

Keywords: nerve regeneration, endoplasmic reticulum stress, apoptosis, caspase-12, caspase-3, traumatic penumbra, traumatic brain injury, neural regeneration
Abstract
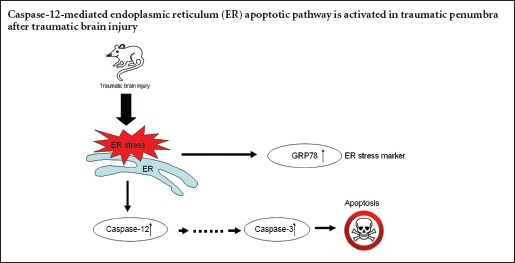
Neuronal apoptosis is mediated by intrinsic and extrinsic signaling pathways such as the membrane-mediated, mitochondrial, and endoplasmic reticulum stress pathways. Few studies have examined the endoplasmic reticulum-mediated apoptosis pathway in the penumbra after traumatic brain injury, and it remains unclear whether endoplasmic reticulum stress can activate the caspase-12-dependent apoptotic pathway in the traumatic penumbra. Here, we established rat models of fluid percussion-induced traumatic brain injury and found that protein expression of caspase-12, caspase-3 and the endoplasmic reticulum stress marker 78 kDa glucose-regulated protein increased in the traumatic penumbra 6 hours after injury and peaked at 24 hours. Furthermore, numbers of terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling-positive cells in the traumatic penumbra also reached peak levels 24 hours after injury. These findings suggest that caspase-12-mediated endoplasmic reticulum-related apoptosis is activated in the traumatic penumbra, and may play an important role in the pathophysiology of secondary brain injury.
Introduction
Traumatic brain injury (TBI) is a leading cause of injury-related long-term disability and death in patients under the age of 45 years (Langlois et al., 2006). In addition to the primary insult, the irreversible deterioration of the traumatic penumbra without timely intervention and treatment at the early stage is, to a large extent, responsible for the poor prognosis. The traumatic penumbra is the potentially salvageable brain tissue surrounding the primary lesion, in which secondary brain injury develops after the primary insult (Stoffel et al., 1997; Harish et al., 2015). Following TBI, brain lesions are not limited to the site of the primary neurotrauma, but expand progressively and centrifugally. There is evidence that focal necrosis increases over time and the volume of necrotic tissue can reach 400% of the initial lesion 24 hours after impact (Stoffel et al., 2002). Clinical studies have also demonstrated that expansion of the penumbra impairs cerebral blood flow and leads to edema and compromised local metabolism, resulting in clinical deterioration (Newcombe et al., 2013; Wu et al., 2013; Sheriff and Hinson, 2015). Pathophysiologically, the traumatic penumbra involves a series of damage cascades such as glutamate excitotoxicity, loss of ionic homeostasis, inflammation, and oxidative stress, and eventually gives rise to neuronal apoptosis (Rosenfeld et al., 2012; Ding et al., 2015; Sheriff and Hinson, 2015).
Apoptosis, or programmed cell death, is one of the main factors affecting the outcome and prognosis of TBI. Previous studies have demonstrated neuronal apoptosis in the contusion site and penumbra in animal models and patients with TBI (Ang et al., 2003; Zhang et al., 2005). Furthermore, approximately two-thirds of cell death might be attributable to apoptosis in traumatic penumbra (Zhang et al., 2005; Ziebell et al., 2011). Intrinsic and extrinsic signaling pathways mediate neuronal apoptosis, including membrane-mediated, mitochondrial, and endoplasmic reticulum (ER) stress pathways (Jin et al., 2015; Li et al., 2015; Chuang et al., 2016). Previous studies have focused on investigating the former two apoptotic pathways in the traumatic penumbra, but few reports have addressed the ER stress-induced pathway (Yoneda et al., 2001; Long et al., 2013; Han et al., 2014; Kabadi and Faden, 2014).
The caspase family of cysteine proteases plays a critical role in the caspase-12-dependent apoptotic pathway, in which caspase-12 and caspase-3 function as an upstream initiator and downstream effector, respectively, of the apoptotic pathway. When cells are exposed to apoptotic stimuli such as oxidative stress, caspase-12 is activated by autoproteolytic modification, and leads directly or indirectly to the cleavage of caspase-3 (Zhao et al., 2013). During late-stage apoptosis, caspase-3 executes the dismantling of the cell and the formation of apoptotic bodies.
The ER-localized molecular chaperone 78 kDa glucose-regulated protein (GRP78) is a marker of the ER, and its upregulation indicates ER stress (Ma et al., 2008; Martínez-Pizarro et al., 2016). It is well known that oxidative stress, including ER stress, is involved in neural cell death. However, the involvement of the ER stress-induced caspase-12-mediated apoptotic pathway in the traumatic penumbra is not completely clear. We hypothesized that this pathway is activated in the traumatic penumbra after TBI. To test this hypothesis, we measured neuronal apoptosis and protein expression of caspase-12, caspase-3 and GRP78 in rats at various time points after TBI.
Materials and Methods
Ethics statement
Animal studies were approved by the Institutional Animal Care and Use Committee of Hebei Medical University (approval No. HbMUEC-130306) and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Precautions were taken to minimize suffering and the number of animals used in each experiment.
Experimental animals
Sixty male healthy Sprague-Dawley rats, aged 9–10 weeks and weighing 300–350 g, were purchased from the Laboratory Animal Center of Hebei Medical University, China (license No. SCXK (Ji) 2013-1-003). Rats were housed in separate cages at 22–25°C and 50–70% humidity under a 12-hour reversed light/dark cycle with food and water ad libitum. The animals were habituated under these conditions for at least 1 week before the start of the experiment. Rats were randomly divided into sham-operated (n = 12) and TBI (n = 48) groups. The TBI group was further divided into four subgroups (n = 12) to be sacrificed at different time points (6, 12, 24 and 72 hours after TBI).
Establishment of TBI models
All surgical procedures were performed under aseptic conditions. TBI models were established using a fluid percussion device (HPD-1700, Dragonfly R&D, Silver Spring, MD, USA). Rats were anesthetized with an intraperitoneal injection of sodium pentobarbital (100 mg/kg) and fixed in a stereotaxic frame in the prone position. A 5 mm craniotomy was performed over the right parietal cortex (3.8 mm posterior to and 2.5 mm lateral to the bregma), keeping the dura intact (Tomura et al., 2011). A plastic Luer Lock hub (BD, Franklin Lakes, NJ, USA) was secured over the skull window with dental acrylic cement. All rats in the TBI group were subjected to fluid percussion injury (FPI) of high grade severity (263–304 kPa, 16 ms in duration). The Luer Lock was removed immediately after FPI. Sham-operated animals were subjected to all surgical procedures except FPI. Following scalp suturing, rats were returned to their home cages with food and water available ad libitum.
Brain tissue preparation
Rats were anesthetized (100 mg/kg sodium pentobarbital intraperitoneally) at 6, 12, 24 or 72 hours after TBI, according to group allocation. For terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) and immunohistochemistry, rats (n = 6 per subgroup) were perfused via the left cardiac ventricle with saline at 4°C followed by 4% neutral-buffered formalin solution. Brains were quickly harvested and placed on a frozen plate. Tissue samples were rapidly taken from the region adjacent to the site of injury (7–12 mm from frontal polar region), preserved overnight in 4% neutral-buffered formalin solution, and embedded in paraffin. For western blot analysis, rats (n = 6 per subgroup) were exsanguinated via cardiac puncture. Brain tissue was harvested as described above, and immediately stored at −80°C in liquid nitrogen until western blot analysis.
Immunohistochemistry
Paraffin-embedded samples were placed in a microtome, serially sliced into 4 μm thick sections, and routinely deparaffinized. H2O2/methanol (3%) was added to block endogenous peroxidase activity. PBS containing 10% normal goat serum was used to block nonspecific antibody binding. Sections were incubated overnight at 4°C with anti-caspase-12 (1:100; Cell Signaling, Beverly, MA, USA) and anti-caspase-3 (1:100; Cell Signaling) rabbit polyclonal antibodies. The sections were washed three times in PBS for 5 minutes each time, and then incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 60 minutes. Finally, 3,3-diaminobenzidine/H2O2 solution was used to visualize caspase-12 and caspase-3 expression. The sections were counterstained with hematoxylin for 4 minutes to visualize the nuclei, and then mounted. Optical density of immunoreactive cells was calculated using Image-Pro Plus 6.0 (Media Cybernetics, Rockville, MD, USA).
Western blot analysis
Frozen brain samples were thawed and homogenized in seven volumes of ice-cold lysate buffer (200 mM HEPES [pH 7.5], 250 mM sucrose, 1 mM dithiothreitol, 1.5 mM MgCl2, 10 mM KCl, 1 mM EDTA, 1 mM EGTA, 0.1 mM phenylmethylsulfonyl fluoride, 10 μg/mL leupeptin, 5 μg/mL pepstatin, 2 μg/mL aprotinin). The homogenates were centrifuged at 3,000 × g for 10 minutes at 4°C and the supernatant was then centrifuged at 8,000 × g for 20 minutes at the same temperature. The resulting supernatant was collected and further centrifuged at 54,000 × g for 60 minutes at 4°C. The pellet was identified as the microsome fraction, which is enriched in ER, and the supernatant as the cytosolic fraction. The Bradford assay (Bio-Rad Laboratories, Hercules, CA, USA) was used to determine the protein concentration of each sample against albumin standards.
For the microsomal samples, aliquots (60 μg) of each sample were subjected to 10% or 12.5% sodium dodecylsulfate-polyacrylamide gel electrophoresis. The separated protein was then transferred from the gel to a polyvinylidene difluoride membrane (Gibco, New York, NY, USA) at 120 V for 50 minutes at room temperature. For immunoblotting, nonspecific protein binding to the membrane was blocked with 5% non-fat dried milk for 2 hours at room temperature, then incubated with the following primary antibodies, all raised in rabbit: anti-mouse GRP78 monoclonal antibody (1:500; Santa Cruz Biotechnology), anti-caspase-12 polyclonal antibody (1:500; Santa Cruz Biotechnology) and anti-caspase-3 polyclonal antibody (1:750; Santa Cruz Biotechnology). The secondary antibody was horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (1:2,000; Cell Signaling, Beverly, MA, USA), and rabbit monoclonal anti-β-actin antibody (1:10,000; Cell Signaling) was used as an endogenous control for all samples. SeeBlue®Plus2 Pre-Stained Standard (Life Technologies, Carlsbad, CA, USA) was used to determine molecular weights. The reaction was visualized using LumiGLO chemiluminescent substrate (Cell Signaling) according to the manufacturer's instructions. The bands were scanned and analyzed using optical density values with background subtraction and normalized to β-actin using ImageJ software (NIH).
TUNEL analysis
Paraffin-embedded brain tissues were sliced into 4 μm thick sections in a microtome (Leica Biosystems, Nussloch, Germany). Apoptotic cells were identified using a TUNEL detection kit (DeadEnd™ TUNEL system, Promega, Madison WI, USA) according to the manufacturer's instructions. In brief, the sections were deparaffinized, rehydrated, and then digested in proteinase K (20 μg/mL) at room temperature for 15 minutes. After being washed with PBS for 5 minutes, sections were incorporated with biotinylated nucleotides at the 30-OH DNA end using terminal deoxyribonucleotide transferase (TdT) for 1 hour at 37°C. Negative controls were incubated with label solution not containing TdT. Sections were then incubated with FITC-conjugated avidin, counterstained for nuclei with DAPI, and positive cells were identified, counted, and analyzed under a fluorescence microscope (Olympus, Tokyo, Japan) by an investigator blinded to the grouping. The apoptotic index was defined as the average percentage of TUNEL-positive cells in each section counted in 10 randomly selected cortical microscopic fields (at 400× magnification).
Statistical analysis
All experimental data are expressed as the mean ± SD. Statistical differences between sham-operated and TBI groups were determined by one-way analysis of variance with Tukey's test (SPSS 13.0 software, SPSS, Chicago, IL, USA). P < 0.05 was considered statistically significant.
Results
GRP78 expression in traumatic penumbra after TBI
The expression of GRP78 increased markedly in the penumbra from 6 hours after TBI, and peaked at 24 hours (P < 0.05); GRP78 expression was significantly elevated compared with the sham value at all time points examined after injury (P < 0.05) (Figure 1).
Figure 1.

Time course of 78 kDa glucose-regulated protein (GRP78) expression in the penumbra at 6, 12, 24 and 72 hours (h) after traumatic brain injury (TBI).
(A) Western blot of GRP78 and β-actin. (B) Quantification of GRP78 expression. Optical density of GRP78 bands was normalized to that of β-actin. Data are expressed as the mean ± SD; n = 6 rats per group. *P < 0.05, vs. sham-operated (sham) group (one-way analysis of variance with Tukey's test).
Caspase-12 expression in penumbra after TBI
Western blot assay showed that caspase-12 protein expression in the traumatic penumbra was significantly greater than that in sham-operated rats at all time points examined, with maximal upregulation observed 24 hours after TBI (P < 0.05; Figure 2A). The same trend was observed with immunohistochemical staining (P < 0.05; Figure 2B, C).
Figure 2.

Time course of caspase-12 expression in penumbra at 6, 12, 24 and 72 hours (h) after traumatic brain injury (TBI).
(A) Western blot. Optical density of caspase-12 bands was normalized to that of β-actin. (B) Representative photomicrographs showing elevated immunoreactivity of caspase-12 in traumatic penumbra compared with sham-operated rats. Arrows: Caspase-12-positive cells. Bar: 50 μm. (C) Quantification of caspase-12 immunoreactivity in traumatic penumbra normalized to average optical density (AOD). Data are expressed as the mean ± SD; n = 6 rats per group. *P < 0.05, vs. sham-operated (sham) group (one-way analysis of variance with Tukey's test).
Caspase-3 expression in penumbra after TBI
Western blot analysis revealed that caspase-3 protein expression in the traumatic penumbra was significantly elevated compared with the sham-operated group at all time points examined (P < 0.05), and peak expression was observed at 24 hours after TBI (P < 0.05; Figure 3A). Immunohistochemistry results were consistent with western blot results (P < 0.05; Figure 3B, C).
Figure 3.

Time course of caspase-3 expression in traumatic penumbra at 6, 12, 24, and 72 hours (h) after traumatic brain injury (TBI).
(A) Western blot. Optical density of caspase-3 bands was normalized to that of β-actin. (B) Representative photomicrographs showing elevated immunoreactivity of caspase-3 in traumatic penumbra after TBI. Arrows: Caspase-3-positive cells. Bar: 50 μm. (C) Quantification of caspase-3 immunonreactivity (average optical density, AOD) in traumatic penumbra. Data are expressed as the mean ± SD, n = 6 rats per group.*P < 0.05, vs. sham-operated (sham) group (one-way analysis of variance with Tukey's test).
Time course of cell apoptosis in penumbra after TBI
There were few TUNEL-positive cells in sham-operated rat brains. However, after TBI, significantly more TUNEL-positive cells were observed in the traumatic penumbra at all time points examined, with the greatest number observed at 24 hours (P < 0.05; Figure 4).
Figure 4.

Cell apoptosis in penumbra after traumatic brain injury (TBI).
(A) Representative images showing TUNEL immunofluorescence in the traumatic penumbra at 6, 12, 24 and 72 hours (h) after TBI. Bar: 50 μm. (B) Apoptotic index, defined as the average percentage of TUNEL-positive cells in each section counted in 10 randomly selected cortical microscopic fields (at 400× magnification). Data are expressed as the mean ± SD; n = 6 rats per group. *P < 0.05, vs. sham-operated (sham) group (one-way analysis of variance with Tukey's test).
Discussion
ER stress activates the intrinsic and extrinsic apoptotic pathways in response to different stimuli (Jin et al., 2015; Li et al., 2015; Chuang et al., 2016). ER stress-induced apoptosis has been implicated in the development of several neurodegenerative conditions, including Alzheimer's and Parkinson's diseases and ischemic damage (Chakrabarti and Mohanakumar, 2016; Li et al., 2016; Wei et al., 2016), and activates the JNK, CAAT/enhancer binding protein homologous protein (CHOP), and GSK3/3β pathways (Liu et al., 2013; Shah et al., 2016). Prolonged ER stress from the abovementioned conditions also induces overexpression of caspase-12. Increased expression of caspase-12 triggers the caspase-12-mediated apoptotic pathway (Brewster et al., 2006; Liu et al., 2013). Enhanced caspase-12 immunoreactivity is also observed in the contusion-damaged region in models of controlled cortical impact TBI and blast-induced TBI (Larner et al., 2004; Logsdon et al., 2014). However, whether the ER stress-induced caspase-12 apoptotic pathway is activated in the traumatic penumbra has not, to date, been confirmed. The findings of the present study show that caspase-12 expression is elevated in the traumatic penumbra. A similar result was observed in rat models of spinal cord injury (Lee et al., 2014). Our results also show that elevated caspase-12 protein expression had the same temporal profile as caspase-3, and the time course of the expression of these proteins was consistent with that of neuronal apoptosis. These data indicate that the caspase-12-mediated ER apoptotic pathway is activated in the traumatic penumbra after TBI.
The ER is highly sensitive to changes in intraluminal calcium and redox status, and some conditions, such as ischemia, hypoxia and nutrient deprivation, give rise to ER stress—a condition in which unfolded or misfolded proteins accumulate. To prevent the adverse effects of ER stress, cells and their organelles have developed a series of protective programs, collectively named the unfolded protein response (UPR), to cope with defective proteins. In one major pathway of the UPR, expression of ER-localized molecular chaperones, such as GRP78, is increased to repair unfolded proteins. Moreover, the induction of GRP78 has been widely identified as a marker for ER stress and initiation of the UPR (Zhao et al., 2013). In the present study, the expression of GRP78 protein was markedly upregulated in the traumatic penumbra, suggesting that TBI actives ER stress in secondary brain injury in rats. Increased GRP78 has recently been reported in ER stress-mediated apoptosis, in cultured hippocampal neurons during glucose deprivation and in hippocampal cell damage induced by recurrent febrile seizures (de la Cadena et al., 2014; Han et al., 2015).
When ER stress conditions persist, three ER stress-mediated apoptotic pathways are triggered, including the transcriptional induction of the caspase-12-dependent pathway. Caspase-12 is specifically localized to the cytoplasmic side of the ER and is the key regulator specific to ER stress-induced caspase-12-dependent apoptosis (Brewster et al., 2006). These distinct but overlapping pathogeneses in ischemia and neurodegenerative diseases result in the imbalances in energy metabolism, redox status and intracellular calcium concentration that occur after the primary insult, and are all factors that initiate ER stress and caspase-12 upregulation (Rosenfeld et al., 2012; Liu et al., 2013; Sheriff and Hinson, 2015). The findings of the present study demonstrate that caspase-12 protein expression and neuronal apoptosis follow the same temporal pattern, initially increasing at 6 hours, and peaking at 24 hours post-injury. This suggests that TBI activates caspase-12, the key factor that initiates ER stress-mediated apoptosis, in the traumatic penumbra.
Activation of caspase-3 is the point of convergence of the intrinsic and extrinsic apoptotic pathways, and activated caspase-3 executes cell apoptosis by cleaving cytoskeletal and nuclear proteins, chromatin-modifying proteins, DNA repair proteins and endonucleases (Morishima et al., 2002). In the present study, increased protein expression of caspase-3 with the same temporal profile as that of caspase-12 strongly suggests that caspase-12, directly or indirectly, activates caspase-3, contributing to the downstream apoptotic pathway in the traumatic penumbra. Previous studies in neuroblastoma cells have shown that caspase-12 activates caspase-9 which, in turn, activates caspase-3, during apoptosis associated with ER stress (Martinez et al., 2010; Borkham-Kamphorst et al., 2016). Recent research has confirmed that caspase-12 can also be activated by the caspase-3 pathway, initiating a positive feedback loop that further activates caspase-3 via caspase-9, thus potentiating apoptosis (Nakagawa et al., 2000; Zhang et al., 2013; Liu et al., 2014). Caspase-12−/− mice are resistant to ER stress-induced apoptosis but sensitive to other death stimuli, indicating that caspase-12 is a specific regulator of ER stress-induced apoptosis (Nakagawa et al., 2000). Together, these data indicate that the caspase-12-mediated ER apoptotic pathway is activated in the penumbra after TBI, and may play an important role in the pathophysiology of secondary brain injury.
This study was unable to provide detailed evidence that caspase-12 gene knockout downregulates the expression of caspase-3 and neuronal apoptosis in the traumatic penumbra, by which the key role of caspase-12 has been previously verified in this apoptotic pathway. Caspase-4 is the human equivalent of rat caspase-12; it is localized to the ER and has the same function as caspase-12 (Hitomi et al., 2004). Rat models of TBI cannot completely embody the activation of the ER stress-induced caspase-12 apoptotic pathway in the traumatic penumbra in humans. Additionally, a proper control would be to examine 6 sham-operated rats at each time point (6, 12, 24 and 72 hours after surgery) and compare TBI rats with sham rats at each time point, which would be more scientific.
In summary, we have outlined the time course of caspase-12 upregulation in the traumatic penumbra for the first time, and revealed that TBI activates the caspase-12-mediated ER apoptotic pathway. The ER stress and caspase-12-mediated ER apoptotic pathway might be a new target in the treatment of TBI.
Footnotes
Funding: This study was supported by the Natural Science Foundation of Hebei Province of China, No. H2014206383; and Foundation for High-Level Personnel Projects in Hebei Province of China, No. A201401041.
Conflicts of interest: None declared.
Plagiarism check: This paper was screened twice using Cross-Check to verify originality before publication.
Peer review: This paper was double-blinded and stringently reviewed by international expert reviewers.
Copyedited by Slone-Murphy J, Raye W, Yu J, Song LP, Zhao M
References
- Ang BT, Yap E, Lim J, Tan WL, Ng PY, Ng I, Yeo TT. Poly(adenosine diphosphate-ribose) polymerase expression in human traumatic brain injury. J Neurosurg. 2003;99:125–130. doi: 10.3171/jns.2003.99.1.0125. [DOI] [PubMed] [Google Scholar]
- Borkham-Kamphorst E, Steffen BT, Van de Leur E, Haas U, Tihaa L, Friedman SL, Weiskirchen R. CCN1/CYR61 overexpression in hepatic stellate cells induces ER stress-related apoptosis. Cell Signal. 2016;28:34–42. doi: 10.1016/j.cellsig.2015.10.013. [DOI] [PubMed] [Google Scholar]
- Brewster JL, Linseman DA, Bouchard RJ, Loucks FA, Precht TA, Esch EA, Heidenreich KA. Endoplasmic reticulum stress and trophic factor withdrawal activate distinct signaling cascades that induce glycogen synthase kinase-3β and a caspase-9-dependent apoptosis in cerebellar granule neurons. Mol Cell Neurosci. 2006;32:242–253. doi: 10.1016/j.mcn.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Mohanakumar KP. Aging and neurodegeneration: a tangle of models and mechanisms. Aging Dis. 2016;7:111–113. doi: 10.14336/AD.2016.0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang WL, Lin PY, Lin HC, Chen YL. The apoptotic effect of ursolic acid on SK-Hep-1 cells is regulated by the PI3K/Akt p38 and JNK MAPK signaling pathways. Molecules. 2016 doi: 10.3390/molecules21040460. doi: 10.3390/molecules21040460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cadena SG, Hernández-Fonseca K, Camacho-Arroyo I, Massieu L. Glucose deprivation induces reticulum stress by the PERK pathway and caspase-7- and calpain-mediated caspase-12 activation. Apoptosis. 2014;19:414–427. doi: 10.1007/s10495-013-0930-7. [DOI] [PubMed] [Google Scholar]
- Ding K, Wang H, Wu Y, Zhang L, Xu J, Li T, Ding Y, Zhu L, He J. Rapamycin protects against apoptotic neuronal death and improves neurologic function after traumatic brain injury in mice via modulation of the mTOR-p53-Bax axis. J Surg Res. 2015;194:239–247. doi: 10.1016/j.jss.2014.09.026. [DOI] [PubMed] [Google Scholar]
- Han Y, Yi W, Qin J, Zhao Y, Zhang J, Chang X. Carbon monoxide offers neuroprotection from hippocampal cell damage induced by recurrent febrile seizures through the PERK-activated ER stress pathway. Neurosci Lett. 2015;585:126–131. doi: 10.1016/j.neulet.2014.11.040. [DOI] [PubMed] [Google Scholar]
- Han Z, Chen F, Ge X, Tan J, Lei P, Zhang J. miR-21 alleviated apoptosis of cortical neurons through promoting PTEN-Akt signaling pathway in vitro after experimental traumatic brain injury. Brain Res. 2014;1582:12–20. doi: 10.1016/j.brainres.2014.07.045. [DOI] [PubMed] [Google Scholar]
- Harish G, Mahadevan A, Pruthi N, Sreenivasamurthy SK, Puttamallesh VN, Keshava Prasad TS, Shankar SK, Srinivas Bharath MM. Characterization of traumatic brain injury in human brains reveals distinct cellular and molecular changes in contusion and pericontusion. J Neurochem. 2015;134:156–172. doi: 10.1111/jnc.13082. [DOI] [PubMed] [Google Scholar]
- Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, Tsujimoto Y, Tohyama M. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Aβ-induced cell death. J Cell Biol. 2004;165:347–356. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Yang YW, Cheng WP, Lu JK, Hou SY, Dong XH, Liu SY. Serine-threonine protein kinase activation may be an effective target for reducing neuronal apoptosis after spinal cord injury. Neural Regen Res. 2015;10:1830–1835. doi: 10.4103/1673-5374.170313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabadi SV, Faden AI. Selective CDK inhibitors: promising candidates for future clinical traumatic brain injury trials. Neural Regen Res. 2014;9:1578–1580. doi: 10.4103/1673-5374.141779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. 2006;21:375–378. doi: 10.1097/00001199-200609000-00001. [DOI] [PubMed] [Google Scholar]
- Larner SF, Hayes RL, McKinsey DM, Pike BR, Wang KK. Increased expression and processing of caspase-12 after traumatic brain injury in rats. J Neurochem. 2004;88:78–90. doi: 10.1046/j.1471-4159.2003.02141.x. [DOI] [PubMed] [Google Scholar]
- Lee JY, Maeng S, Kang SR, Choi HY, Oh TH, Ju BG, Yune TY. Valproic acid protects motor neuron death by inhibiting oxidative stress and endoplasmic reticulum stress-mediated cytochrome C release after spinal cord injury. J Neurotrauma. 2014;31:582–594. doi: 10.1089/neu.2013.3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HF, Zhao SX, Xing BP, Sun ML. Ulinastatin suppresses endoplasmic reticulum stress and apoptosis in the hippocampus of rats with acute paraquat poisoning. Neural Regen Res. 2015;10:467–472. doi: 10.4103/1673-5374.153698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Liu K, Zhao J, Holscher C, Li GL, Liu YZ. Neuroprotective role of (Val(8))GLP-1-Glu-PAL in an in vitro model of Parkinson's disease. Neural Regen Res. 2016;11:326–331. doi: 10.4103/1673-5374.177742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Zhang M, Yin H. Signaling pathways involved in endoplasmic reticulum stress-induced neuronal apoptosis. Int J Neurosci. 2013;123:155–162. doi: 10.3109/00207454.2012.746974. [DOI] [PubMed] [Google Scholar]
- Liu H, Wang Z, Nowicki MJ. Caspase-12 mediates carbon tetrachloride-induced hepatocyte apoptosis in mice. World J Gastroenterol. 2014;20:18189–18198. doi: 10.3748/wjg.v20.i48.18189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon AF, Turner RC, Lucke-Wold BP, Robson MJ, Naser ZJ, Smith KE, Matsumoto RR, Huber JD, Rosen CL. Altering endoplasmic reticulum stress in a model of blast-induced traumatic brain injury controls cellular fate and ameliorates neuropsychiatric symptoms. Front Cell Neurosci. 2014;8:421. doi: 10.3389/fncel.2014.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J, Cai L, Li J, Zhang L, Yang H, Wang T. JNK3 involvement in nerve cell apoptosis and neurofunctional recovery after traumatic brain injury. Neural Regen Res. 2013;8:1491–1499. doi: 10.3969/j.issn.1673-5374.2013.16.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Han L, Gao Y, Li L, Shang X, Hu W, Xue C. The endoplasmic reticulum stress-mediated apoptosis signal pathway is involved in sepsis-induced abnormal lymphocyte apoptosis. Eur Surg Res. 2008;41:219–225. doi: 10.1159/000135631. [DOI] [PubMed] [Google Scholar]
- Martínez-Pizarro A, Desviat LR, Ugarte M, Pérez B, Richard E. Endoplasmic reticulum stress and autophagy in homocystinuria patients with remethylation defects. PLoS One. 2016;11:e0150357. doi: 10.1371/journal.pone.0150357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez JA, Zhang Z, Svetlov SI, Hayes RL, Wang KK, Larner SF. Calpain and caspase processing of caspase-12 contribute to the ER stress-induced cell death pathway in differentiated PC12 cells. Apoptosis. 2010;15:1480–1493. doi: 10.1007/s10495-010-0526-4. [DOI] [PubMed] [Google Scholar]
- Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem. 2002;277:34287–34294. doi: 10.1074/jbc.M204973200. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- Newcombe VF, Williams GB, Outtrim JG, Chatfield D, Gulia Abate M, Geeraerts T, Manktelow A, Room H, Mariappen L, Hutchinson PJ, Coles JP, Menon DK. Microstructural basis of contusion expansion in traumatic brain injury: insights from diffusion tensor imaging. J Cereb Blood Flow Metab. 2013;33:855–862. doi: 10.1038/jcbfm.2013.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JV, Maas AI, Bragge P, Morganti-Kossmann MC, Manley GT, Gruen RL. Early management of severe traumatic brain injury. Lancet. 2012;380:1088–1098. doi: 10.1016/S0140-6736(12)60864-2. [DOI] [PubMed] [Google Scholar]
- Shah A, Vaidya NK, Bhat HK, Kumar A. HIV-1 gp120 induces type-1 programmed cell death through ER stress employing IRE1α JNK and AP-1 pathway. Sci Rep. 2016;6:18929. doi: 10.1038/srep18929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheriff FG, Hinson HE. Pathophysiology and clinical management of moderate and severe traumatic brain injury in the ICU. Semin Neurol. 2015;35:42–49. doi: 10.1055/s-0035-1544238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffel M, Eriskat J, Plesnila M, Aggarwal N, Baethmann A. The penumbra zone of a traumatic cortical lesion: a microdialysis study of excitatory amino acid release. Acta Neurochir Suppl. 1997;70:91–93. doi: 10.1007/978-3-7091-6837-0_28. [DOI] [PubMed] [Google Scholar]
- Stoffel M, Rinecker M, Graf R, Baethmann A, Plesnila N. Nitric oxide in the penumbra of a focal cortical necrosis in rats. Neurosci Lett. 2002;324:201–204. doi: 10.1016/s0304-3940(02)00196-9. [DOI] [PubMed] [Google Scholar]
- Tomura S, Nawashiro H, Otani N, Uozumi Y, Toyooka T, Ohsumi A, Shima K. Effect of decompressive craniectomy on aquaporin-4 expression after lateral fluid percussion injury in rats. J Neurotrauma. 2011;28:237–243. doi: 10.1089/neu.2010.1443. [DOI] [PubMed] [Google Scholar]
- Wei XW, Hao LY, Qi SH. Inhibition on the S-nitrosylation of MKK4 can protect hippocampal CA1 neurons in rat cerebral ischemia/reperfusion. Brain Res Bull. 2016;124:123–128. doi: 10.1016/j.brainresbull.2016.04.005. [DOI] [PubMed] [Google Scholar]
- Wu HM, Huang SC, Vespa P, Hovda DA, Bergsneider M. Redefining the pericontusional penumbra following traumatic brain injury: evidence of deteriorating metabolic derangements based on positron emission tomography. J Neurotrauma. 2013;30:352–360. doi: 10.1089/neu.2012.2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, Tohyama M. Activation of Caspase-12 an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2 (TRAF2) dependent mechanism in response to the ER stress. J Biol Chem. 2001;276:13935–13940. doi: 10.1074/jbc.M010677200. [DOI] [PubMed] [Google Scholar]
- Zhang H, Li H, Liu X, Bi J. Effect of caspase-9 inhibition on endoplasmic reticulum stress induced cortical neuronal injury in rats. Int J Clin Exp Med. 2013;6:546–551. [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen Y, Jenkins LW, Kochanek PM, Clark RSB. Bench-to-bedside review: Apoptosis/programmed cell death triggered by traumatic brain injury. Crit Care. 2005;9:66–75. doi: 10.1186/cc2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Xiong Z, Mao X, Meng D, Lei Q, Li Y, Deng P, Chen M, Tu M, Lu X, Yang G, He G. Atmospheric pressure room temperature plasma jets facilitate oxidative and nitrative stress and lead to endoplasmic reticulum stress dependent apoptosis in HepG2 cells. PLoS One. 2013;8:e73665. doi: 10.1371/journal.pone.0073665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebell JM, Bye N, Semple BD, Kossmann T, Morganti-Kossmann MC. Attenuated neurological deficit, cell death and lesion volume in Fas-mutant mice is associated with altered neuroinflammation following traumatic brain injury. Brain Res. 2011;1414:94–105. doi: 10.1016/j.brainres.2011.07.056. [DOI] [PubMed] [Google Scholar]