

Abstract
Epigenetic mechanisms underlie the phenotypic plasticity of cells, while aberrant epigenetic regulation through genetic mutations and/or misregulated expression of epigenetic factors leads to aberrant cell fate determination, which provides a foundation for oncogenic transformation. Lysine‐specific demethylase‐1 (LSD1, KDM1A) removes methyl groups from methylated proteins, including histone H3, and is frequently overexpressed in various types of solid tumors and hematopoietic neoplasms. While LSD1 is involved in a wide variety of normal physiological processes, including stem cell maintenance and differentiation, it is also a key player in oncogenic processes, including compromised differentiation, enhanced cell motility and metabolic reprogramming. Here, we present an overview of how LSD1 epigenetically regulates cellular plasticity through distinct molecular mechanisms in different biological contexts. Targeted inhibition of the context‐dependent activities of LSD1 may provide a highly selective means to eliminate cancer cells.
Keywords: Cancer metabolism, chromatin, histone demethylation, lysine‐specific demethylase‐1, stemness
Epigenetic gene regulation plays a central role not only in maintaining cell identity but also in reprogramming a cell's phenotype in response to environmental fluctuations.1 Because cancer cells exhibit epigenomic signatures that are distinct from their normal counterparts, it is likely that their phenotypic plasticity is controlled in a unique way.2
Methylation of specific lysine residues in the N‐terminal tails of histone proteins underlie diverse gene regulatory responses, including transcriptional activation and repression.3 In general, methyl modifications at histone H3 lysine 4 (H3K4me) reflects transcriptional competency, while those at lysine 9 and 27 (H3K9me and H3K27me, respectively) are components of repressive chromatin structure.4 These marks are dynamically regulated by specific methyltransferases and demethylases, both in steady‐state cells and during cellular transitions. The proper regulation of these marks is essential for the maintenance of cell identity as well as for differentiation, and their misregulation is often linked to the development of cancer.5
Lysine‐specific demethylase‐1 (LSD1) was the first histone demethylase to be identified that demethylates histone H3K4 and H3K9. Extensive studies have established that LSD1 is essential for stem cell function and animal development. In addition, overexpression of LSD1 has been found in many types of cancer, and has been experimentally demonstrated to be a critical player in cancer development. Here, we provide an overview of how LSD1 contributes to phenotypic plasticity in cancer and normal stem cells through chromatin regulation. A number of proteins other than histones have also been identified as substrates of LSD1‐mediated demethylation. A detailed review of LSD1 in non‐histone protein demethylation can be found elsewhere.6
Molecular structure and function of lysine‐specific demethylase‐1
To date, according to the HUGO database (www.genenames.org), 21 lysine demethylases have been identified in the human genome, most of which target histones in a residue‐selective manner.5 Nineteen demethylases belong to the jumonji domain‐containing dioxygenase family, while LSD1 and LSD2 (KDM1B) are the only members of the flavin‐dependent amine oxidase family, which require flavin adenine dinucleotide for their enzymatic activity (Fig. 1a). Biochemical and cell‐based analyses revealed that H3K4me was the endogenous substrate of LSD1 (Fig. 1b).7, 8 Up to three methyl groups form cohesive bonds with H3K4, which are generally recognized as an “active” chromatin signature in cells. Mono‐methylated H3K4 is a hallmark of enhancer regions, while di‐methylated and tri‐methylated H3K4 are enriched in active promoters.4 Because a lone pair of electrons in the nitrogen atom in the methylated lysine is required for catalytic activity, LSD1 is capable of demethylating mono‐methylated and di‐methylated lysine but not tri‐methylated lysine.9 In addition to the catalytic amine oxidase domain, LSD1 contains two unique structural domains that are closely associated with its molecular function (Fig. 1a). The tower domain contains two anti‐parallel helices forming a protruding structure, which serves as a platform for SANT (Swi3, Ada2, Ncor and TFIIIB) domain proteins, such as CoREST and metastasis‐associated protein, MTA.10 Association of LSD1 with these proteins facilitates demethylation activity.11, 12 Because these binding partners form HDAC‐containing repressor complexes, LSD1‐mediated H3K4 demethylation is linked to the transcriptionally repressive chromatin structure.12, 13
Figure 1.
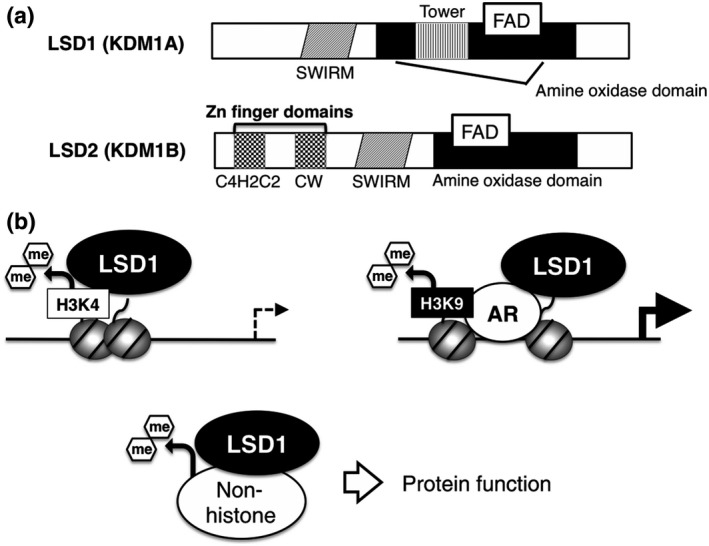
Basic characteristics of lysine‐specific demethylase‐1 (LSD1). (a) Domain structures of LSD1 and LSD2 proteins. FAD, flavin adenine dinucleotide. (b) Substrate‐selectivity of LSD1. LSD1 is involved in the repression of promoter and enhancer activities through the regulation of H3K4me, while activating transcription through H3K9 demethylation in cooperation with the androgen receptor (AR). A number of non‐histone substrates have also been identified.
Lysine‐specific demethylase‐1 has also been implicated in transcriptional activation through H3K9 demethylation activity but in limited circumstances. Upon stimulation by androgen receptor (AR) agonist, LSD1 facilitates the demethylation of H3K9 and, thus, augments AR‐mediated transcriptional activation in prostate cancer cells (Fig. 1b).14 A recent study by Laurent et al. demonstrated that a splice variant of LSD1 containing an extra four amino acids, which is selectively expressed in the neural cell lineage, mediates H3K9 demethylation possibly through an indirect mechanism.15, 16 Structural models have revealed a highly selective recognition of H3K4 by LSD1.17, 18, 19 Mechanistic insight into LSD1‐mediated H3K9 demethylation awaits further investigation.
Misregulated expression of lysine‐specific demethylase‐1 in cancer
Increased expression of LSD1 has been reported in various types of cancer. In particular, many types of hematopoietic and lymphatic neoplasm, including acute myeloid leukemia (AML), acute lymphoblastic leukemia, myelodysplastic syndromes, T cell non‐Hodgkin lymphoma and Hodgkin lymphoma, exhibit LSD1 overexpression.20, 21 In addition, solid tumors in bladder, liver (hepatocellular carcinoma), colon, prostate and lung (small cell cancer) show elevated levels of LSD1 mRNA and/or protein.22, 23, 24 Although the overexpression of LSD1 may be a shared feature across cancer types, the regulatory mechanism of LSD1 gene transcription, both in normal and oncogenic contexts, is poorly understood. A recent study proposed that increased protein stability might also contribute to the gain‐of‐function of LSD1. A deubiquitinase, USP28, has been experimentally shown to protect LSD1 from proteasomal degradation through direct deubiquitination.25 In breast cancer cells, the depletion of USP28 resulted in a reduced level of LSD1 protein and elevated levels of H3K4me at LSD1‐target genes, which was accompanied by the loss of stem cell properties. Importantly, the protein levels of LSD1 and USP28 were positively correlated in human breast tumors. This evidence strongly indicates the involvement of LSD1 in shaping the epigenomic landscape in cancer.
Role of lysine‐specific demethylase‐1 in stem cell maintenance
Lysine‐specific demethylase‐1 is essential for embryonic development in mice. When the Lsd1 gene was conventionally deleted, no viable embryo could be found after E7.5.26, 27 Moreover, conditional deletion of Lsd1 in the pituitary, hematopoietic system and adipose tissue led to severe dysplastic phenotypes, suggesting the requirement of LSD1 for stem cell maintenance and/or differentiation.26, 28, 29 LSD1‐KO embryonic stem (ES) cells have been generated by several groups, exhibiting somewhat different phenotypic outcomes. Wang et al. 27 report that Lsd1‐deleted mouse ES cells exhibited impaired growth, with an increased rate of apoptosis and the failure of embryoid body formation. In contrast, Foster et al.,30 using a gene trap method, demonstrated that LSD1 deletion did not cause any defect in proliferation, while showing an increased apoptosis rate when embryoid body formation was induced. Interestingly, a ChIP‐seq analysis of LSD1‐bound sites in mouse ES cells revealed that the vast majority of active enhancers and promoters were occupied by LSD1.31 However, LSD1 was not required for the maintenance of stemness. Instead, LSD1 was essential for H3K4 demethylation and the silencing of ES cell‐enriched genes upon differentiation, a process called “enhancer decommissioning.”31 Moreover, in human ES cells, a reduced level of LSD1 has been linked to impaired cell cycle progression and aberrant expression of developmentally regulated genes.32 The difference in phenotypic outcomes may reflect the different methods used for gene manipulation, cell line differences or species variation.
In neural stem cells (NSC) in mice, LSD1 is required for the maintenance of proliferative capacity through interaction with an NSC maintenance factor, TLX.33 In contrast, in human fetal NSC, LSD1 mediates neuronal differentiation by repressing a Notch‐target gene, HEYL.34 Overall, these data indicate the importance of LSD1 function both in embryonic and somatic stem cells.
Lysine‐specific demethylase‐1 in hematopoiesis and leukemogenesis
Important roles for LSD1 in both normal hematopoiesis and leukemogenesis have been characterized (Fig. 2). In an early study, LSD1 was identified as a binding partner of growth factor independence (Gfi)‐1 and (Gfi)‐1b, transcription factors (TF), which are involved in multiple steps of hematopoiesis.35 RNAi‐mediated depletion of LSD1 resulted in the compromised differentiation in erythroid and megakaryocytic cells. Mechanistically, LSD1 occupies the promoter region of Gfi‐1b‐target genes, and represses their expression most likely through H3K4 demethylation. Another transcriptional regulator of hematopoiesis, TAL1, has also been shown to bind LSD1.36 Differentiation stage‐dependent interaction of these proteins is essential for the timely expression of genes associated with the erythroid lineage. A later study, using genetic approaches in mice, revealed that LSD1 was required for early and late differentiation processes in the hematopoietic lineage.28 Both pre‐natal and post‐natal deletion of Lsd1 resulted in a dramatic reduction of mature blood cells accompanied by a fatally severe anemia. Specifically, Lsd1‐deficient mice lacked mature myeloid progenitor cells, but the colony forming potential of hematopoietic stem cells was reserved. In addition, numbers of terminally differentiated granulocytes and erythrocytes were reduced, while their precursors were accumulated. Transcriptomic and epigenomic data indicates that LSD1 represses stem and progenitor cell‐associated genes through H3K4 demethylation at their promoter and enhancer regions. These studies indicate that LSD1 is important for hematopoietic differentiation, especially in the erythroid lineage.
Figure 2.
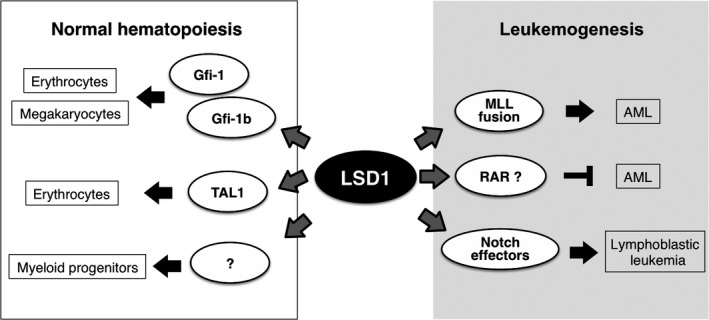
Pivotal role of lysine‐specific demethylase‐1 (LSD1) in hematopoiesis and leukemogenesis. Gfi‐1 and ‐1b, growth factor independence‐1 and ‐1b; TAL1, T‐cell acute lymphocytic leukemia 1; MLL, myeloid/lymphoid or mixed‐lineage leukemia; RAR, retinoic acid receptor.
The increased expression of LSD1 in different types of human hematopoietic neoplasm indicates its possible involvement in leukemogenesis. This prediction has been shown to be true, most prominently in the case of AML. In acute promyelocytic leukemia, which harbors the PML‐RARA gene fusion, treatment with all‐trans‐retinoic acid (ATRA) efficiently induces cellular differentiation and growth arrest, but this therapeutic effect has not been achieved in other types of AML.37 Schenk et al.38 demonstrated that inhibition of LSD1 activity in combination with ATRA exposure promoted the differentiation of AML cells with different genetic backgrounds. Upon LSD1 inhibition and ATRA treatment, the expression of genes associated with myeloid differentiation was upregulated with a concomitant increase of H3K4me2 levels at these genes. LSD1 inhibitor exerts synergistic effects with other anti‐cancer agents, such as Ara‐C or an inhibitor of H3K27 methyltransferase, on the induction of AML cell death, indicating the multifaceted function of LSD1.39 Moreover, Harris et al. report the contribution of LSD1 in maintaining stem cell properties in a subtype of AML harboring an MLL gene translocation.40 Increased expression of LSD1 was detected in MLL‐mutant leukemia cells, especially in cells expressing the MLL‐AF9 fusion protein, which acts as an oncogenic transcriptional regulator. Genome‐wide transcriptomic and epigenomic analyses revealed that LSD1 is enriched at MLL‐AF9‐target genes. Interestingly, LSD1 and MLL‐AF9 cooperatively promoted the expression of these genes, although MLL itself is a H3K4 methyltransferase normally counteracting LSD1 to dynamically remodel H3K4 methylation status. These findings indicate a distinct mode of epigenetic regulation in leukemia cells with specific genetic backgrounds.
Direct evidence that the increased expression of LSD1 can support malignant transformation of HSC has been reported.21 Among the four reported LSD1 splice variants, the transgenic expression of the shortest, and perhaps the most well‐known, isoform induced lymphocyte hyperplasia in mice, and when exposed to γ‐irradiation, the mice developed T‐lymphoblastic leukemia (T‐LBL). LSD1 is a key epigenetic effector downstream of notch signaling, which is frequently activated in lymphoid malignancies.41, 42 Considering that LSD1 is often overexpressed in human T‐LBL,21 LSD1 may be a strong driver of epigenetic disruption that paves the way to leukemogenesis.
Lysine‐specific demethylase‐1 in epithelial‐to‐mesenchymal transition and cell motility
Lysine‐specific demethylase‐1 is a key epigenetic regulator of the cellular state; therefore, it is plausible that it also contributes to the environmental adaptation of cancer cells. Indeed, a number of reports have shown that LSD1 is critically involved in the regulation of the epithelial‐to‐mesenchymal transition (EMT). EMT confers mesenchymal cell properties on tumor cells, including the cell motility that is required for invasion and metastasis.43, 44 EMT is also associated with the acquisition of cancer stem cell‐like properties, such as self‐renewal and colony forming capacities.43 EMT involves highly ordered transcriptional regulation, in which several master TF, including SNAIL family proteins, repress epithelial marker genes and activate mesenchymal markers.44, 45 Two groups independently demonstrated that LSD1 physically associates with SNAIL1 in breast cancer cells.46, 47 LSD1 is recruited to the E‐cadherin gene promoter in a SNAIL1‐dependent manner, and represses its expression via H3K4 demethylation (Fig. 3). Interestingly, an inhibitor of LSD1 enzymatic activity abolished the LSD1/SNAIL1 interaction, leading to impaired cell motility.46 The expression of LSD1 was highly correlated with that of SNAIL1 in human breast tumor specimens, indicating the cooperativity of these proteins during tumor development.46 The LSD1/SNAIL1 complex has also been shown to enhance bone marrow homing activity in AML cells, indicating its conserved regulatory role in cell motility across different cell types.48 Moreover, the expression of LSD1 was increased during transforming growth factor (TGF)‐β‐induced EMT of non‐cancerous hepatocytes.49 This EMT process was accompanied by an increase of gross H3K4 methylation and a decrease of H3K9 methylation, which was reversed by LSD1 depletion. Although the mechanism for this is not clear, the data indicate that LSD1 is a major determinant of genome‐scale epigenetic reprogramming during EMT. Other reports have demonstrated that LSD1 is a negative regulator of cell motility. Wang et al.12 show that LSD1 cooperates with the NuRD complex to repress a set of genes associated with TGF‐β signaling, which, in turn, inhibits cell migration (Fig. 3). In agreement with this molecular mechanism, the loss of LSD1 enhanced the metastatic behavior of breast cancer cells transplanted into mice. It has also been reported that LSD1 represses the expression of SNAIL1 and other EMT‐associated genes.50 Another Snail family TF, SLUG/SNAI2, also binds to LSD1.51 This protein complex co‐localized at and transcriptionally repressed lineage‐specific genes to maintain an undifferentiated state in breast cancer cells.52
Figure 3.
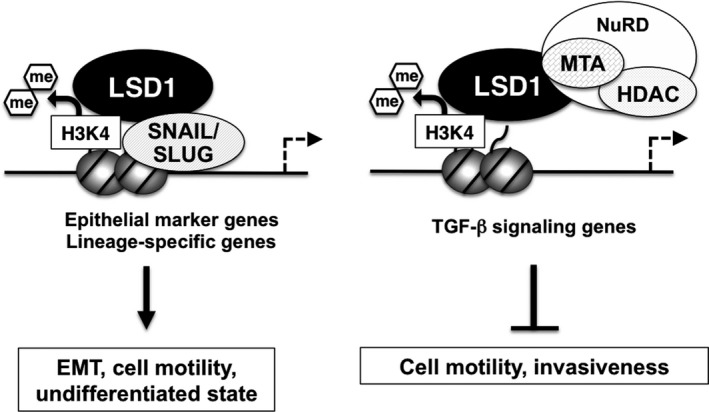
Lysine‐specific demethylase‐1 (LSD1) regulates cell motility and EMT in cancer cells. H3K4 demethylation activity of LSD1 exerts opposite effects on cell motility and epithelial‐to‐mesenchymal transition (EMT) depending on interacting partners.
These lines of evidence suggest that LSD1 is a pivotal regulator of the phenotypic plasticity of cancer cells. It can either promote or inhibit EMT and cell motility, presumably depending on the genetic background of the cells and/or environmental cues that can influence the behavior of LSD1. It is also important to note that remodeling of H3K4me status, either local or global, is intimately associated with the progression of EMT.
Lysine‐specific demethylase‐1 is an integrative regulator of the glycolytic shift in cancer cells
Cancer cells undergo a rewiring of their energy metabolism pathways, a process known as metabolic reprogramming, in order to adapt to their microenvironment and to support their proliferative potential.53 A hallmark of cancer cell metabolism is glycolysis‐shifted energy production rather than mitochondrial respiration. Such an energy strategy enables not only survival under hypoxic conditions but also efficient production of macromolecules, including lipids and nucleotides, which serve as building blocks for cell division.54 Although the expression of metabolic genes is dramatically remodeled in cancer cells, the underlying epigenetic mechanism is poorly understood.55, 56 We have previously demonstrated that LSD1 is an integrative regulator of the glycolytic shift in hepatocellular carcinoma (HCC) cells (Fig. 4).57 Mechanistically, LSD1 represses mitochondrial respiration‐associated genes such as PPARGC1A, ACADM and EHHADH through H3K4 demethylation at their promoter regions. Moreover, LSD1 promotes the expression of most of the glycolytic genes, including GLUT1, HK2 and PKM2, by facilitating hypoxia‐inducible factor‐1α (HIF‐1α)‐mediated transcriptional activation. Interestingly, LSD1 was required to sequester HIF‐1α from proteasomal degradation, and, thus, contributes to the stabilization of HIF‐1α protein under hypoxia. Consistent with this gene regulatory function, loss of LSD1 resulted in increased oxidative phosphorylation (OXPHOS) capacity as well as reduced glucose uptake and glycolytic activity. Using an HCC xenograft in mice, we demonstrated that tumor growth was severely impaired by LSD1 depletion. We also observed a significant correlation between LSD1 and GLUT1 expression in human HCC specimens. These data highlight a key role for LSD1 in metabolic reprogramming of HCC cells. Highly similar results were observed in human esophageal cancer (EC), in which LSD1 expression levels were significantly correlated with glucose uptake as detected by fluorodeoxyglucose‐positron emission tomography.58 In EC cells, LSD1 was essential for the maintenance of glycolytic gene expression. Intriguingly, the reduction of glycolytic activity by LSD1 inhibition was accompanied by compromised motility rather than a proliferative defect or cell death, indicating the requirement of metabolic adaptation for cell migration. Moreover, an LSD1‐dependent glycolytic shift has also been demonstrated in pancreatic cancer cells.59 These results suggest a conserved role of LSD1 in the control of metabolic reprogramming across different types of cancer. LSD1 also regulates energy metabolism in normal cells. Of note, LSD1 suppresses mitochondrial respiration but does not influence glycolytic activity in adipose cells,60 suggesting that cancer‐specific conditions, such as oncogenic signaling and/or the tumor microenvironment, contribute to selective gene regulation by LSD1. In addition, it is not clear how H3K4 demethylation by LSD1 can be triggered by specific signaling and TF during metabolic reprogramming. This point should be clarified in future studies.
Figure 4.
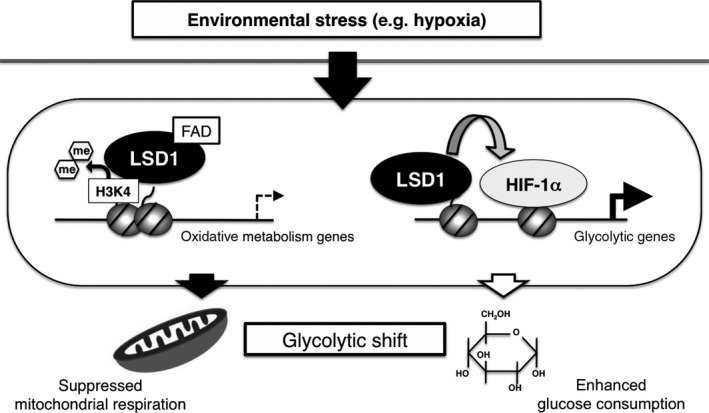
Lysine‐specific demethylase‐1 (LSD1) as an integrative regulator of the glycolytic shift in cancer cells. LSD1 promotes the glycolytic shift by directly suppressing mitochondrial respiration and by activating glycolysis via hypoxia‐inducible factor‐1α (HIF‐1α).
Although not much is known about the biological function of LSD2, some reports have described its role in the regulation of cellular metabolism. In hepatic cells, LSD2 represses the expression of genes associated with lipid metabolism and transport directly through the demethylation of H3K4 at their enhancers.61 LSD2‐depleted cells exhibited an increased rate of fatty acid uptake and an accumulation of large lipid metabolites, such as cholesterol and phospholipids. The growth of LSD2‐depleted cells was markedly impaired by fatty acid exposure, indicating that LSD2 protects the cell from lipotoxic damage. A recent study has shown that LSD2 represses the expression of glycolytic genes.62 miR‐215, a micro‐RNA whose level is increased in glioma‐initiating cells, represses the expression of LSD2, leading to reduced expression of glycolytic genes. Of note, the low level of LSD2 expression and the high level of miR‐215 expression coexisted in glioblastoma patients. These findings indicate that LSD1 and LSD2 have non‐redundant roles in regulating energy metabolism and in the development of cancer. Because both LSD1 and LSD2 show relatively ubiquitous expression patterns across cell and tissue types, these proteins may work either cooperatively or competitively in certain circumstances.
Conclusions
Lysine‐specific demethylase‐1 plays a pivotal role in various biological processes, including the maintenance of stemness, cell motility, EMT and glycolysis‐shifted metabolism, all of which are typically associated with oncogenesis. Indeed, the increased expression of LSD1 in many types of cancer is consistent with the hypothesis that LSD1 gain‐of‐function leads to aberrant epigenomic regulation. Because LSD1 regulates the H3K4me status of key genes both in normal and cancer cells, it is important for future studies to elucidate whether the overexpression of LSD1 causes a redistribution of H3K4me marks in cancer. It is also tempting to examine whether somatic mutation and/or sequence variation of LSD1 could contribute to aberrant epigenome formation in cancer.
It is essential to consider the methylation–demethylation dynamics in order to link the LSD1 function to epigenetic plasticity. However, it is mostly unclear how LSD1 and specific H3K4 methyltransferases counteract to establish a certain H3K4me equilibrium. Because multiple H3K4 methyltransferases exist, LSD1 may exert different impacts on the epigenetic plasticity and stability, depending on the co‐working methyltransferase.
Monoamine oxidase inhibitors have a potent inhibitory effect on LSD1 demethylase activity.63 Moreover, recently developed LSD1 inhibitors with increased potency and selectivity exert marked anti‐carcinogenic effects.39, 40, 64, 65 Because LSD1 participates in diverse biological processes depending on the cellular context and partner proteins, LSD1 inhibition in combination with the perturbation of other pathways and molecules might confer selective effects against desired target cells.
Disclosure Statement
The authors have no conflict of interest to declare.
Cancer Sci 107 (2016) 1187–1192
Funding InformationThis work was supported by the JSPS KAKENHI (JP15H04707 and JP15K15068), by the Japan Agency for Medical Research and Development (CREST) to M.N., and by a grant from the Takeda Science Foundation to M.N and S.H. S.H. was also supported by the JSPS KAKENHI (JP16K07215), by the Kanae Foundation for the Promotion of Medical Science and by the Ono Medical Research Foundation
Contributor Information
Shinjiro Hino, Email: s-hino@kumamoto-u.ac.jp.
Mitsuyoshi Nakao, Email: mnakao@gpo.kumamoto-u.ac.jp.
References
- 1. Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet 2011; 13: 97–109. [DOI] [PubMed] [Google Scholar]
- 2. Stergachis AB, Neph S, Reynolds A et al Developmental fate and cellular maturity encoded in human regulatory DNA landscapes. Cell 2013; 154: 888–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell 2012; 48: 491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet 2011; 12: 7–18. [DOI] [PubMed] [Google Scholar]
- 5. Hojfeldt JW, Agger K, Helin K. Histone lysine demethylases as targets for anticancer therapy. Nat Rev Drug Discov 2013; 12: 917–30. [DOI] [PubMed] [Google Scholar]
- 6. Hamamoto R, Saloura V, Nakamura Y. Critical roles of non‐histone protein lysine methylation in human tumorigenesis. Nat Rev Cancer 2015; 15: 110–24. [DOI] [PubMed] [Google Scholar]
- 7. Forneris F, Binda C, Vanoni MA, Mattevi A, Battaglioli E. Histone demethylation catalysed by LSD1 is a flavin‐dependent oxidative process. FEBS Lett 2005; 579: 2203–7. [DOI] [PubMed] [Google Scholar]
- 8. Shi Y, Lan F, Matson C et al Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004; 119: 941–53. [DOI] [PubMed] [Google Scholar]
- 9. Forneris F, Battaglioli E, Mattevi A, Binda C. New roles of flavoproteins in molecular cell biology: histone demethylase LSD1 and chromatin. FEBS J 2009; 276: 4304–12. [DOI] [PubMed] [Google Scholar]
- 10. Hou H, Yu H. Structural insights into histone lysine demethylation. Curr Opin Struct Biol 2010; 20: 739–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell 2005; 19: 857–64. [DOI] [PubMed] [Google Scholar]
- 12. Wang Y, Zhang H, Chen Y et al LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 2009; 138: 660–72. [DOI] [PubMed] [Google Scholar]
- 13. Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 2005; 437: 432–5. [DOI] [PubMed] [Google Scholar]
- 14. Metzger E, Wissmann M, Yin N et al LSD1 demethylates repressive histone marks to promote androgen‐receptor‐dependent transcription. Nature 2005; 437: 436–9. [DOI] [PubMed] [Google Scholar]
- 15. Laurent B, Ruitu L, Murn J et al A specific LSD1/KDM1A isoform regulates neuronal differentiation through H3K9 demethylation. Mol Cell 2015; 57: 957–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zibetti C, Adamo A, Binda C et al Alternative splicing of the histone demethylase LSD1/KDM1 contributes to the modulation of neurite morphogenesis in the mammalian nervous system. J Neurosci 2010; 30: 2521–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stavropoulos P, Blobel G, Hoelz A. Crystal structure and mechanism of human lysine‐specific demethylase‐1. Nat Struct Mol Biol 2006; 13: 626–32. [DOI] [PubMed] [Google Scholar]
- 18. Forneris F, Binda C, Dall'Aglio A, Fraaije MW, Battaglioli E, Mattevi A. A highly specific mechanism of histone H3‐K4 recognition by histone demethylase LSD1. J Biol Chem 2006; 281: 35289–95. [DOI] [PubMed] [Google Scholar]
- 19. Forneris F, Binda C, Adamo A, Battaglioli E, Mattevi A. Structural basis of LSD1‐CoREST selectivity in histone H3 recognition. J Biol Chem 2007; 282: 20070–4. [DOI] [PubMed] [Google Scholar]
- 20. Niebel D, Kirfel J, Janzen V, Holler T, Majores M, Gutgemann I. Lysine‐specific demethylase 1 (LSD1) in hematopoietic and lymphoid neoplasms. Blood 2014; 124: 151–2. [DOI] [PubMed] [Google Scholar]
- 21. Wada T, Koyama D, Kikuchi J, Honda H, Furukawa Y. Overexpression of the shortest isoform of histone demethylase LSD1 primes hematopoietic stem cells for malignant transformation. Blood 2015; 125: 3731–46. [DOI] [PubMed] [Google Scholar]
- 22. Hayami S, Kelly JD, Cho HS et al Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int J Cancer 2011; 128: 574–86. [DOI] [PubMed] [Google Scholar]
- 23. Magerl C, Ellinger J, Braunschweig T et al H3K4 dimethylation in hepatocellular carcinoma is rare compared with other hepatobiliary and gastrointestinal carcinomas and correlates with expression of the methylase Ash2 and the demethylase LSD1. Hum Pathol 2010; 41: 181–9. [DOI] [PubMed] [Google Scholar]
- 24. Kahl P, Gullotti L, Heukamp LC et al Androgen receptor coactivators lysine‐specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res 2006; 66: 11341–7. [DOI] [PubMed] [Google Scholar]
- 25. Wu Y, Wang Y, Yang XH et al The deubiquitinase USP28 stabilizes LSD1 and confers stem‐cell‐like traits to breast cancer cells. Cell Rep 2013; 5: 224–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang J, Scully K, Zhu X et al Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 2007; 446: 882–7. [DOI] [PubMed] [Google Scholar]
- 27. Wang J, Hevi S, Kurash JK et al The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet 2009; 41: 125–9. [DOI] [PubMed] [Google Scholar]
- 28. Kerenyi MA, Shao Z, Hsu YJ et al Histone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturation. Elife 2013; 2: e00633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Duteil D, Metzger E, Willmann D et al LSD1 promotes oxidative metabolism of white adipose tissue. Nat Commun 2014; 5: 4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Foster CT, Dovey OM, Lezina L et al Lysine‐specific demethylase 1 regulates the embryonic transcriptome and CoREST stability. Mol Cell Biol 2010; 30: 4851–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Whyte WA, Bilodeau S, Orlando DA et al Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 2012; 482: 221–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adamo A, Sese B, Boue S et al LSD1 regulates the balance between self‐renewal and differentiation in human embryonic stem cells. Nat Cell Biol 2011; 13: 652–9. [DOI] [PubMed] [Google Scholar]
- 33. Sun G, Alzayady K, Stewart R et al Histone demethylase LSD1 regulates neural stem cell proliferation. Mol Cell Biol 2010; 30: 1997–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hirano K, Namihira M. LSD1 mediates neuronal differentiation of human fetal neural stem cells by controlling the expression of a novel target gene HEYL. Stem Cells 2016; 34: 1872–82. [DOI] [PubMed] [Google Scholar]
- 35. Saleque S, Kim J, Rooke HM, Orkin SH. Epigenetic regulation of hematopoietic differentiation by Gfi‐1 and Gfi‐1b is mediated by the cofactors CoREST and LSD1. Mol Cell 2007; 27: 562–72. [DOI] [PubMed] [Google Scholar]
- 36. Hu X, Li X, Valverde K et al LSD1‐mediated epigenetic modification is required for TAL1 function and hematopoiesis. Proc Natl Acad Sci U S A 2009; 106: 10141–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Petrie K, Zelent A, Waxman S. Differentiation therapy of acute myeloid leukemia: past, present and future. Curr Opin Hematol 2009; 16: 84–91. [DOI] [PubMed] [Google Scholar]
- 38. Schenk T, Chen WC, Gollner S et al Inhibition of the LSD1 (KDM1A) demethylase reactivates the all‐trans‐retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med 2012; 18: 605–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McGrath JP, Williamson KE, Balasubramanian S et al Pharmacological inhibition of the histone lysine demethylase KDM1A suppresses the growth of multiple acute myeloid leukemia subtypes. Cancer Res 2016; 76: 1975–88. [DOI] [PubMed] [Google Scholar]
- 40. Harris WJ, Huang X, Lynch JT et al The histone demethylase KDM1A sustains the oncogenic potential of MLL‐AF9 leukemia stem cells. Cancer Cell 2012; 21: 473–87. [DOI] [PubMed] [Google Scholar]
- 41. Yatim A, Benne C, Sobhian B et al NOTCH1 nuclear interactome reveals key regulators of its transcriptional activity and oncogenic function. Mol Cell 2012; 48: 445–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lobry C, Oh P, Mansour MR, Look AT, Aifantis I. Notch signaling: switching an oncogene to a tumor suppressor. Blood 2014; 123: 2451–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Scheel C, Weinberg RA. Cancer stem cells and epithelial–mesenchymal transition: concepts and molecular links. Semin Cancer Biol 2012; 22: 396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat Rev Mol Cell Biol 2014; 15: 178–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barrallo‐Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development 2005; 132: 3151–61. [DOI] [PubMed] [Google Scholar]
- 46. Lin Y, Wu Y, Li J et al The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine‐specific demethylase 1. EMBO J 2010; 29: 1803–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lin T, Ponn A, Hu X, Law BK, Lu J. Requirement of the histone demethylase LSD1 in Snai1‐mediated transcriptional repression during epithelial–mesenchymal transition. Oncogene 2010; 29: 4896–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ferrari‐Amorotti G, Fragliasso V, Esteki R et al Inhibiting interactions of lysine demethylase LSD1 with snail/slug blocks cancer cell invasion. Cancer Res 2013; 73: 235–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McDonald OG, Wu H, Timp W, Doi A, Feinberg AP. Genome‐scale epigenetic reprogramming during epithelial‐to‐mesenchymal transition. Nat Struct Mol Biol 2011; 18: 867–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Choi HJ, Park JH, Park M et al UTX inhibits EMT‐induced breast CSC properties by epigenetic repression of EMT genes in cooperation with LSD1 and HDAC1. EMBO Rep 2015; 16: 1288–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wu ZQ, Li XY, Hu CY, Ford M, Kleer CG, Weiss SJ. Canonical Wnt signaling regulates Slug activity and links epithelial–mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc Natl Acad Sci U S A 2012; 109: 16654–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Phillips S, Prat A, Sedic M et al Cell‐state transitions regulated by SLUG are critical for tissue regeneration and tumor initiation. Stem Cell Reports 2014; 2: 633–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324: 1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 2012; 21: 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hino S, Nagaoka K, Nakao M. Metabolism–epigenome crosstalk in physiology and diseases. J Hum Genet 2013; 58: 410–5. [DOI] [PubMed] [Google Scholar]
- 56. Wang X, Jin H. The epigenetic basis of the Warburg effect. Epigenetics 2010; 5: 566–8. [DOI] [PubMed] [Google Scholar]
- 57. Sakamoto A, Hino S, Nagaoka K et al Lysine demethylase LSD1 coordinates glycolytic and mitochondrial metabolism in hepatocellular carcinoma cells. Cancer Res 2015; 75: 1445–56. [DOI] [PubMed] [Google Scholar]
- 58. Kosumi K, Baba Y, Sakamoto A et al Lysine‐specific demethylase‐1 contributes to malignant behavior by regulation of invasive activity and metabolic shift in esophageal cancer. Int J Cancer 2016; 138: 428–39. [DOI] [PubMed] [Google Scholar]
- 59. Qin Y, Zhu W, Xu W et al LSD1 sustains pancreatic cancer growth via maintaining HIF1alpha‐dependent glycolytic process. Cancer Lett 2014; 347: 225–32. [DOI] [PubMed] [Google Scholar]
- 60. Hino S, Sakamoto A, Nagaoka K et al FAD‐dependent lysine‐specific demethylase‐1 regulates cellular energy expenditure. Nat Commun 2012; 3: 758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nagaoka K, Hino S, Sakamoto A et al Lysine‐specific demethylase 2 suppresses lipid influx and metabolism in hepatic cells. Mol Cell Biol 2015; 35: 1068–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hu J, Sun T, Wang H et al MiR‐215 is induced post‐transcriptionally via HIF‐Drosha complex and mediates glioma‐initiating cell adaptation to hypoxia by targeting KDM1B. Cancer Cell 2016; 29: 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee MG, Wynder C, Schmidt DM, McCafferty DG, Shiekhattar R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem Biol 2006; 13: 563–7. [DOI] [PubMed] [Google Scholar]
- 64. Mimasu S, Umezawa N, Sato S, Higuchi T, Umehara T, Yokoyama S. Structurally designed trans‐2‐phenylcyclopropylamine derivatives potently inhibit histone demethylase LSD1/KDM1. Biochemistry 2010; 49: 6494–503. [DOI] [PubMed] [Google Scholar]
- 65. Neelamegam R, Ricq EL, Malvaez M et al Brain‐penetrant LSD1 inhibitors can block memory consolidation. ACS Chem Neurosci 2012; 3: 120–8. [DOI] [PMC free article] [PubMed] [Google Scholar]