression of the HIV-1 tat gene in Jurkat cells. Journal of human virology. 1998;1:430–440. [PubMed] [Google Scholar]
- 17.Balicki D, Reisfeld RA, Pertl U, Beutler E, Lode HN. Histone H2A-mediated transient cytokine gene delivery induces efficient antitumor responses in murine neuroblastoma. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:11500–11504. doi: 10.1073/pnas.210382997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balicki D, Beutler E. Histone H2A significantly enhances in vitro DNA transfection. Mol Med. 1997;3:782–787. [PMC free article] [PubMed] [Google Scholar]
- 19.Balicki D, Putnam CD, Scaria PV, Beutler E. Structure and function correlation in histone H2A peptide-mediated gene transfer. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:7467–7471. doi: 10.1073/pnas.102168299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwartz B, Ivanov MA, Pitard B, Escriou V, Rangara R, Byk G, et al. Synthetic DNA-compacting peptides derived from human sequence enhance cationic lipid-mediated gene transfer in vitro and in vivo. Gene therapy. 1999;6:282–292. doi: 10.1038/sj.gt.3300795. [DOI] [PubMed] [Google Scholar]
- 21.Haberland A, Knaus T, Zaitsev SV, Buchberger B, Lun A, Haller H, et al. Histone H1-mediated transfection: serum inhibition can be overcome by Ca2+ ions. Pharmaceutical research. 2000;17:229–235. doi: 10.1023/a:1007581700996. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Mangipudi SS, Canine BF, Hatefi A. A designer biomimetic vector with a chimeric architecture for targeted gene transfer. J Control Release. 2009;137:46–53. doi: 10.1016/j.jconrel.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Canine BF, Wang Y, Hatefi A. Biosynthesis and characterization of a novel genetically engineered polymer for targeted gene transfer to cancer cells. J Control Release. 2009;138:188–196. doi: 10.1016/j.jconrel.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mangipudi SS, Canine BF, Wang Y, Hatefi A. Development of a genetically engineered biomimetic vector for targeted gene transfer to breast cancer cells. Mol Pharm. 2009;6:1100–1109. doi: 10.1021/mp800251x. [DOI] [PubMed] [Google Scholar]
- 25.Li W, Nicol F, Szoka FC., Jr GALA: a designed synthetic pH-responsive amphipathic peptide with applications in drug and gene delivery. Advanced drug delivery reviews. 2004;56:967–985. doi: 10.1016/j.addr.2003.10.041. [DOI] [PubMed] [Google Scholar]
- 26.Banerjee R, Tyagi P, Li S, Huang L. Anisamide-targeted stealth liposomes: a potent carrier for targeting doxorubicin to human prostate cancer cells. Int J Cancer. 2004;112:693–700. doi: 10.1002/ijc.20452. [DOI] [PubMed] [Google Scholar]
- 27.Graham MJ, Freier SM, Crooke RM, Ecker DJ, Maslova RN, Lesnik EA. Tritium labeling of antisense oligonucleotides by exchange with tritiated water. Nucleic acids research. 1993;21:3737–3743. doi: 10.1093/nar/21.16.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grigsby CL, Leong KW. Balancing protection and release of DNA: tools to address a bottleneck of non-viral gene delivery. Journal of the Royal Society, Interface/the Royal Society. 2010;7(Suppl 1):S67–S82. doi: 10.1098/rsif.2009.0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tseng YC, Mozumdar S, Huang L. Lipid-based systemic delivery of siRNA. Advanced drug delivery reviews. 2009;61:721–731. doi: 10.1016/j.addr.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Y, Tseng YC, Huang L. Biodistribution studies of nanoparticles using fluorescence imaging: a qualitative or quantitative method? Pharmaceutical research. 2012;29:3273–3277. doi: 10.1007/s11095-012-0818-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lollo CP, Banaszczyk MG, Mullen PM, Coffin CC, Wu D, Carlo AT, et al. Poly-L-lysine-based gene delivery systems. Synthesis, purification, and application. Methods in molecular medicine. 2002;69:1–13. doi: 10.1385/1-59259-141-8:001. [DOI] [PubMed] [Google Scholar]
- 32.Song H, Wang G, He B, Li L, Li C, Lai Y, et al. Cationic lipid-coated PEI/DNA polyplexes with improved efficiency and reduced cytotoxicity for gene delivery into mesenchymal stem cells. International journal of nanomedicine. 2012;7:4637–4648. doi: 10.2147/IJN.S33923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nam HY, Nam K, Lee M, Kim SW, Bull DA. Dendrimer type bio-reducible polymer for efficient gene delivery. J Control Release. 2012;160:592–600. doi: 10.1016/j.jconrel.2012.04.025. [DOI] [PubMed] [Google Scholar]
- 34.Zhao D, Zhuo RX, Cheng SX. Modification of calcium carbonate based gene and drug delivery systems by a cell-penetrating peptide. Molecular bioSystems. 2012;8:3288–3294. doi: 10.1039/c2mb25233c. [DOI] [PubMed] [Google Scholar]
- 35.Torchilin VP. Cell penetrating peptide-modified pharmaceutical nanocarriers for intracellular drug and gene delivery. Biopolymers. 2008;90:604–610. doi: 10.1002/bip.20989. [DOI] [PubMed] [Google Scholar]
- 36.Matschke J, Bohla A, Maucksch C, Mittal R, Rudolph C, Rosenecker J. Characterization of Ku70(2)-NLS as bipartite nuclear localization sequence for non-viral gene delivery. PloS one. 2012;7:e24615. doi: 10.1371/journal.pone.0024615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bremner KH, Seymour LW, Logan A, Read ML. Factors influencing the ability of nuclear localization sequence peptides to enhance nonviral gene delivery. Bioconjugate chemistry. 2004;15:152–161. doi: 10.1021/bc034140k. [DOI] [PubMed] [Google Scholar]
- 38.Moffatt S, Wiehle S, Cristiano RJ. Tumor-specific gene delivery mediated by a novel peptide-polyethylenimine-DNA polyplex targeting aminopeptidase N/CD13. Human gene therapy. 2005;16:57–67. doi: 10.1089/hum.2005.16.57. [DOI] [PubMed] [Google Scholar]
- 39.Amiram M, Quiroz FG, Callahan DJ, Chilkoti A. A highly parallel method for synthesizing DNA repeats enables the discovery of 'smart' protein polymers. Nature materials. 2011;10:141–148. doi: 10.1038/nmat2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li SD, Huang L. Stealth nanoparticles: high density but sheddable PEG is a key for tumor targeting. J Control Release. 2010;145:178–181. doi: 10.1016/j.jconrel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hunter AC, Moghimi SM. Cationic carriers of genetic material and cell death: a mitochondrial tale. Biochim Biophys Acta. 2010;1797:1203–1209. doi: 10.1016/j.bbabio.2010.03.026. [DOI] [PubMed] [Google Scholar]
- 42.Lee AY, Gulnik SV, Erickson JW. Conformational switching in an aspartic proteinase. Nature structural biology. 1998;5:866–871. doi: 10.1038/2306. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.