

INTRODUCTION
It is now known through the various studies that the endocannabinoids have dual role in human health. On the one hand, they contribute to the etiology of some diseases; on other they play “protective role” in some other disease states. This evoked the interest in developing exogenous drugs that can modulate cannabinoid system.
Drugs which enhance the activity of endocannabinoids such as cannabinoid receptor agonists, agents modifying cannabinoid transport or inhibiting their metabolism has the capacity to be used as analgesics,[1] hypnotics, antiemetics, antihypertensive, antiasthmatics, antiepileptics, neuroprotectives,[2] immunomodulatory, anti-inflammatory, alcohol withdrawal, and eating disorders.[1,2,3]
One of the major advances in this field was the generation of fatty acid amide hydrolase (FAAH) inhibitors.[3,4] FAAH hydrolyses the endocannabinoids with amide bonds including anandamide (AEA). Inhibition of FAAH would lead to extended endocannabinoid activity at its site of synthesis resulting in tissue selective activation of CB1 receptors. This enhanced endocannabinoid activity is suggested to be useful in the treatment of several clinical conditions. At present, a lot of research is being carried out to establish their role in the management of neuropathic pain.
A recent report of tragic mishap in Phase I clinical trial of BIA 10–2474, an FAAH inhibitor came as a major setback to researchers.[5] Portugal's Bial Pharmaceuticals was conducting Phase I clinical trials in France with this FAAH inhibitor. The Investigator Brochure for BIA 10–2474 states that it was developed “for the treatment of medical conditions in which there is advantage in enhance the levels of endogenous AEA and tonically increase the drive of the endocannabinoid system.” Bial later on after the incident confirmed that the BIA 10–2474 was being developed for neuropathic pain.
A total of 128 participants were enrolled in this trial, out of which ninety were dosed with compound, and the others were given placebo. Volunteers who were subjected to multiple doses of test drug were adversely affected of them were admitted to hospital, out of which one of the volunteer was declared brain dead and other four, out of remaining five were said to have irreversible brain damage.[5] The magnetic resonance imaging (MRI) of the affected patients showed evidence of deep cerebral hemorrhage and necrosis.
Besides this, the administration of the next dose of the test drug to the remaining persons after the occurrence of serious adverse event is not ethical. Bial did not suspect that the acute symptom was due to the test drug and thus gave the next dose to remaining five persons on the following day. It should have waited for the results of tests, especially the MRI scan of on the affected volunteer. The Trial was discontinued only after the first volunteer who was hospitalized went into coma; but by that time, the next dose was already administered to the remaining five persons of that cohort. These persons were also hospitalized later due to the occurrence of adverse effects. If the trial was stopped immediately after the occurrence of the first serious adverse event; then, the remaining five persons would not have suffered.
STUDY DETAILS
As per the protocol, the primary objectives of the study were to assess the safety and tolerability of BIA 10–2474 after single and multiple oral doses and to investigate the effect of food on the pharmacokinetic (PK) and pharmacodynamics (PD) of BIA 10–2474.[6] The secondary objectives were to characterize the PK profile of BIA 10–2474 (and its metabolites) after single and multiple oral doses, to characterize not only its PD profile (mainly FAAH) activity inhibition but also concentrations of N-arachidonoyl-ethanolamine (AEA) and related fatty acid amides, and to assess several potential PD effects.
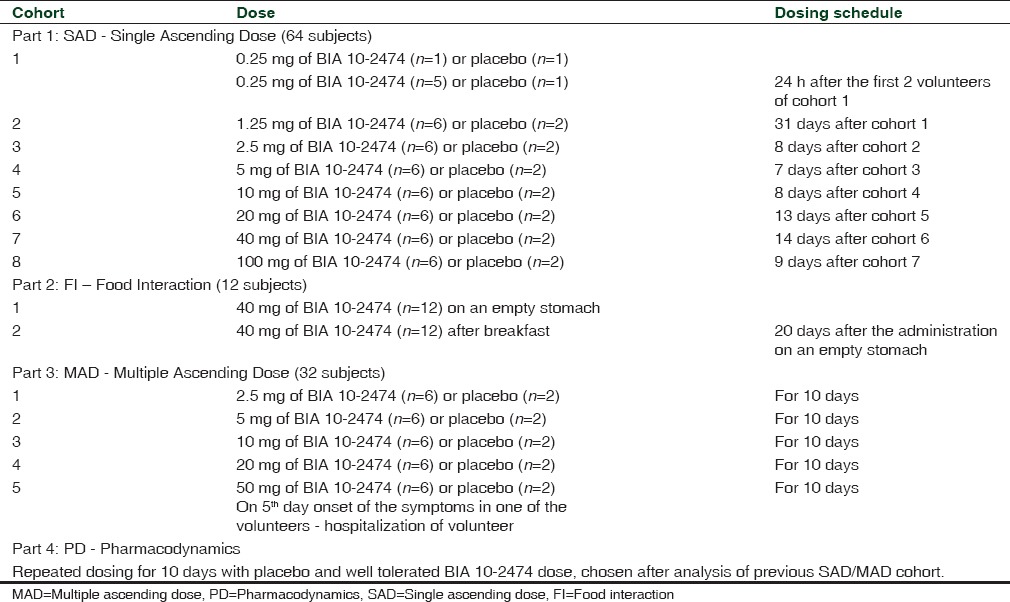
It was a double-blind, randomized, placebo-controlled combined single ascending dose (SAD) and multiple ascending dose (MAD) study, including an additional food interaction part (which is an open-label design), and a PD part as shown in Table 1.[6] The doses and dosing schedule details of these four groups are given in Table 2. The study was conducted at the single center, and the persons were to participate for the maximum of 13 weeks.
Table 1.
Study design and characteristics of Phase I trial of BIA 10-2474

Table 2.
Study parts with volunteers and dosing schedule
Other trials on fatty acid amide hydrolase inhibitors
As mentioned earlier in the article, FAAH inhibitors are being researched extensively, but none of them has reached market yet. Other clinical trials that are conducted on FAAH inhibitors are Merck's MK-4409, Pfizer's PF-04457845,[7] and Vernalis’ V158866.[8] None of these trials had reported any adverse effect with this group of agents, and they were considered safe in humans. Thus, it gives a speculation that the incidents related to BIA 10–2474 occurred because the drug may have hit the wrong and unexpected target.
Animal studies
Bial performed studies on four animal species: rat, mice, dog, and monkey. As per the Temporary Specialist Scientific Committee (TSSC), the use of four different species of animals for testing BIA 10–2474 was unusual, and this was not followed during any other study with FAAH inhibitor. The possible cause suggestive for this could be due to poor tolerance result in the first species due to which the study was switched to other species. The reason behind conducting studies in mice could be to determine the doses for the carcinogenesis studies to be conducted in future. The explained given by Bial for these extensive animal studies was due to delay in the start of clinical development; they continued additional toxicology studies (i.e., carcinogenesis studies).
Animal toxicological studies were carried out in mice, dogs, and monkeys for 13 weeks and rats for 26 weeks.[6] In case of a novel compound to have better perception of its effect's in humans, it should be evaluated in primates close to humans such as cynomolgus monkeys, chimpanzees, or Bonobos as high-risk molecules.[9] Bial conducted BIA 10–2474 toxicity study in cynomolgus monkeys and the medulla oblongata damage observed at higher doses of 100 mg/kg/24 h should have been evaluated further. These primates can very well predict species-specific effects as observed by increase in cytokines and interferon levels at higher doses used in cynomolgus monkeys in preclinical part of TGN1412 trial,[10] conducted by TeGenero.
POSSIBLE CAUSES FOR THE MISHAP
Health agencies such as the United States Food and Drug Administration, European Medicines Agency, National Agency for the Safety of Medicines, and Health Products (L’Agence nationale de sécurité du médicament et des produits de santé or ANSM) has joined hands to investigate the cause for this tragedic trial. Researchers all over the world are suggesting possible underlying cause for it. Broadly, the possible causes can be divided into two categories: human error and off-target action of the investigational drug. Although exact mechanism for this event could only be determined by the experts, who are closely investigating the event. This article is discussing few of the possible causes responsible for this mishap which is being suggested by the researchers worldwide.
Reversibility and low specificity
As per the report by the TSSC, set by the ANSM, the health agency of France, FAAH inhibition should be considered as irreversible process as it forms a covalent bond between hydrolase serine 241 and the carbamate or urea electrophilic carbon. This inhibition is extremely prolonged one, and complete inhibition is observed even after 8 h and in humans, this inhibition persisted for up to 24 h even when the plasma concentrations of the drug had fallen below the quantifiable limits of the test method used by Bial.
In in vitro studies, the inhibition of other enzymes by BIA 10—2474 occurred at about 50 to 100 times of concentration that is required for FAAH inhibition. Besides this, there are possibilities that this ratio is even lower with other cerebral hydrolases. The other FAAH inhibitors such as PF-3845 by Pfizer and JNJ-42165279 by Janssen were highly specific to FAAH compared to those inhibiting a panel of around twenty other hydrolases.
The information available so far does not give any information regarding the specificity of BIA 10–2474 for FAAH compared to other hydrolases. The availability of specificity data for BIA 10–2474 will help to determine if the serious adverse event was due to off-target effect of the compound.
Impact on immune system
BIA 10–2474 is claimed to be reversible inhibitor of FAAH enzyme. However, as mentioned earlier causes a prolonged inhibition, and there is a possibility that the drug may be acting as an irreversible inhibitor of some other enzymes, this may signal the immune system to recognize it as foreign body and there are chances that the immune system initiates an inflammatory response against it. This can lead to hypersensitivity or autoimmune reaction in areas where this enzyme is present in the body.
Drugs such as halothane can cause autoimmune drug reaction known as halothane hepatitis.[11] On metabolism of halothane by the liver, trifluoroacetyl chloride and trifluoroacetic acid metabolites are produced as intermediate compounds. These metabolites bind with liver proteins and in genetically predisposed individuals, antibodies are formed to this metabolite-protein complex which mediates type II hypersensitivity. Similarly, penicillin's can cause hypersensitive reaction in certain individuals by similar mechanism. Thus, such autoimmune reaction in the brain could not be ruled out as possible cause of brain death and other irreversible damage caused by BIA 10–2474 in affected subjects of its Phase I clinical trial.
Dose calculation
There is no established experience of the use of FAAH inhibitors in humans, and it is a new chemical entity and novel compound, and lesser is known about its target distribution, signaling pathways, and pharmacological effects or systemic activity. Besides this, it might have potential for amplification, supralinear, or threshold dose-response and there is a lack of biomarkers of effect/toxicity. In such cases, animal models are of limited relevance to study pharmacology and toxicology. Thus, it should be treated as a high-risk molecule and minimal anticipated biologic effect level (MABEL)-based approach should have been followed rather than no observed adverse effect level (NOAEL) dosing approach for calculating safe starting dose followed by dose escalations.[9] The NOAEL-based approach suggests 96 mg as the maximum dose to be used in a study on humans for BIA 10–2474, which was close to 100 mg, the highest dose tested in humans in single or multiple doses. Based on the alleged mechanism of the pharmacological activity of BIA 10–2474, the FAAH inhibition caused by it is achieved in humans at 1.25 mg and is almost complete at 5 mg. Therefore, 100 mg is equivalent to testing a dose 20 to 50 times higher than that presumed to be effective.[12] If MABEL approached would have been used the starting dose would have been 100 times lower than that of NOAEL-based approach which could have prevented the mishap as was the case with TGN1412.
A Phase I clinical trial conducted in 2006 with TGN1412, a CD28 superagonist, was evaluated for safety. Six previously healthy subjects who received this drug in a first-in-man trial caused a “cytokine storm” and multiple organ failures. In the TGN1412 study, subclinical dose of 0.1 mg per kg – based on NOAEL (500 times lower than the dose found safe in animals) – was used. If the dose is calculated using the MABEL approach; then, the safe starting dose is 0.001 mg/kg, administration of this dose would probably not have caused the mishaps in this trial.
Similarly, for BIA 10–2474, if MABEL approach for dose calculation was followed then this mishap could have been avoided.
Cumulative toxicity
The mishap occurred in Part 2: MAD with 50 mg of BIA 10–2474 on the 5th and 6th day of administration of this dose. This dose in almost 10 times higher than that required to fully inhibit the FAAH activity (as per NOAEL approach). At this dose, the tissue concentrations increased beyond that completely inhibits the FAAH enzyme, and this may have led to binding with other serine hydrolases that are also facilitated by the low specificity of BIA 10–2474 for the target enzyme. This binding of the test drug to other enzymes by some mechanism could have led to brain damage in the affected subjects. Such damage was not observed in the trial subjects that received single 100 mg dose of test drugs. Thus, this indicates that the damage occurred due to cumulative toxicity. BIA 10–2474 is known to have nonproportional PKs and large volume of distribution, and this could have contributed to the gradual accumulation of the drug in the brain tissue. Another possibility is that some metabolite of BIA 10–2474 with longer half-life than the parent compound could have accumulated in the tissues and led to brain damage. The concentrations of test drug or responsible metabolite reached the damaging level on the 5th or 6th day of administration and led to serious adverse events.[12]
The clinical presentation of affected volunteers included headache, cerebellar syndrome, loss of consciousness, memory impairment, diplopia, and hemiparesis with tremor of one side of the body. In four volunteers, MRI scan exhibited “anomalies of highly variable intensity, affecting hippocampus, and pons (protuberance) predominant in the anterior part (extending at times to the bulb or to the mesencephalon), bilaterally and symmetrically.” In deceased volunteer, thalamus, and cerebral cortex were involved. These findings suggest the presence of microstructural changes with vascular component, i.e., micro bleeds which are nonspecific. The bilateral and symmetrical topography and very early appearance of hypo signal in Susceptibility-weighted imaging rule out inflammatory process and involvement of hippocampus, pons, thalamus, and cortex make primary vascular mechanism unlikely and therefore, the findings are more suggestive of toxic/metabolic process.[12] The clinical symptoms consistent with findings of MRI and progression of neurological symptoms suggest that higher doses of the product resulted in higher concentrations of product or metabolites are possibly responsible for the adverse event. Similar findings of cerebral damage, especially in hippocampus with gliosis and inflammatory cell infiltration were observed in rats and mice at very high doses which were not observed in other FAAH inhibitor trials. In some monkeys receiving 100 mg/kg/24 h medulla oblongata (spinal bulb) damage in the form of axonal dystrophy was observed and not seen with lower doses.[12]
Two volunteers from the 10 mg MAD cohort presented with blurred vision (lasted between 10 and 30 min) on two occasions, although blurred vision was not observed in volunteer of higher dose cohorts as well as those receiving placebo. As mentioned earlier, the nonlinear relationship of area under curve (AUC) and dose would have resulted in higher concentration of drug in these subjects. Therefore, considering them irrelevant by investigator and monitoring committee is questionable.[12] The frequency of adverse events which were mainly cardiovascular (orthostatic hypotension, PR, and QT prolongation), dizziness, and headache were higher as compared to other first in human or Phase I trial of other FAAH inhibitors. Even the PK data in volunteers with SAD cohorts suggest that there is nonlinear relationship with regard to AUC and doses, i.e., AUC reflecting exposure increases more rapidly as compared to increase in dose. Elimination half-life of BIA 10–2474 gradually increases at higher doses suggesting a possibility of saturation of elimination or metabolic process at doses between 40 and 100 mg.[12] This should have treated as warning signal for avoiding the use of higher doses in their volunteers.
Other off-target effects
Another speculation is that the drug might have exhibited some off-target effect. FAAH enzymes belong to group of serine hydrolases. Serine hydrolases family of enzymes have about 200 enzymes. Functions of all these enzymes are not yet well understood. There is a possibility that in higher doses, this drug is binding to other enzymes or some cellular proteins ultimately leading to decreased blood flow in the brain.
There are nine known metabolites of BIA 10–2474: Compounds BIA 10–2639, 10–2583, 10–3258, 10–3827, 10–2445, 10–2631, 10–3844, 10–2580, and 10–3764. The structure of these metabolites is very similar to the BIA 10–2474. Three of these metabolites have potential to inhibit FAAH with the same intensity as the mother compound. These metabolites can be detected in very small quantities even after the 14 days of administration of parent compound in animals. However, BIA 10-2631 is found in larger quantities in primates. There is a possibility that there is an unknown metabolite that is responsible for the mishap, or there is the probability of high concentrations of known/unknown metabolite/s accumulating in the brain tissue. Another very important aspect is that BIA 10–2474 consists of imidazole nucleus in the position adjacent to the molecule's electrophilic site that potentially makes it a “leaving group” that may produce an isocyanate to which many brain proteins could bind.[12]
The sensitivity of the assay methods used during toxicology studies only identified five peripheral (plasma compartment) metabolites out of the nine produced by BIA 10–2474. These metabolites are theoretically identical to those found in humans and also produced in very small quantities about 1% of the parent product.[12] Thus, the toxicity studies specifically for these metabolites were not legally compulsory and were also not conducted.
Nonclinical studies and species specificity
In 2006, another life-threatening incident occurred in all six healthy volunteers during the Phase I trial of the CD28 super agonist monoclonal antibody TGN1412, conducted by now bankrupt German company TeGenero. TGN1412 was expected to treat conditions such as rheumatoid arthritis and leukemia. The systemic inflammatory immune response which was termed as “cytokine storm” occurred after 1 h of infusion of TGN1412. It was not predicted by preclinical safety testing. This immune response led to life-threatening multiorgan failure in all six participants.
It is suggested that this cytokine storm caused by TGN1412 is due to activation of CD4+ effector memory T-cells by TGN1412. The absence of CD28 expression on the CD4+ effector memory T-cells of species except cynomolgus monkeys which were used for preclinical safety testing of TGN1412 could be a possible explanation for the failure to predict “cytokine storm” in humans. Another aspect is that the peak serum concentrations of cytokines (interleukin) were found to be raised in the cynomolgus monkeys used as one of the species for the nonclinical study, and this was overlooked by the scientists involved with research on TGN1412. If these abnormal serum levels of cytokines were not ignored during preclinical studies then it would have used as an alarm for further investigation of the compound then probably the suffering of six volunteers Phase I clinical trial of TGN1412 could have avoided.
Drugs with proven safety and efficacy in nonclinical animal models may exhibit different pharmacological properties when used in humans. Therefore, it is essential to develop proper nonclinical models which can effectively predict the drug response in humans. Moreover, for the first-in-man trials (Phase I clinical trials), the drug should be initially tested on the single human subject or on the very less number of human subjects before higher number of individuals are exposed to the drug. In BIA 10–2474 Phase I trial, all six subjects were simultaneously administered to multiple higher doses of FAAH inhibitor under investigation. This led to the death of one and serious adverse events in four other volunteers. To make Phase I trial safer for participants, initially, only one person should be exposed to the test drug. After a lag period of five half-lives which ensures that most of the drug is eliminated from the body and ensuring that no adverse event occurred, only then the drug should be considered safe to be administered in other subjects. Therefore, in our opinion, instead of 6 + 2 design, N of 1 randomized control trials design should be practiced to make Phase I trial safer for human population.
Similar could be case for BIA 10–2474 where the safety studies done in animal studies predicted it to be safe for human use but due to species variation some of the adverse effects could not be very well predicted. Besides this, primates close to humans such as cynomolgus monkeys and chimpanzee which can predict species-specific response in humans more accurately were not used for nonclinical studies.
Drug impurities
Another hypothesis made my some researchers is derived from the chemical structure that is exhibited on the US patent application for BIA 10–2474.
The synthesis of BIA 10–2474 requires an additional step to remove “N-acetylated aniline impurity.”[13] If this impurity is not properly removed, then the aniline present in the drug could convert hemoglobin to methemoglobin and impair oxygen supply to various tissues. Another assumption is that the aniline might be produced as one the metabolite of the drug. There are established reports that aniline in higher concentration is a toxic compound which affects the tissues with high oxygen supply such as brain and heart. British scientist at Pfizer, Dennis A. Smith, Ph.D., wrote in his textbook, Metabolism, PKs, and Toxicity of Functional Groups that,[14] “Compounds containing aromatic amines (anilines) induce a variety of toxicological responses including carcinogenicity and hepatotoxicity. Several drugs containing an aniline moiety, which have been withdrawn from the market, have a black box warning on their labels. Therefore, anilines have been put on the blacklist of functional groups that the medicinal chemists generally avoid.”
Production/manufacture error
Since several pharmaceutical companies (Janssen, Pfizer, Merck, Sanofi) have conducted Phase I and Phase II clinical trials with FAAH inhibitors and no such adverse event were reported/occurred.[7,8] Thus, there is possibility that the error was at the level of production/manufacture of that particular batch which was administered to the affected persons. Human errors such as mislabeling or contamination also cannot be ruled out.
RECOMMENDATIONS FOR FUTURE PHASE I CLINICAL TRIALS
Till February 25, 2016, 209,194 global clinical trials have been registered on clinicaltrial.gov.[15] History of clinical trials has been very troubled throughout. Every incident came as a lesson for regulatory bodies and the researching agencies. To safeguard the participants’, time to time new laws and guidelines were framed. This incident with BIA 10–2474 is one of the two most bizarre in the last 10 years, another being with TGN1412.
Once the investigation by health agency proves that BIA 10–2474 used at the trial was formulated correctly, complied with its product specification and was free from pro-inflammatory contaminants. Then, the other possibilities for failure of trials such as off-target effect, dose calculation, unexpected immune response, species variation, and cumulative dose toxicity would be sought.
Such incidents not only alarm the regulatory authorities and the pharmaceutical researchers but also it gives a discouraging message to human volunteers to participate in the clinical trials. One lesson that regulatory authorities should learn after TGN1412 and BIA 10–1412 mishaps that no drug in perfectly safe and thus amend regulations to include patients instead of healthy volunteers in Phase I trial as done for anticancer drugs.
The second recommendation is to give importance to preclinical data by the sponsor, investigator, ethics committee, and regulatory authorities. Sufficient preclinical pharmacology studies should be conducted on a sufficiently broad dose range which is fairly predictive of real life future therapeutic efficacy. Specifically, the toxicological studies even for the “low risk” novel compounds should be evaluated in primates close to humans such as cynomolgus monkeys, chimpanzees, or Bonobos to predict the possible toxic effects.
The third recommendation to make the patient population more suitable and safe, for compounds which act through central nervous system, it is advised that for the subject selection, inclusion, and follow-up the neuropsychological assessment of the subjects with clinical interview and cognitive tests should be performed. Such tests were performed for other FAAH inhibitor trials but not for BIA 10–2474.
The fourth recommendation that all the drugs used for the first time in humans should be treated as “high risk” molecules and MABEL, not NOAEL should be used for the calculation of first in human dose. The fifth recommendation is that even for “low risk” molecules, MABEL should be used for calculation of dose for Phase I studies.
The sixth recommendation would be with regard to the design of Phase I trials, N of 1 randomized control trial[16] should be used in place of 6 + 2 design. N of 1 trials are trial design in which the patient undergoes pairs of treatment periods organized so that one period involves the use of the experimental treatment and the other involves the use of an alternate or placebo therapy [Figure 1].[17] It can be double blinded, i.e., both the patient and physician are blinded and outcomes are monitored, if possible. Treatment periods are replicated until the clinician and patient are convinced that the treatments are definitely different or definitely not different. Usually, the pair of interventions varies from two to seven and is not prespecified. This design will help as each subject will act as his or her own control decreasing the inter-individual variability which is a drawback of randomized control trials. Second, it will be helpful in determining when symptoms or adverse event may be caused by medication. The design, characteristics, and follow-up of participants in N of 1 trials in shown in Figure 1.
Figure 1.
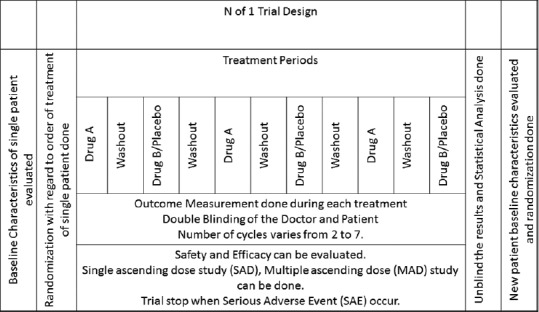
Flowchart of N of 1 trials showing the design, characteristics, and follow-up of participants
CONCLUSION
No FAAH inhibitor is yet approved for therapeutic use. Incident with BIA 10–2474 is the only tragic happening with FAAH inhibitors so far, other trials involving FAAH inhibitors were discontinued due to lack of efficacy and no safety concern was found with them. Therefore, it cannot be predicted that in future safety issues would be with all FAAH inhibitors. Maybe in future, FAAH inhibitors which are reversible, highly specific, with shorter duration of action, and better safety profile are developed. Although rules, regulations, and guidelines need to be modified to make trials more damage proof and avoid such mishaps in future.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Lau BK, Vaughan CW. Targeting the endogenous cannabinoid system to treat neuropathic pain. Front Pharmacol. 2014;5:28. doi: 10.3389/fphar.2014.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fowler CJ, Jonsson KO, Tiger G. Fatty acid amide hydrolase: Biochemistry, pharmacology, and therapeutic possibilities for an enzyme hydrolyzing anandamide, 2-arachidonoylglycerol, palmitoylethanolamide, and oleamide. Biochem Pharmacol. 2001;62:517–26. doi: 10.1016/s0006-2952(01)00712-2. [DOI] [PubMed] [Google Scholar]
- 3.Cravatt BF, Lichtman AH. Fatty acid amide hydrolase: An emerging therapeutic target in the endocannabinoid system. Curr Opin Chem Biol. 2003;7:469–75. doi: 10.1016/s1367-5931(03)00079-6. [DOI] [PubMed] [Google Scholar]
- 4.Otrubova K, Ezzili C, Boger DL. The discovery and development of inhibitors of fatty acid amide hydrolase (FAAH) Bioorg Med Chem Lett. 2011;21:4674–85. doi: 10.1016/j.bmcl.2011.06.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scientists Speculate on What Caused the Bial Drug Testing Tragedy in France – Forbes. [Last cited on 2016 Feb 24]. Available from: http://www.forbes.com/sites/davidkroll/2016/01/18/scientists-speculate-on-what-caused-the-bial-drug-testing-tragedy-in-france/#604bbceb301f .
- 6. [Last cited on 2016 Feb 24]. Available from: http://www.ansm.sante.fr/content/download/84681/1069223/version/1/file/protocole_BIAL_102474+101.22012016131259.pdf .
- 7.A Study to Investigate Whether PF.04457845 is Effective in Treating Pain, is Safe and Tolerable in Patients with Osteoarthritis of the Knee – Full Text View. [Last cited on 2016 Feb 18]. Available from: https://www.clinicaltrials.gov/ct2/show/NCT00981357 .
- 8.A Safety, Tolerability and Efficacy Study of V158866 in Central Neuropathic Pain Following Spinal Cord Injury – Full Text View. [Last cited on 2016 Feb 24]. Available from: https://www.clinicaltrials.gov/ct2/show/NCT01748695 .
- 9.Guideline on Requirements for First-in-Man Clinical Trials for Potential High-Risk Medicinal Products. [Last cited on 2016 Feb 24]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002989.pdf .
- 10.Attarwala H. TGN1412: From discovery to disaster. J Young Pharm. 2010;2:332–6. doi: 10.4103/0975-1483.66810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calvey N, Williams N. Principles and Practice of Pharmacology for Anaesthetists. Oxford, London: Blackwell publishing, John Wiley & Sons; 2009. p. 375. [Google Scholar]
- 12.Report by the Temporary Specialist Scientific Committee (TSSC), “FAAH (Fatty Acid Amide Hydrolase)”, on the Causes of the Accident during a Phase 1 Clinical Trial. [Last cited on 2016 May 24]. Available from: http://www.ansm.sante.fr/var/ansm_site/storage/original/application/744c7c6daf96b141bc9509e2f85c227e.pdf .
- 13.Scientists Speculate on What Caused the Bial Drug Testing Tragedy in France. Forbes. [Last cited on 2016 Feb 25]. Available from: http://www.forbes.com/sites/davidkroll/2016/01/18/scientists-speculate-on-what-caused-the-bial-drug-testing-tragedy-infrance/
- 14.Smith DA. (RSC Drug Discovery) London, UK: The Royal Society of Chemistry; 2010. Metabolism, Pharmacokinetics and Toxicity of Functional Groups; p. P001. [Google Scholar]
- 15. [Last cited on 2016 Feb 25]. Available from: https://www.clinicaltrials.gov/
- 16.Lillie EO, Patay B, Diamant J, Issell B, Topol EJ, Schork NJ. The n-of-1 clinical trial: The ultimate strategy for individualizing medicine? Per Med. 2011;8:161–73. doi: 10.2217/pme.11.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Straus SE, Glasziou P, Richardson WS, Haynes RB, editors. Evidence-Based Medicine: How to Practice and Teach It. 4th ed. Edinburgh, UK: Churchill Livingstone Elsevier; 2011. Therapy; pp. 67–136. [Google Scholar]