

Abstract
High‐linear energy transfer (LET) heavy ions have been increasingly employed as a useful alternative to conventional photon radiotherapy. As recent studies suggested that high LET radiation mainly affects the nonhomologous end‐joining (NHEJ) pathway of DNA double strand break (DSB) repair, we further investigated this concept by evaluating the combined effect of an NHEJ inhibitor (NU7441) at a non‐toxic concentration and carbon ions. NU7441‐treated non‐small cell lung cancer (NSCLC) A549 and H1299 cells were irradiated with X‐rays and carbon ions (290 MeV/n, 50 keV/μm). Cell survival was measured by clonogenic assay. DNA DSB repair, cell cycle distribution, DNA fragmentation and cellular senescence induction were studied using a flow cytometer. Senescence‐associated protein p21 was detected by western blotting. In the present study, 0.3 μM of NU7441, nontoxic to both normal and tumor cells, caused a significant radio‐sensitization in tumor cells exposed to X‐rays and carbon ions. This concentration did not seem to cause inhibition of DNA DSB repair but induced a significant G2/M arrest, which was particularly emphasized in p53‐null H1299 cells treated with NU7441 and carbon ions. In addition, the combined treatment induced more DNA fragmentation and a higher degree of senescence in H1299 cells than in A549 cells, indicating that DNA‐PK inhibitor contributes to various modes of cell death in a p53‐dependent manner. In summary, NSCLC cells irradiated with carbon ions were radio‐sensitized by a low concentration of DNA‐PK inhibitor NU7441 through a strong G2/M cell cycle arrest. Our findings may contribute to further effective radiotherapy using heavy ions.
Keywords: DNAPK protein, G2 cell cycle arrest, heavy ions, non‐small cell lung cancer, p53 genes
Heavy‐ion radiation therapy has been increasingly utilized due to its lesser side effects compared to conventional photon radiation therapy;1, 2 tumor tissue can be well targeted by physically adjusting the Bragg peak on the tumor area, and a high‐linear energy transfer (LET) beam can induce more effective tumor cell killing than low‐LET beams.3, 4, 5 Another strategy to improve the outcome of radiotherapy would be to use a radio‐sensitizer, which enhances radiation‐induced cell killing by targeting DNA damage response.6, 7, 8, 9 Although many studies are available on low‐LET radiation combined with sensitizers, further biological studies are necessary in the combined regimen of radio‐sensitizer and heavy ions for future clinical applications.
There are two main pathways for DNA double‐strand break (DSB) repair: homologous recombination (HR) and nonhomologous end joining (NHEJ). NHEJ is a dominant pathway in DSB repair induced by low‐LET radiation, such as X‐rays and γ‐rays,10 while NHEJ has been shown to be suppressed to some extent and the HR pathway utilized more in DSB induced by high‐LET radiation.11, 12, 13
Furthermore, Takahashi et al.14 reported that when using HR and NHEJ pathway‐deficient mouse embryonic fibroblasts, high‐LET radiation caused significant radio‐sensitization even in NHEJ‐defective cells. Their study suggests that the NHEJ pathway is still dominant in DSB repair induced by high‐LET radiation. Therefore, it is important to study how cells with reduced or inhibited NHEJ respond to high‐LET heavy‐ion radiation. DNA‐PKcs is one of the key proteins in the NHEJ pathway, and many studies report radio‐sensitization as a consequence of DNA‐PK inhibition.15, 16, 17, 18 In particular, NU7441 has often been studied as an effective specific DNA‐PK inhibitor; this specificity was confirmed by the use of DNA‐PKcs defective cells, which did not show any radio‐sensitization with NU7441 treatment.9
In this study, we investigate the combined effect of NU7441 and carbon ion radiation in human non‐small cell lung cancer (NSCLC) cells. We find that the nontoxic concentration of NU7441 causes significant radio‐sensitization with X‐rays and carbon ions in NSCLC cells, with little effect on DSB repair; strong G2/M arrest is found to be associated with this sensitization.
Materials and Methods
Cell culture
Non‐small cell lung cancer cell lines A549 and H1299 cells were obtained from Riken BRC (Tsukuba, Japan) and the American Type Culture Collection (Manassas, VA, USA), respectively. A549 and H1299 were grown in MEMα and RPMI‐1640, respectively, and supplemented with 10% (v/v) FBS. Normal human fibroblast cell line HFL1 cells were obtained from Riken BRC and were grown in MEMα supplemented with 15% FBS. All cells were maintained in a 37°C incubator with 5% CO2. Exponentially growing cells were used in the present study.
Irradiation and drug treatment
Cells were irradiated with 290 MeV/n carbon ions (LET: 50 keV/μm) at the Heavy Ion Medical Accelerator in Chiba (HIMAC). The dose rate for carbon ions was 1 Gy/min. X‐ray irradiation was performed using a TITAN‐320 (200 kV, 20 mA, Shimadzu, Kyoto, Japan) at a dose rate of 1 Gy/min. NU7441, a specific DNA‐PK inhibitor, was purchased from Cayman Chemical (Ann Arbor, MI, USA). NU7441 was dissolved in DMSO and stored at −20°C in a freezer. Cells were pretreated with NU7441 1 h before irradiation, and the drug was kept throughout the experiment.
Clonogenic survival assay
Cells were seeded at concentrations that may yield approximately 50–100 colonies per dish after 10–12 days. The colonies were fixed with 100% ethanol and stained with 0.1% crystal violet solution. The colonies containing more than 50 cells were scored.
Flow cytometry
γ‐H2AX (a DSB marker), cell cycle distribution and senescence‐associated beta‐galactosidase (SA‐βGal) signals were analyzed using a FACS Calibur (BD Biosciences, Franklin Lakes, NJ, USA) flow cytometer. For detection of DSB, the γ‐H2AX signal was measured according to protocol described elsewhere.19 Cells were trypsinized, washed with cold PBS twice and stained with γ‐H2AX antibody FITC conjugate (0.6 μg/mL; Merck Millipore, Billerica, MA, USA) on ice overnight. The cells were then suspended and analyzed. For cell cycle analysis, cells were trypsinized, washed with cold PBS, fixed in 70% ethanol and stored at −20°C. Cells were stained with propidium iodide (PI) at 50 μg/mL in the presence of RNase (1 mg/mL) for 30 min on ice and then analyzed. For the SA‐βGal assay, the SA‐βGal signal was measured according to a protocol described elsewhere.20 Cells were labeled with C12FDG (20 μg/mL; Setareh Biotech, Eugene, OR, USA) as fluorescent substrates of beta‐galactosidase for 2 h in culture medium 3 days after irradiation, washed with PBS twice, trypsinized, resuspended in cold PBS and then analyzed. At least 10 000 cells were counted for analysis in all assays.
Western blotting
Anti‐p21 and anti‐GAPDH antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). For immunoblot analysis, proteins were separated by SDS‐PAGE, transferred onto Immobilon‐P membranes (Merck Millipore), and detected using the Western‐Light Plus Chemiluminescent Detection System (Tropix, Bedford, MA, USA) using appropriate secondary antibodies. Quantification was carried out using an LAS‐4000 mini luminoimaging analyzer (Fujifilm, Tokyo, Japan).
Statistical analysis
All data were analyzed using GraphPad Prism 6 (GraphPad, La Jolla, CA, USA). Statistical comparison of the mean values was performed using Student's t‐test and P < 0.05 was considered to be statistically significant.
Results
Non‐toxic concentration of NU7441 induces radio‐sensitization in non‐small cell lung cancer cells irradiated with low‐LET and high‐LET radiation
Cellular toxicity of NU7441 was evaluated in normal HFL1 cells and NSCLC cells using a clonogenic survival assay. HFL1 cells showed a concentration‐dependent decrease in plating efficiency, and this was more distinct for the concentrations higher than 1 μM (Fig. 1a). NSCLC cells showed a clear drop in plating efficiency at 3 μM (Fig. 1b). These results indicate that 0.3 μM of NU7441 is nontoxic in both normal and cancer cells. A clonogenic survival assay was performed with this nontoxic concentration of NU7441. Significant radio‐sensitization was confirmed in NU7441‐treated NSCLC cells, not only with X‐rays but also with carbon ions (Fig. 1c,d). The relative biological effectiveness (RBE) of carbon ions (50 keV/μm) compared to X‐rays and the sensitization enhancement ratio (SER) of NU7441 (0.3 μM) were calculated based on the D 10 values. Carbon ions showed an RBE of 2.15 in A549 cells and 1.87 in H1299 cells. NU7441 gave an SER of 1.77 (X‐rays) and 1.55 (carbon ions) in A549 cells and 1.94 (X‐ray) and 1.58 (carbon ions) in H1299 cells. Judging from these analyses, carbon ions caused more effective cell killing in A549 than H1299 cells, while NU7441 induced higher radio‐sensitization with X‐rays than carbon ions in H1299 cells, and the degree of sensitization was smaller for A549 cells.
Figure 1.
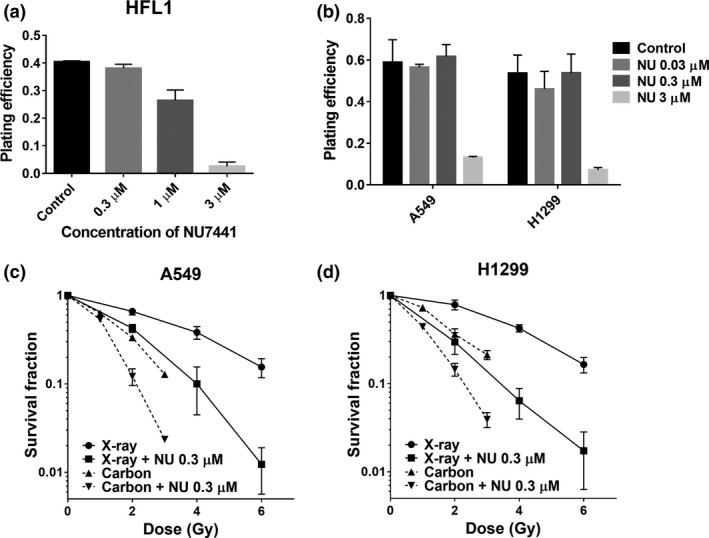
Both X‐rays and carbon ions induce radio‐sensitization in non‐small cell lung cancer (NSCLC) cells with nontoxic conce ntrations of NU7441 treatment. The cellular toxicities of NU7441 in HFL1 cells (a) and NSCLC cells (b) were determined by clonogenic survival assay. The survival fraction in A549 (c) and H1299 (d) cells after indicated treatments were determined by clonogenic survival assay. Colonies containing more than 50 cells were scored. Data represent mean ± SD from three independent experiments.
Low concentration of NU7441 does not seem to show double strand break‐repair inhibition in irradiated cells
Double strand break repair after irradiation was evaluated by analyzing γ‐H2AX signals. Three micromolar of NU7441 showed significantly increased persistent γ‐H2AX signals compared to the control, even in carbon‐irradiated cells showing clear DSB‐repair inhibition. In contrast, 0.3 μM of NU7441 did not seem to show a significant shift from the control in γ‐H2AX signals, except for a very slight increase in the 24‐h sample in some cases (Fig. 2a–d). Our results suggest that the low concentration of NU7441 (0.3 μM) did not induce obvious DSB repair inhibition in spite of its marked radio‐sensitization effect at the cell survival level (Fig. 1c,d).
Figure 2.
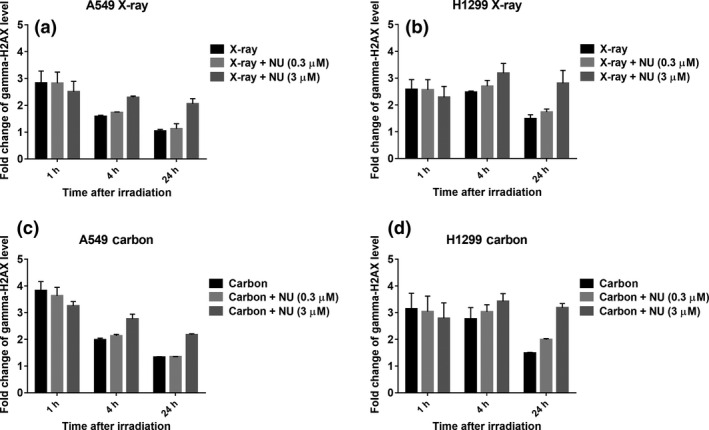
Low concentration of NU7441 does not seem to show double strand break (DSB) repair inhibition in X‐ray‐irradiated and carbon‐irradiated non‐small cell lung cancer (NSCLC) cells. The quantification of DSB was analyzed by γ‐H2AX signal using flow cytometry 1, 4 and 24 h after 4 Gy of irradiation. The fold change of the γ‐H2AX level at the indicated time after irradiation was calculated with respect to control (without irradiation and NU7441). DSB repair kinetics with 0.3 and 3 μM NU7441 were analyzed in A549 cells irradiated with X‐rays (a) and carbon ions (c), and in H1299 cells irradiated with X‐rays (b) and carbon ions (d). Data represent mean ± SD from at least two independent experiments.
Low concentration of NU7441 causes significant G2/M arrest in irradiated H1299 cells
To study the mechanism of radio‐sensitization at the low concentration (0.3 μM) of NU7441, we investigated the cell cycle distribution in tumor cells 24 h after the combined treatment. As shown in Figure 3, both NU7441 and X‐rays alone did not seem to cause a significant change in cell cycle distribution compared to the control. However, G2/M arrest was clearly observed in the combined treatment of NU7441 and radiation. The most intense G2/M arrest was induced by the combination of NU7441 and carbon ions in both cell lines. Interestingly, with carbon irradiation alone, A549 cells showed more G2/M arrest than H1299 cells, but after NU7441 and carbon irradiation, G2/M arrest was more distinct in H1299 cells than A549 cells.
Figure 3.
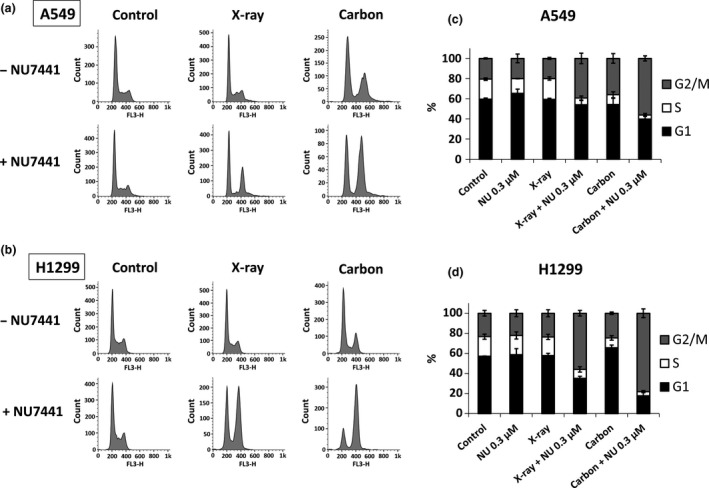
Non‐small cell lung cancer (NSCLC) cells treated with NU7441 (0.3 μM) and radiation show significant G2/M arrest. Cell cycle distribution after indicated treatments was analyzed 24 h after 4 Gy of irradiation using flow cytometry. The figures show the representative histogram and the stacked chart of cell cycle distribution in A549 cells (a, c) and in H1299 cells (b, d). Data represent mean ± SD from at least three independent experiments.
Irradiated H1299 cells were more effectively killed by NU7441
To investigate the mode of cell death, DNA fragmentation and cellular senescence were studied. For DNA fragmentation analysis, the sub‐G1 population in NSCLC cells was evaluated for 3 days. A very small portion of X‐irradiated A549 cells became sub‐G1 phase even with NU7441, while carbon irradiation increased the sub‐G1 population regardless of NU7441 presence 3 days after treatment (Fig. 4a). We were not able to confirm a difference between A549 and H1299 cells 1 day after irradiation; however, H1299 cells treated with NU7441 (0.3 μM) and exposed to X‐rays and carbon ions showed a remarkable increase in the sub‐G1 population after day 2 (Fig. 4b). For cellular senescence analysis, an SA‐βGal assay in NSCLC cells was performed 3 days after irradiation. SA‐βGal–positive cells increased significantly in carbon‐irradiated A549 cells and in H1299 cells treated with NU7441 and carbon irradiation (Fig. 5a,b). Because the p53‐p21 signal pathway is an important senescence pathway, and the p53 status in A549 and H1299 cells are wild type and null, respectively, p21 protein expression was measured in these cells. As shown in Figure 5(c), p21 expression in A549 cells was consistent with the result of the SA‐βGal assay; in contrast, no activation was observed in H1299 cells. These results indicate that cellular senescence was observed in p53‐null H1299 cells, suggesting that this process does not depend on p53 status.
Figure 4.
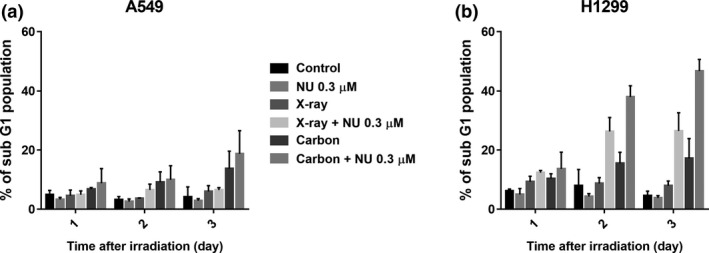
NU7441 (0.3 μM) causes a remarkable increase of DNA fragmentation in irradiated H1299 cells. DNA fragmentation after indicated treatments was determined by analyzing the sub‐G1 population in A549 cells (a) and H1299 cells (b) 1, 2 and 3 days after 4 Gy of irradiation using flow cytometry. Data represent mean ± SD from at least three independent experiments.
Figure 5.
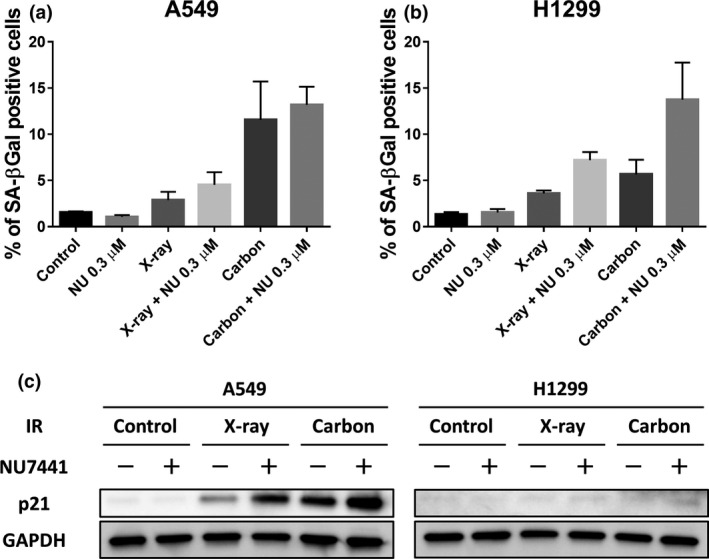
NU7441 (0.3 μM) enhances cellular senescence in irradiated H1299 cells. Cellular senescence after indicated treatments was determined by analyzing SA‐βGal using flow cytometry in A549 cells (a) and H1299 cells (b) and p21 expression in both cells (c) using western blotting 3 days after 4 Gy of irradiation. Data represent mean ± SD from at least three independent experiments.
Discussion
In this report, we found that a nontoxic concentration of DNA‐PK inhibitor, NU7441, caused significant radio‐sensitization both in X‐irradiated and carbon‐irradiated NSCLC cells; for x‐rays, SER are 1.77 and 1.94, and for carbon ions SER are 1.55 and 1.58 depending on cell lines (Fig. 1). In previous reports,9, 21, 22, 23, 24 much higher concentrations of this drug were used to induce radio‐sensitization. The drug concentration of 0.3 μM used in this study, 1/3 to 1/30 the concentrations used in the abovementioned publications, did not affect cell viability. Therefore, 0.3 μM of NU7441 is a useful strategy for radio‐sensitization. Our strategy is particularly beneficial for particle radiation therapy including heavy ions as its irradiation beams mostly hit the tumor tissue with little sensitizing effect on surrounding normal tissues. The sensitization ratio is lower in cells irradiated with carbon ions than X‐rays. Our results may not be in conflict with a partial diminution of the role for NHEJ in high‐LET irradiated cells.14, 25 Our results also indicate that p53‐deficient cells, which may be resistant to carbon ions, might become a good candidate for the combination regimen of NU7441 and heavy ions.
Inhibition of DNA‐PK, a key protein for NHEJ pathway, is known to impair DSB repair.9, 21 The nontoxic concentration (0.3 μM) of NU7441 did not seem to cause DNA repair inhibition in irradiated tumor cells, whereas 3 μM of NU7441, toxic to normal cells, clearly showed DSB repair inhibition (Fig. 2). We can suggest that a low and nontoxic concentration of DNA‐PK inhibitor might cause radio‐sensitization via a pathway other than inhibition of DSB repair. Our results somewhat reflect a previous study indicating that DNA‐PKcs knockdown cells induced radio‐sensitization, while there was no significant difference in DSB repair kinetics.24 These authors also reported that smaller amounts of DNA‐PKcs protein were accumulated at DSB sites in DNS‐PKcs knockdown cells and that this amount might be sufficient for DSB repair function. The detailed mechanism needs to be further examined, but DNA‐PK inhibition might have another role leading to an increase in cell death.
One of the remarkable effects that the low concentration of NU7441 caused was the marked G2/M cell cycle arrest following irradiation. It is known that DNA‐PK inhibition increases the percentage of cells in the G2/M phase with low‐LET radiation.26 There is a possibility that DNA‐PK inhibition partially regulates the G2/M checkpoint pathway without affecting NHEJ. G2/M delay was more clearly manifested in p53‐null H1299 cells. G1 arrest is not normally observed in p53‐deficient cells27 and, thus, we think that with NU7441 treatment, more cells can be accumulated at G2/M in irradiated H1299 cells. In particular, this phenomenon was noted in carbon‐irradiated cells. These results suggest high‐LET heavy‐ion‐irradiated cells with a low concentration of NU7441 might be arrested in the G2/M phase regardless of NHEJ suppression.
We also evaluated DNA fragmentation and cellular senescence. Irradiated H1299 cells with NU7441 showed a dramatic increase in the sub‐G1 population, especially 2 days after irradiation and beyond (Fig. 4). p53‐deficient H1299 cells induced a higher number of sub‐G1 cells than p53 wild‐type A549 cells in our case. Interestingly, these tendencies are similar to those of G2/M arrest and subsequent DNA fragmentation.28 We also believe that the marked G2/M arrest induced by DNA‐PK inhibition might cause DNA fragmentation. In addition, we found that NU7441 also promoted cellular senescence in irradiated H1299 cells (Fig. 5). Cellular senescence is a form of cellular growth control and this has been suggested as a strategy to deter tumor cell proliferation. The p53‐p21 pathway is the most essential senescence pathway,29 and we showed no p21 expression in H1299 cells. Several studies also indicated p53 independent senescence.30, 31 Thus, we also investigated p16 levels in H1299 cells as an important senescence pathway; however, the levels had no correlation with senescence induction (data not shown). Therefore, the enhanced senescence in H1299 cells via DNA‐PK inhibition might be caused by telomere dysfunction32, 33, 34 and mitosis skipping.35 In this senescence analysis, H1299 cells showed resistance to carbon ions compared to A549 cells (Fig. 5); however, NU7441 treatment caused more effective cell deaths in carbon‐irradiated H1299 cells. These results are consistent with cell survival, and indicate that p53 status would be an important factor when treatment options are considered.
In summary, we found that a low concentration of DNA‐PK inhibitor NU7441, nontoxic to normal cells, caused a significant radio‐sensitization in irradiated tumor cells, independent of their p53 status. This is probably not from DSB repair inhibition but rather a consequence of strong G2/M arrest. A more detailed mechanism of this sensitization may be determined in the future. NU7441 caused effective radio‐sensitization in irradiated H1299 cells, which showed a slight radio‐resistant phenotype. These findings would provide a basis for future clinical application of a DNA‐PK inhibitor in radiation therapy including heavy ion treatment.
Disclosure Statement
The authors have no conflict of interest to declare.
Acknowledgments
We would like to thank Mr M. Hama (National Institute of Radiological Sciences) for technical assistance in our experiments. We are grateful to the HIMAC operators for their technical support during carbon irradiation. This work was supported by JSPS KAKENHI grant 24249067 and a JSPS Research Fellowship.
Cancer Sci 107 (2016) 1250–1255
Funding Information
Japan Society for the Promotion of Science (Grant/Award Number: 15J09331, 24249067).
References
- 1. Marx Vivien. Sharp shooters. Nature 2014; 508: 133. [DOI] [PubMed] [Google Scholar]
- 2. Kamada T, Tsujii H, Blakely EA et al Carbon ion radiotherapy in Japan: an assessment of 20 years of clinical experience. Lancet Oncol 2015; 16: e93–e100. [DOI] [PubMed] [Google Scholar]
- 3. Kagawa K, Murakami M, Hishikawa Y et al Preclinical biological assessment of proton and carbon ion beams at Hyogo Ion Beam Medical Center. Int J Radiat Oncol Biol Phys 2002; 54: 928–938. [DOI] [PubMed] [Google Scholar]
- 4. Matsufuji N, Kanai T, Kanematsu N et al Specification of carbon ion dose at the National Institute of Radiological Sciences (NIRS). J Radiat Res 2007; 48(Suppl. A): A81–A86. [DOI] [PubMed] [Google Scholar]
- 5. Asaithamby A, Chen DJ. Mechanism of cluster DNA damage repair in response to high‐atomic number and energy particles radiation. Mutat Res 2011; 711: 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roos WP, Kaina B. DNA damage‐induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett 2013; 332: 237–248. [DOI] [PubMed] [Google Scholar]
- 7. Hickson I, Zhao Y, Richardson CJ et al Identification and characterization of a novel and specific inhibitor of the ataxia‐telangiectasia mutated kinase ATM identification and characterization of a novel and specific inhibitor of the ataxia‐telangiectasia mutated kinase ATM. Cancer Res 2004; 64: 9152–9159. [DOI] [PubMed] [Google Scholar]
- 8. Reaper PM, Griffiths MR, Long JM et al Selective killing of ATM‐ or p53‐deficient cancer cells through inhibition of ATR. Nat Chem Biol 2011; 7: 428–430. [DOI] [PubMed] [Google Scholar]
- 9. Zhao Y, Thomas HD, Batey MA et al Preclinical evaluation of a potent novel DNA‐dependent protein kinase inhibitor NU7441. Cancer Res 2006; 66: 5354–5362. [DOI] [PubMed] [Google Scholar]
- 10. Li Y, Reynolds P, O'Neill P, Cucinotta FA. Modeling damage complexity‐dependent non‐homologous end‐joining repair pathway. PLoS ONE 2014; 9: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Okayasu R, Okada M, Okabe A, Noguchi M, Takakura K, Takahashi S. Repair of DNA damage induced by accelerated heavy ions in mammalian cells proficient and deficient in the non‐homologous end‐joining pathway. Radiat Res 2006; 165: 59–67. [DOI] [PubMed] [Google Scholar]
- 12. Yajima H, Fujisawa H, Nakajima NI et al The complexity of DNA double strand breaks is a critical factor enhancing end‐resection. DNA Repair (Amst) 2013; 12: 936–946. [DOI] [PubMed] [Google Scholar]
- 13. Wang H, Zhang X, Wang P et al Characteristics of DNA‐binding proteins determine the biological sensitivity to high‐linear energy transfer radiation. Nucleic Acids Res 2010; 38: 3245–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takahashi A, Kubo M, Ma H et al Nonhomologous end‐joining repair plays a more important role than homologous recombination repair in defining radiosensitivity after exposure to high‐LET radiation. Radiat Res 2014; 182: 338–344. [DOI] [PubMed] [Google Scholar]
- 15. Collis SJ, DeWeese TL, Jeggo PA, Parker AR. The life and death of DNA‐PK. Oncogene 2005; 24: 949–961. [DOI] [PubMed] [Google Scholar]
- 16. Okayasu R, Suetomi K, Ullrich RL. Wortmannin inhibits repair of DNA double‐strand breaks in irradiated normal human cells. Radiat Res 1998; 149: 440–445. [PubMed] [Google Scholar]
- 17. Rosenzweig KE, Youmell MB, Palayoor ST, Price BD. Radiosensitization of human tumor cells by the phosphatidylinositol 3‐kinase inhibitors wortmannin and LY294002 correlates with inhibition of DNA‐dependent protein kinase and prolonged G2‐M Delay. Clin Cancer Res 1997; 11: 1149–1156. [PubMed] [Google Scholar]
- 18. Veuger SJ, Curtin NJ, Richardson CJ, Smith GCM. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA‐dependent protein kinase and poly (ADP‐ribose) polymerase‐1. Cancer Res 2003; 63: 6008–6015. [PubMed] [Google Scholar]
- 19. Muslimovic A, Ismail IH, Gao Y, Hammarsten O. An optimized method for measurement of gamma‐H2AX in blood mononuclear and cultured cells. Nat Protoc 2008; 3: 1187–1193. [DOI] [PubMed] [Google Scholar]
- 20. Debacq‐Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence‐associated beta‐galactosidase (SA‐βgal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc 2009; 4: 1798–1806. [DOI] [PubMed] [Google Scholar]
- 21. Ciszewski WM, Tavecchio M, Dastych J, Curtin NJ. DNA‐PK inhibition by NU7441 sensitizes breast cancer cells to ionizing radiation and doxorubicin. Breast Cancer Res Treat 2014; 143: 47–55. [DOI] [PubMed] [Google Scholar]
- 22. Tavecchio M, Munck JM, Cano C, Newell DR, Curtin NJ. Further characterisation of the cellular activity of the DNA‐PK inhibitor, NU7441, reveals potential cross‐talk with homologous recombination. Cancer Chemother Pharmacol 2012; 69: 155–164. [DOI] [PubMed] [Google Scholar]
- 23. Yu L, Shang Z‐F, Hsu F‐M et al NSCLC cells demonstrate differential mode of cell death in response to the combined treatment of radiation and a DNA‐PKcs inhibitor. Oncotarget 2015; 6: 3848–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gustafsson AS, Abramenkovs A, Stenerlöw B. Suppression of DNA‐dependent protein kinase sensitize cells to radiation without affecting DSB repair. Mutat Res 2014; 769: 1–10. [DOI] [PubMed] [Google Scholar]
- 25. Okayasu R. Repair of DNA damage induced by accelerated heavy ions–a mini review. Int J Cancer 2012; 130: 991–1000. [DOI] [PubMed] [Google Scholar]
- 26. Sturgeon CM, Knight ZA, Shokat KM, Roberge M. Effect of combined DNA repair inhibition and G2 checkpoint inhibition on cell cycle progression after DNA damage. Mol Cancer Ther 2006; 5: 885–892. [DOI] [PubMed] [Google Scholar]
- 27. Nagasawa H, Li CY, Maki CG, Imrich AC, Little JB. Relationship between radiation‐induced G1 phase arrest and p53 function in human tumor cells. Cancer Res 1995; 55: 1842–1846. [PubMed] [Google Scholar]
- 28. Sleiman RJ, Catchpoole DR, Stewart BW. Drug‐induced death of leukaemic cells after G2/M arrest: higher order DNA fragmentation as an indicator of mechanism. Br J Cancer 1998; 77: 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: the critical roles of p53. Oncogene 2013; 32: 5129–5143. [DOI] [PubMed] [Google Scholar]
- 30. Prieur A, Besnard E, Babled A, Lemaitre J. p53 and p16INK4A independent induction of senescence by chromatin‐dependent alteration of S‐phase progression. Nat Commun 2011; 2: 473. [DOI] [PubMed] [Google Scholar]
- 31. Wang M, Morsbach F, Sander D et al EGF receptor inhibition radiosensitizes NSCLC cells by inducing senescence in cells sustaining dna double‐strand breaks. Cancer Res 2011; 71: 6261–6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gilley D, Tanaka H, Hande MP et al DNA‐PKcs is critical for telomere capping. Proc Natl Acad Sci USA 2001; 98: 15084–15088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou X, Zhang X, Xie Y, Tanaka K, Wang B, Zhang H. DNA‐PKcs inhibition sensitizes cancer cells to carbon‐ion irradiation via telomere capping disruption. PLoS ONE 2013; 8: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Williams ES, Klingler R, Ponnaiya B et al Telomere dysfunction and DNA‐PKcs deficiency: characterization and consequence. Cancer Res 2009; 69: 2100–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Davoli T, Denchi EL, De LangeT. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell 2010; 141: 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]