

Abstract
Genetic alterations in myelodysplastic syndromes (MDS) are critical for pathogenesis. We previously showed that peripheral blood cell‐free DNA (PBcfDNA) may be more sensitive for genetic/epigenetic analyses than whole bone marrow (BM) cells and mononuclear cells in peripheral blood (PB). Here we analyzed the detailed features of PBcfDNA and its utility in genetic analyses in MDS. The plasma‐PBcfDNA concentration in MDS and related diseases (N = 33) was significantly higher than that in healthy donors (N = 14; P = 0.041) and in International Prognostic Scoring System higher‐risk groups than that in lower‐risk groups (P = 0.034). The concentration of plasma‐/serum‐PBcfDNA was significantly correlated with the serum lactate dehydrogenase level (both P < 0.0001) and the blast cell count in PB (P = 0.034 and 0.025, respectively). One nanogram of PBcfDNA was sufficient for one assay of Sanger sequencing using optimized primer sets to amplify approximately 160‐bp PCR products. PBcfDNA (approximately 50 ng) can also be utilized for targeted sequencing. Almost all mutations detected in BM‐DNA were also detected using corresponding PBcfDNA. Analyses using serially harvested PBcfDNA from an RAEB‐2 patient showed that the somatic mutations and a single nucleotide polymorphism that were detected before allogeneic transplantation were undetectable after transplantation, indicating that PBcfDNA likely comes from MDS clones that reflect the disease status. PBcfDNA may be a safer and easier alternative to obtain tumor DNA in MDS.
Keywords: Biomarkers, genetic mutations, myelodysplastic syndromes, peripheral blood cell‐free DNA, targeted sequencing
Myelodysplastic syndromes (MDS) are a group of hematopoietic stem cell disorders characterized by peripheral cytopenia, dysplasia and a risk of progression to acute myeloid leukemia (AML).1 Recently, several somatic mutations in genes that affect DNA methylation (e.g. DNMT3A, TET2 and IDH1/2), chromatin modification (e.g. EZH2 and ASXL1), RNA splicing (e.g. SF3B1, U2AF1, SRSF2 and ZRSR2), cohesion complexes (e.g. STAG2, SMC3 and RAD21) and others2, 3, 4, 5, 6, 7, 8 have been identified in more than 90% of MDS patients.9, 10 Some of these genetic abnormalities are closely related to specific phenotypes, such as ring sideroblasts,6 hypo‐lobulated granulocytes11, 12 and micro‐megakaryocytes13 as well as prognosis.9, 10, 14 In AML, several specific genetic mutations act as initiating and/or driver mutations and result in disease onset and progression via a growth advantage and clonal expansion of leukemic stem cells.15, 16, 17, 18 Similar molecular mechanisms may also occur in MDS.19, 20, 21 Researchers have speculated that accumulation of specific genetic abnormalities, such as mutations in BCOR,22 SETBP1,23 ASXL1,10 STAG2 and SMC3,24 may be critical for relapse, progression and acquisition of drug resistance.25
Thus, genetic analyses using serially harvested samples at various time points during the clinical course have become more important not only for diagnosis but also for recognizing the disease status and selecting the optimal molecular targeted therapies. Genomic DNA from bone marrow (BM) mononuclear cells (MNC) obtained by BM aspiration is usually used for genetic analyses. However, repeated BM aspirations should be avoided as much as possible because they are difficult for patients.
Recently, peripheral blood circulating cell‐free DNA (PBcfDNA), which is fragmented DNA detected in plasma and serum, has received attention as an alternative DNA source that originates from malignant tumors. Utilization of PBcfDNA for genetic analyses has been reported mainly for solid tumors, such as breast,26, 27, 28 colorectal29 and lung cancer,30 especially in advanced stage patients. To date, few studies on PBcfDNA in MDS have been published.31, 32 Previously, we reported the usefulness of PBcfDNA for detecting genetic and epigenetic abnormalities in a limited number of MDS cases. We showed that genetic mutations detected in BM CD34(+)/CD38(−) blast cells accumulate in PBcfDNA and that epigenetic changes after using azacitidine, a DNA methyltransferase inhibitor, can be detected more sensitively in serially harvested PBcfDNA than PBMNC.31 However, detailed information about the clinical and molecular features of PBcfDNA has not been reported.
In this manuscript, we report the molecular characteristics of PBcfDNA from MDS patients and healthy donors, and the correlation of PBcfDNA with laboratory data and clinical features. We also show that PBcfDNA can be utilized as a biomarker of the disease status and as an alternative source of tumor DNA instead of BMMNC for genetic analyses.
Materials and Methods
Patients’ characteristics
After obtaining appropriate informed consent, the study population included 33 patients with MDS‐related diseases (MDS; N = 24, MDS/MPN; N = 2, and MDS/tAML; N = 7) diagnosed at Nagoya University Hospital. Diagnosis and prognostic stratification were assigned according to the World Health Organization classification33 and International Prognostic Scoring System (IPSS),34 respectively. Patient characteristics are summarized in Table S1.
Collection of plasma, serum and mononuclear cells from peripheral blood
Bone marrow and PB samples were harvested from these patients at or close to the same disease period. In one patient with MDS refractory anemia with excess blasts‐2 (RAEB‐2) at diagnosis, BM and PB samples were serially collected during the clinical course, and a total of 34 BM and PB paired samples were analyzed. Plasma samples were collected in all 33 patients, and serum samples were harvested from 28 patients. Plasma from 14 healthy volunteer donors was also harvested after obtaining appropriate informed consent. Blood collection tubes with EDTA‐2Na were mainly utilized for plasma collection. For serum, tubes with a separating agent were used. Collected blood samples were stored at 4°C until further use. After centrifugation for 10 min at 430 g for plasma and 1710 g for serum, plasma and serum were aliquoted into 1.7‐mL tubes and stored at −80°C.
Mononuclear cells from PB (PBMNC) and BM were collected using Ficoll‐paque (GE Healthcare, Uppsala, Sweden) as indicated previously.35 CD3‐positive T cells were purified from PBMNC or BMMNC using CD3 MicroBeads/human (Miltenyi Biotec, Bergisch Gladbach, Germany) and Dynabeads Human T‐activator CD3/CD28 (Invitrogen, Carlsbad, CA, USA).36 DNA from buccal mucosa was obtained for use as germline control DNA.
Extraction of PBcfDNA and DNA from peripheral blood/bone marrow mononuclear cells
Genomic DNA from PBMNC and BMMNC was extracted using the QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA, USA). PBcfDNA from plasma (plasma‐PBcfDNA) and serum (serum‐PBcfDNA) was extracted using the QIAamp MinElute Virus Vacuum Kit (Qiagen) or QIAamp Circulating Nucleic Acid Kit (Qiagen). For germline control DNA, genomic DNA was extracted from CD3+ T cells and/or buccal mucosa using the QIAamp DNA Blood Mini Kit.
Confirmation of PBcfDNA concentration
Plasma‐/serum‐PBcfDNA extracted from 45 μL plasma and/or serum was visualized with 1% agarose gel electrophoresis. Fragmented bands of PBcfDNAs were quantitated by using DNA analysis software (CS Analyzer ver. 3.0; ATTO, Tokyo, Japan), and the size and total amount of PBcfDNA were calculated. In some cases, the concentration and quality of PBcfDNA were also analyzed by BioAnalyzer (Agilent Technologies, Santa Clara, CA, USA). The concentration of PBcfDNA was compared with the patients’ clinical data.
Detection of genetic mutations
Using genomic DNA from BMMNC (BM‐DNA), PBMNC (PBMNC‐DNA) and PBcfDNA, the nucleotide sequences of IDH2 (exon 4), SETBP1 (exon 4), U2AF1 (exon 2 and 6), SRSF2 (exon 2) and NRAS (exon 2 and 3) were determined by PCR followed by Sanger sequencing. TET2 mutation (exons 3 to 11) was analyzed in patients showing 4q uni‐parental disomy in single nucleotide polymorphism (SNP) array analysis (unique patient number [UPN] #3 and #6) as indicated previously.31 All mutations were determined to be somatic mutations following comparison with the corresponding germline control DNA. The PCR primers utilized are shown in Table S2. Targeted sequencing was performed for two patients (UPN #2‐2 and #33) using the TruSight Myeloid Sequencing Panel (Illumina, San Diego, CA, USA). Detection of FLT3 internal tandem duplication (FLT3‐ITD) by PCR was carried out as reported previously.37
Statistical analyses
Statistical analysis with the unpaired t‐test and nonlinear regression were performed with Prism version 5 software (Graph Pad Software, La Jolla, CA, USA). P‐values < 0.05 were considered statistically significant.
Results
Characteristics of PBcfDNA in myelodysplastic syndromes and healthy donors
To determine the characteristics of PBcfDNA in MDS, plasma‐/serum‐PBcfDNA from a clinically well‐characterized cohort of MDS patients was analyzed by agarose gel electrophoresis, and the concentration was measured by using gel imaging software. All plasma‐PBcfDNA (MDS; N = 34, healthy donors; N = 14) and serum‐PBcfDNA (MDS; N = 29) samples were successfully visualized in 1% agarose gels (Fig. S1a). As indicated previously,31 PBcfDNA was detected as a ladder pattern of 160–180 bp following 1% agarose gel electrophoresis and analysis using BioAnalyzer, a capillary electrophoresis system (Fig. S1b). Considering the fragmented size of PBcfDNA, PBcfDNA may exist in nucleosomal structures. The red arrows in Figure S1(a) may correspond to DNA fragments from mono‐nucleosomes to penta‐nucleosomes. Note that large variations in PBcfDNA concentrations were detected among patients (Fig. S1a).
Comparison of the PBcfDNA concentrations from plasma and serum
PBcfDNA concentrations (ng/mL of plasma or serum) are depicted in Figure 1(a). The median plasma‐PBcfDNA and serum‐PBcfDNA concentrations were 60.7 ng/mL (range; 0–915.9) and 30.7 ng/mL (range; 0–465.2), respectively. The plasma‐/serum‐PBcfDNA concentration from MDS patients was significantly higher than the plasma‐PBcfDNA concentration from healthy donors (P = 0.041 and 0.048, respectively). The plasma‐PBcfDNA and serum‐PBcfDNA concentrations were significantly correlated (r: 0.892, P < 0.0001) (Fig. 1b).
Figure 1.
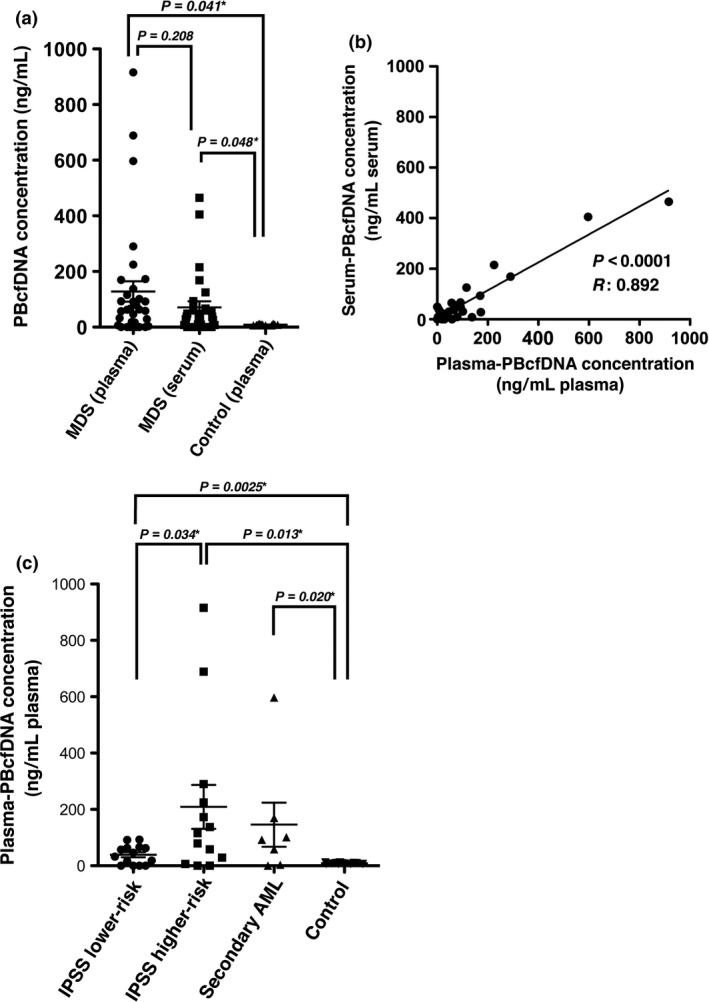
Plasma‐PBcfDNA and serum‐PBcfDNA concentration in myelodysplastic syndromes (MDS), and correlation of the PBcfDNA concentration with the International Prognostic Scoring System (IPSS) risk group. (a) Plasma‐/serum‐PBcfDNA from MDS patients (N = 33) and healthy donors (plasma) (N = 14) was analyzed. The plasma‐PBcfDNA and serum‐PBcfDNA concentrations in MDS were significantly higher than those in healthy donors. The P‐value marked with an asterisk indicates a significant difference with the unpaired t‐test. (b) Comparison of the plasma‐/serum‐PBcfDNA concentration shows a significant linear correlation following linear regression analysis. (c) The plasma‐PBcfDNA concentrations in the lower‐/higher‐risk IPSS score groups and secondary AML were compared. Plasma‐PBcfDNA from the higher‐risk IPSS group was significantly higher than that in the lower‐risk IPSS group, and the concentration in both the lower‐risk and higher‐risk IPSS groups was significantly higher than that in normal donors. Asterisks indicate a significant difference in the unpaired t‐test.
The PBcfDNA concentration is significantly higher in higher‐risk International Prognostic Scoring System patients than in lower‐risk International Prognostic Scoring System patients
International Prognostic Scoring System scores34 in this cohort are shown in Table S1. Of the 33 MDS patients, 14 and 12 cases were stratified into lower (low and intermediate‐1) and higher (intermediate‐2 and high) IPSS scores, respectively. Seven cases showed transformation to secondary AML at the time of PB sample collection. The concentrations of plasma‐PBcfDNA were significantly higher in the higher‐risk IPSS score group compared to the lower‐risk IPSS score group (Fig. 1c, P = 0.034). The plasma‐PBcfDNA concentration in secondary AML was not significantly higher than that in MDS lower‐risk and higher‐risk IPSS groups (P = 0.069 and 0.61, respectively). The concentrations of serum‐PBcfDNA from the lower‐risk and higher‐risk IPSS groups were not significantly different (P = 0.121) (data not shown).
Correlation of PBcfDNA concentration with laboratory data
To determine the biochemical characteristics of PBcfDNA, we compared the PBcfDNA concentrations with laboratory data (Fig. 2). The plasma‐PBcfDNA and serum‐PBcfDNA concentrations were significantly correlated with the serum lactate dehydrogenase (LDH) level (plasma: P < 0.0001, serum: P < 0.0001) and the PB blast count (plasma: P = 0.034, serum: P = 0.025). However, we found no significant correlations between the PBcfDNA concentration and the following PB laboratory data: C‐reactive protein (CRP) (plasma: P = 0.994, serum: P = 0.477), white blood cell counts (WBC) (plasma: P = 0.4656, serum: P = 0.498), and hemoglobin (Hb) (plasma: P = 0.651, serum: P = 0.956). We also found no correlation between the PBcfDNA concentration and the following laboratory data using BM samples: BM cell count (plasma: P = 0.248, serum: P = 0.281), BM blast percentage (plasma: P = 0.947, serum: P = 0.731) and BM blast count (plasma: P = 0.5510, serum: P = 0.8172). These data suggest that the PBcfDNA originates from MDS clones that were likely more fragile than normal blood cells, and the PBcfDNA concentration may be correlated with the disease status and reflects the LDH level and PB blast cell counts, as well as IPSS risk stratification.
Figure 2.
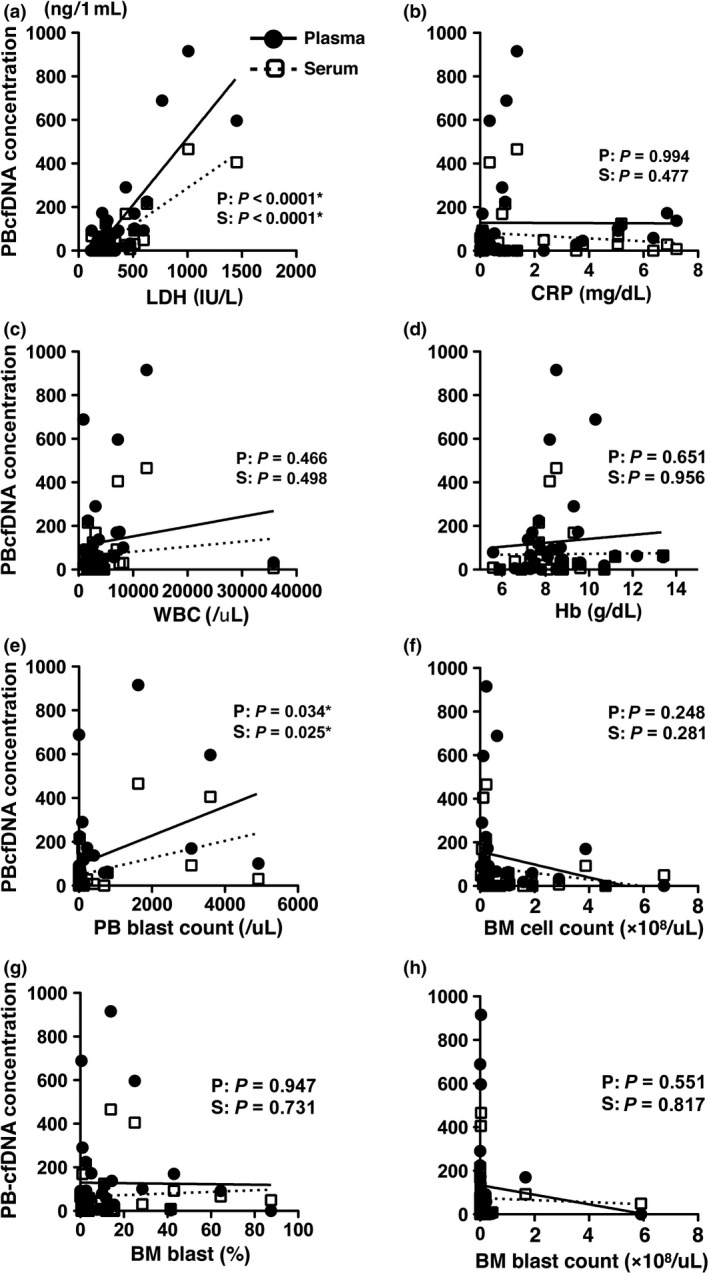
Correlation of the plasma‐PBcfDNA and serum‐PBcfDNA concentration with laboratory data. Correlation of the plasma‐PBcfDNA and serum‐PBcfDNA concentrations with laboratory data was analyzed. (a) Serum lactate dehydrogenase (LDH), (b) C‐reactive protein (CRP), (c) white blood cell counts (WBC), (d) hemoglobin (Hb), (e) peripheral blood (PB) blast count, (f) bone marrow (BM) cell count, (g) BM blast percentage and (h) BM blast count. The PBcfDNA concentration was significantly correlated with the LDH level and the PB blast count. The P‐value was confirmed in linear regression analysis, and the asterisks indicate significant data. Black circles, plasma‐PBcfDNA; white squares, serum‐PBcfDNA.
If the fragility of MDS clones is closely related to the PBcfDNA concentration, the strategy for sample preparation in laboratories is quite critical. To investigate the possibility that genomic DNA may be released from MDS clones in sample tubes after sample collection and storage in refrigerators, plasma from whole blood samples of a patient that had been stored in a refrigerator for different time periods, from 15 min to 24 h after blood collection, was prepared, and then PBcfDNA extraction was performed. No significant elevation of PBcfDNA concentration was observed in this case, even after preservation for 24 h in a refrigerator (Fig. S2). These data suggest that the PBcfDNA concentration may depend on ineffective hematopoiesis resulting from increasing apoptosis in a patient's body rather than release from cells in sample collection tubes.
PCR amplification using PBcfDNA for Sanger sequencing
For mutation analyses using the Sanger sequencing strategy, we first optimized the PCR, because genomic DNA in PBcfDNA is fragmented and is quite different from total genomic DNA extracted from whole cells. Two different primer pairs were used to amplify U2AF1 exon 2, resulting in PCR products of different sizes (primer set (i)): 324 bp and (ii): 161 bp (Fig. 3a). When using BM‐DNA, the PCR amplification efficacy was similar between primer sets (i) and (ii) (Fig. 3b; BM‐DNA), even when using a lower concentration (0.2 ng) of template DNA. When using PBcfDNA as a PCR template, the amplification efficacy was significantly higher when using primer set b) than a) (Fig. 3b; PBcfDNA). The difference was obvious when using a lower concentration (1 ng or less). Considering these data, we designed all primer sets to amplify approximately 160 bp for Sanger sequencing of the target genes (Table S2).
Figure 3.
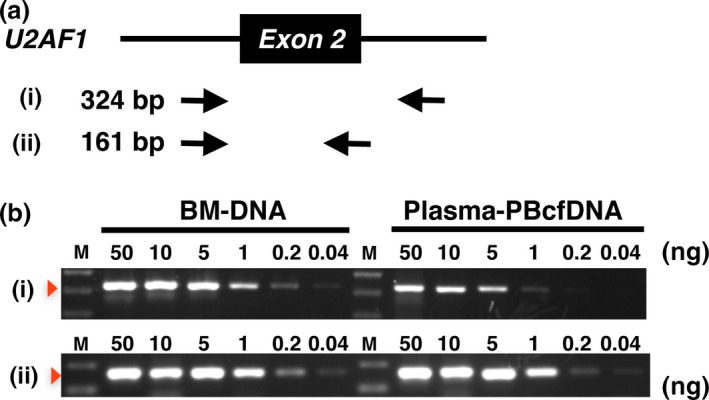
Utilization of PBcfDNA for PCR amplification. (a) Schematic representation of exon 2 and the adjacent introns of U2AF1. Two primer sets for PCR, (i) and (ii), and the size of those products are indicated. Black arrows indicate the primers. (b) Semi‐quantitative PCR was performed using (i) and (ii). BM‐DNA and plasma‐PBcfDNA were used as template DNA. The template DNA concentration in each reaction is also indicated. Note that amplification efficiencies were obviously higher with primer set (ii) than with (i) when using PBcfDNA as the PCR template.
Detection of genetic mutations with Sanger sequencing and targeted sequencing strategies in BM‐DNA and PBcfDNA
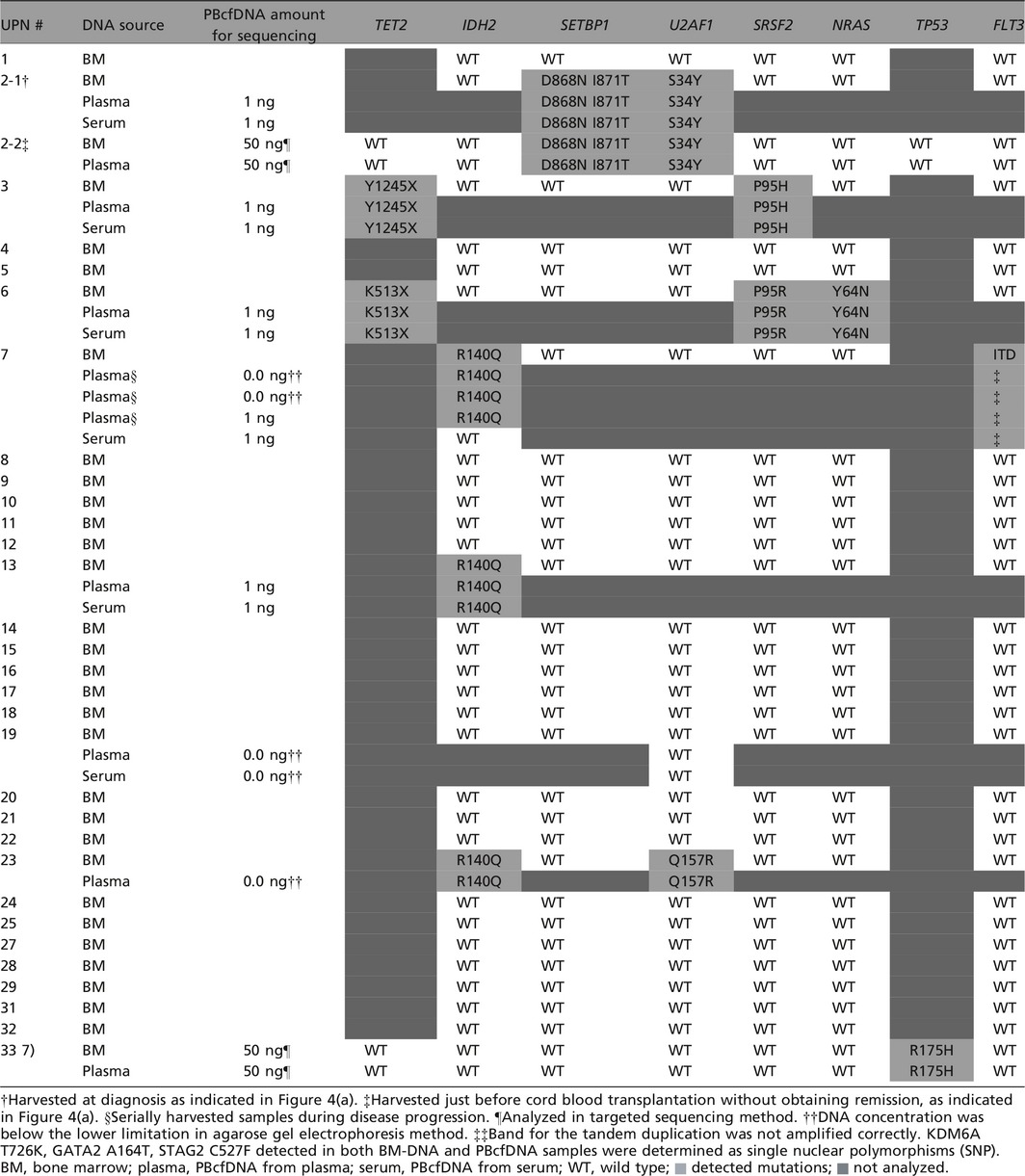
To confirm whether the genetic mutations detected in BM‐DNA are also detectable in PBcfDNA, Sanger sequencing was performed for IDH2, U2AF1 and SRSF2 using BM‐DNA for all samples. For 2 patients (UPN #2 and 6) that showed 4q uni‐parental disomy in SNP array analysis, TET2 mutation was analyzed with Sanger sequencing. The FLT3‐ITD mutation was also analyzed by PCR using BM‐DNA for all samples. In patients in whom some genetic mutations were detected in BM‐DNA, genetic mutations were also analyzed in PBcfDNA. Targeted sequencing, a next‐generation sequencing strategy, was performed in two patients (UPN #2‐2 and #33) using both BM‐DNA and PBcfDNA, and the concordance was confirmed.
The genetic mutations detected in BM‐DNA and PBcfDNA from the 33 MDS patients are summarized in Table 1. Almost all genetic mutations detected by Sanger and targeted sequencing in BM‐DNA were also detected in the corresponding PBcfDNA. Representative data from Sanger sequencing are shown in Figure S3. In one case (UPN #7), a guanine (G) to cytosine (C) mutation resulting in an R140Q substitution in IDH2 was detected in BM‐DNA and three serially harvested plasma‐PBcfDNA samples, but was not detected in serum‐PBcfDNA (Table 1 and Fig. S4). FLT3‐ITD was detected in one patient (UPN #7) in BM‐DNA, but was not detected in plasma‐/serum‐PBcfDNA in the same patient (PCR data not shown). These data suggest that most genetic mutations detected in BM‐DNA are also detectable in plasma‐/serum‐PBcfDNA, and that plasma‐PBcfDNA may be more suitable for mutational analysis than serum‐PBcfDNA. To detect FLT‐ITD in PBcfDNA, optimization of the PCR conditions may be necessary because of the larger size (>180 bp) of the PCR product.
Table 1.
Comparison of genetic mutations detected in BMDNA and PBcfDNA
Utilization of serially harvested PBcfDNA to detect disease status
To determine the usefulness of PBcfDNA for confirming disease status, such as disease progression and/or remission, PBcfDNA was harvested serially from several patients. The clinical course of a representative patient is shown in Figure 4. The patient was a 29‐year‐old man with MDS‐RAEB‐2, in whom a U2AF1 mutation was detected (TCT to TAT substitution in exon 2, resulting in S34Y) (Fig. 4a). After 5‐Aza treatment for 7 days, cord blood transplantation (CBT) was performed after a myeloablative conditioning regimen, and complete remission (CR) was obtained. PB and BM samples were obtained at time points a, b and c, as indicated (Fig. 4a). A chimeric screening test using BM cells to confirm the engraftment efficiency was carried out at time point c, and the complete chimera (donor type) was confirmed.
Figure 4.
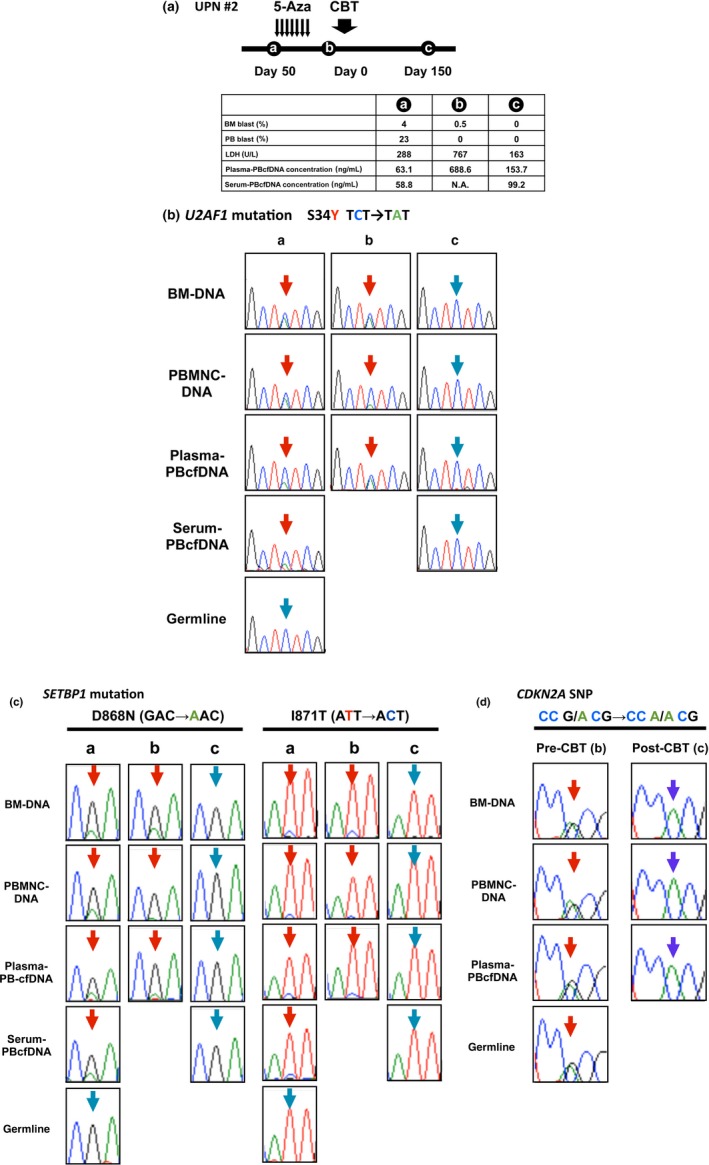
Mutational analyses using serially harvested PBcfDNA samples at different time points of disease status. (a) The clinical course of the MDS‐RAEB2 patient (UPN #2) is indicated. After one course of 5‐Aza, cord blood transplantation (CBT) was performed at Day 0, and complete remission (CR) was obtained. Peripheral blood (PB) and bone marrow (BM)samples were obtained at time points a, b and c. PB and BM blast percentage, serum lactate dehydrogenase (LDH) level and PBcfDNA concentration are also indicated. At time point b, disease progression was clinically observed. (b) U2AF1 and (c) SETBP1 mutations were confirmed by Sanger sequencing of BM‐DNA, PBMNC‐DNA and plasma‐/serum‐PBcfDNA. Note that those two mutations were not detected at time point c. (d) An SNP in CDKN2A detected by Sanger sequencing in BM‐DNA, PBMNC‐DNA, plasma‐/serum‐PBcfDNA and germline cells at time point b was not confirmed after CBT at time point c. Blue arrows, wild‐type sequence; red arrows, mutated sites. N.A., sample not available.
U2AF1 and SETBP1 mutation were detected in BM‐DNA, PBMNC‐DNA and plasma‐/serum‐PBcfDNA before CBT (Fig. 4b, time points a and b, and Fig. 4c, time points a and b). The PBcfDNA concentration was elevated at time point b, showing clinically refractory disease with tumor fever and LDH elevation. The frequency of the U2AF1 mutation in plasma‐PBcfDNA at time point b was also increased (Fig. 4b, time points a and b). However, the blast percentage in BM and PB and the mutation frequency in PBMNC‐DNA at time point b decreased compared to time point a. After CBT at time c, U2AF1 and SETBP1 mutations were not detected in any of the DNA samples (Fig. 4b, time point c, and Fig. 4c, time point c). These data suggest that the concentration of PBcfDNA and the existing frequency of genetic mutations may correlate more accurately with disease status than the blast percentage in PB and BM and reflect the tumor burden in the patient's entire body.
Interestingly, an SNP in CDKN2A confirmed in all samples harvested before CBT was not detected after CBT in BM‐DNA, PBMNC‐DNA or plasma‐PBcfDNA (Fig. 4d). This phenomenon clearly indicated that PBcfDNA is mainly derived from allogeneic transplanted donor blood cells, not germline cells from non‐hematopoietic organs in the host body.
Discussion
For quantification of PBcfDNA, we performed agarose gel electrophoresis followed by semi‐quantification with gel imaging software. We also used BioAnalyzer (Agilent Technologies), which is a capillary electrophoresis system, and NanoVue (GE Healthcare), which is a micro‐volume UV‐Vis spectrophotometer. The lower limits of sensitivity are 2 and 0.1 ng/μL, respectively, according to the manufacturers’ instructions. Unexpectedly, these two strategies were not useful for accurately detecting the DNA, probably because of the low concentration and/or fragmentation of DNA. Agarose gel electrophoresis followed by quantification using imaging software is a conventional strategy for confirming the concentration and DNA fragmentation pattern, even with low concentrations. However, it takes time, and the background is sometimes problematic. Further improvement of strategies for measuring the DNA concentration is still needed.
The PBcfDNA concentration varied among the MDS patients (Figs 1a,S1a) and, importantly, the concentration was significantly lower in healthy donors. The PBcfDNA concentration was significantly correlated with the serum LDH level and the PB blast count (Fig. 2a,e), and was significantly higher in the higher‐risk IPSS score group than in the lower‐risk IPSS score group (Fig. 1c). Furthermore, the PBcfDNA concentration in serially harvested samples at different time points of the disease status in a patient appeared to be closely related to disease progression and/or regression (Fig. 4a–c). Importantly, MDS‐specific genetic mutations seen in BM cells were also confirmed in PBcfDNA. Our previous data indicated that the ratio of the mutated allele in BM CD34(+)/CD38(−) blast cells is much higher than that in CD34(+)/CD38(+) and CD34(−) cells, and the mutant ratio in PBcfDNA was almost equivalent to or much higher than that in CD34(+)/CD38(−) cells.31 These data strongly suggest that PBcfDNA is mainly derived from MDS clones rather than normal clones and normal tissue. This phenomenon may suggest that genomic DNA release occurs much more readily from MDS clones than from normal hematologic cells. MDS clones are more fragile in the patients’ bodies than normal clones due to ineffective hematopoiesis, aberrant membrane structure, and accelerated apoptosis.34
In contrast, the plasma‐PBcfDNA concentration in secondary AML was significantly higher than that in normal controls, but was not significantly higher than that in lower‐risk and higher‐risk MDS patients. This phenomenon suggest that the DNA release from tumor cells in secondary AML cannot be estimated simply by PB blast cell count but that fragility also impacts DNA release. Further accumulation of patients and detailed analyses of the backgrounds of secondary AML in each patient may be required.
The serum‐PBcfDNA concentration tended to be lower compared to plasma‐PBcfDNA (Fig. 1a), and the difference in concentration between lower‐risk and higher‐risk MDS patients was not significant when using serum‐PBcfDNA (data not shown). One possible reason for this phenomenon is that a significant amount of PBcfDNA can be trapped in blood clots, decreasing the precision for determination of the PBcfDNA concentration.
An SNP in CDKN2A (Fig. 4d), which was confirmed in recipient PBcfDNA before CBT, was not detected in PBcfDNA after CBT with CR. This finding suggests that a large proportion of PBcfDNA in a healthy person originates from blood cells, rather than from other tissues such as the liver, intestine and others. Moreover, the PBcfDNA concentration was still high in the condition of CR after CBT, as shown in Figure 4(a). This phenomenon indicated that the PBcfDNA concentration may not reflect only the pathologic state but also certain aspects of the physiological state such as proliferation of hematopoietic cells after engraftment following stem cell transplantation. Further accumulation of samples from patients who have undergone stem cell transplantation is also required to investigate this possibility.
As shown in Figure 2(f–h), we found no correlations between the PBcfDNA concentration and BM data, including BM cell count, BM blast percentage and BM blast count. As we experienced in the clinical setting, results of BM aspiration, such as cellular density, are sometimes different according to the location of BM aspiration and because the aspiration skills may not be the same for all clinicians. Furthermore, the definition of BM blast cells, especially in MDS, is sometimes difficult because of the variety of dysplasia and the regional heterogeneity of genetic abnormalities in MDS clones.15, 21 However, PB collection is usually much easier than BM aspiration, and PB is likely more homogeneous regardless of the collection site. Our data and speculation about sample collection suggest that PBcfDNA may be much more accurate for detection of genetic abnormalities that exist in a patient's entire hematopoietic cell population than BM‐DNA if a high enough DNA concentration for sequencing can be obtained from PBcfDNA.
Analyses using serially harvested PBcfDNA samples from a patient who was treated with 5‐Aza followed by CBT indicated that the DNA concentration was markedly elevated (63.1–688.6 ng/mL) at the time of disease progression as defined by the clinical findings of documented tumor fever and LDH elevation. The ratio of the U2AF1 mutated allele in plasma‐PBcfDNA was also increased (Fig. 4a,b), probably due to significant expansion of MDS clones in his entire body. These data suggest that the PBcfDNA concentration and the ratio of the mutated allele can be utilized as biomarkers to predict the disease status and confirm minimal residual disease. We tried to perform pyro‐sequencing analysis to quantitate the U2AF1 mutation, but, unfortunately, the assay was not successful. Inappropriate assay conditions probably related to fragmentation of the template DNA may explain this lack of success. Further improvement of the assay system and/or introducing allele‐specific quantitative PCR are required.
Because PBcfDNA showed a fragmented pattern that likely reflects nucleosomal structures, we optimized the PCR conditions for Sanger sequencing with primer sets that amplified approximately 160‐bp DNA products (Fig. 3). Most of the genetic mutations were confirmed by Sanger sequencing after obtaining specific PCR products (Table 1). However, to detect FLT3‐ITD, PCR amplification using previously reported primer sets did not work well (data not shown, Table 1), probably because the size of the PCR products was >200 bp.38 We tried to design PCR primers on the flanking position of the duplicated sequences, but this was quite difficult because the duplicated sequences are sometimes longer than 200 bp, and also because the duplicated sequences vary from patient to patient. Further improvement of the detection strategy for FLT3‐ITD using PBcfDNA is required.
PBcfDNA can be obtained from both plasma and serum,31, 39, 40, 41 but which sample is more suitable for detecting genetic abnormalities in malignant situations, especially MDS, was unclear. In healthy individuals, the concentration of serum‐PBcfDNA tended to be higher than that of plasma‐PBcfDNA.31, 39 In our experiment, the concentration of plasma‐PBcfDNA in MDS tended to be higher than that of serum‐PBcfDNA, but the difference was not significant (Fig. 1a). Our previous limited data for genetic analyses using PBcfDNA indicate that both plasma‐PBcfDNA and serum‐PBcfDNA can be utilized for Sanger sequencing, and the mutant allele frequencies in plasma‐PBcfDNA and serum‐PBcfDNA are almost the same and relatively higher compared with the frequency in DNA from BM CD34(+)/38(−) blast fraction cells.31 Mohamedali et al.32 report that serum‐PBcfDNA can be utilized for Sanger sequencing in some cases. However, the concordance with BM‐DNA was only 42%, and Sanger sequencing could be performed in only 9 (75%) of 12 cases. In our experiment, a genetic mutation in IDH2 that was confirmed in BM‐DNA and plasma‐PBcfDNA was not detected in serum‐PBcfDNA in one case (Table 1 and Fig. S4). Furthermore, Board et al.39 report that the serum‐PBcfDNA concentration increases in a time‐dependent manner after blood coagulation, suggesting release of genomic DNA from white blood cells containing normal clones. This phenomenon may be a reasonable explanation for the increasing contamination with the wild‐type genome in serum‐PBcfDNA. Thus, plasma‐PBcfDNA may be more suitable for genetic analysis in MDS than serum‐PBcfDNA.
Our data strongly suggest that PBcfDNA is a useful alternative source of tumor DNA in MDS, not only for conventional sequencing strategies but also for newer, comprehensive genetic analysis. Collection of PB is much safer, easier and relatively less painful compared to BM aspiration, and serial sample collection is also feasible. Furthermore, storing plasma/serum samples in the clinical setting is much easier and faster than storing PBMNC and BMMNC. PBcfDNA may also become a useful biomarker that closely reflects the patient's clinical situation. Further optimization and improvement of the sensitivity of sequencing analyses is still required for this strategy to be widely utilized in the clinical setting, especially for patients with a low PBcfDNA concentration.
Disclosure Statement
H. K. received research funding from Bristol‐Myers Squibb, Chugai Pharmaceutical, Kyowa Hakko Kirin, Dainippon Sumitomo Pharma, Zenyaku Kogyo and FUJIFILM. The other authors have no relevant conflicts to disclose.
Supporting information
Figure S1. Confirmation of the fragmentation pattern of plasma‐/serum‐PBcfDNA in agarose gel electrophoresis.
Figure S2. The difference of PBcfDNA concentration in peripheral blood samples harvested at the different time points.
Figure S3. Detection of genetic mutations in PBcfDNA.
Figure S4. Detection of IDH2 mutation using plasma‐/serum‐PBcfDNA in UPN #7.
Table S1. Patients’ characteristics.
Table S2. PCR primers.
Acknowledgments
This work was supported in part by Grants‐in‐Aid from the National Cancer Center Research and Development Fund (26‐A‐4), and the Ministry of Education, Culture, Sports, Science and Technology (24591388, 15K09473), Japan. We thank Dr Seishi Ogawa and Dr Masashi Sanada for performing the SNP array analysis. We thank Chika Wakamatsu, Yoko Matsuyama, Yukie Konishi, Manami Kira, Rie Kojima, Yuko Kojima, Emi Kono and Saya Kojima for valuable laboratory assistance.
Cancer Sci 107 (2016) 1329–1337
Funding Information
National Cancer Center Research and Development Fund (26‐A‐4); Ministry of Education, Culture, Sports, Science and Technology, Japan (24591388, 15K09473).
References
- 1. Tefferi A, Vardiman JW. Myelodysplastic syndromes. N Engl J Med 2009; 361: 1872–85. [DOI] [PubMed] [Google Scholar]
- 2. Sanada M, Suzuki T, Shih LY et al Gain‐of‐function of mutated C‐CBL tumour suppressor in myeloid neoplasms. Nature 2009; 460: 904–8. [DOI] [PubMed] [Google Scholar]
- 3. Makishima H, Cazzolli H, Szpurka H et al Mutations of e3 ubiquitin ligase cbl family members constitute a novel common pathogenic lesion in myeloid malignancies. J Clin Oncol 2009; 27: 6109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bejar R, Levine R, Ebert BL. Unraveling the molecular pathophysiology of myelodysplastic syndromes. J Clin Oncol 2011; 29: 504–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yoshida K, Sanada M, Shiraishi Y et al Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011; 478: 64–9. [DOI] [PubMed] [Google Scholar]
- 6. Makishima H, Visconte V, Sakaguchi H et al Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 2012; 119: 3203–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yamazaki J, Issa JP. Epigenetic aspects of MDS and its molecular targeted therapy. Int J Hematol 2013; 97: 175–82. [DOI] [PubMed] [Google Scholar]
- 8. Kon A, Shih LY, Minamino M et al Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet 2013; 45: 1232–7. [DOI] [PubMed] [Google Scholar]
- 9. Haferlach T, Nagata Y, Grossmann V et al Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014; 28: 241–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bejar R, Lord A, Stevenson K et al TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood 2014; 124: 2705–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Muto T, Sashida G, Oshima M et al Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. J Exp Med 2013; 210: 2627–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim E, Ilagan JO, Liang Y et al SRSF2 mutations contribute to myelodysplasia by mutant‐specific effects on exon recognition. Cancer Cell 2015; 27: 617–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schneider RK, Adema V, Heckl D et al Role of casein kinase 1A1 in the biology and targeted therapy of del(5q) MDS. Cancer Cell 2014; 26: 509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bejar R, Stevenson KE, Caughey BA et al Validation of a prognostic model and the impact of mutations in patients with lower‐risk myelodysplastic syndromes. J Clin Oncol 2012; 30: 3376–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Welch JS, Ley TJ, Link DC et al The origin and evolution of mutations in acute myeloid leukemia. Cell 2012; 150: 264–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. The Cancer Genome Atlas Research Network. . Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013; 368: 2059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kihara R, Nagata Y, Kiyoi H et al Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia 2014; 28: 1586–95. [DOI] [PubMed] [Google Scholar]
- 18. Naoe T, Kiyoi H. Gene mutations of acute myeloid leukemia in the genome era. Int J Hematol 2013; 97: 165–74. [DOI] [PubMed] [Google Scholar]
- 19. Walter MJ, Shen D, Shao J et al Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia 2013; 27: 1275–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Papaemmanuil E, Gerstung M, Malcovati L et al Clinical, biological implications of driver mutations in myelodysplastic syndromes. Blood 2013; 122: 3616–27; quiz 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cazzola M, Della Porta MG, Malcovati L. The genetic basis of myelodysplasia and its clinical relevance. Blood 2013; 122: 4021–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Damm F, Chesnais V, Nagata Y et al BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood 2013; 122: 3169–77. [DOI] [PubMed] [Google Scholar]
- 23. Makishima H, Yoshida K, Nguyen N et al Somatic SETBP1 mutations in myeloid malignancies. Nat Genet 2013; 45: 942–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thota S, Viny AD. Genetic alterations of the cohesin complex genes in myeloid malignancies. Blood 2014; 124: 1790–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xie M, Lu C, Wang J et al Age‐related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014; 20: 1472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murtaza M, Dawson SJ, Tsui DW et al Non‐invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013; 497: 108–12. [DOI] [PubMed] [Google Scholar]
- 27. Dawson SJ, Tsui DW, Murtaza M et al Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med 2013; 368: 1199–209. [DOI] [PubMed] [Google Scholar]
- 28. Schwarzenbach H. Circulating nucleic acids as biomarkers in breast cancer. Breast Cancer Res 2013; 15: 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thierry AR, Mouliere F, El Messaoudi S et al Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat Med 2014; 20: 430–5. [DOI] [PubMed] [Google Scholar]
- 30. Wang S, An T, Wang J et al Potential clinical significance of a plasma‐based KRAS mutation analysis in patients with advanced non‐small cell lung cancer. Clin Cancer Res 2010; 16: 1324–30. [DOI] [PubMed] [Google Scholar]
- 31. Iriyama C, Tomita A, Hoshino H et al Using peripheral blood circulating DNAs to detect CpG global methylation status and genetic mutations in patients with myelodysplastic syndrome. Biochem Biophys Res Commun 2012; 419: 662–9. [DOI] [PubMed] [Google Scholar]
- 32. Mohamedali AM, Alkhatabi H, Kulasekararaj A et al Utility of peripheral blood for cytogenetic and mutation analysis in myelodysplastic syndrome. Blood 2013; 122: 567–70. [DOI] [PubMed] [Google Scholar]
- 33. Swerdlow SH, Campo E, Harris NL et al WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th edn IARC: Lyon, 2008. [Google Scholar]
- 34. Greenberg P, Cox C, LeBeau MM et al International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997; 89: 2079–88. [PubMed] [Google Scholar]
- 35. Goto E, Tomita A, Hayakawa F, Atsumi A, Kiyoi H, Naoe T. Missense mutations in PML‐RARA are critical for the lack of responsiveness to arsenic trioxide treatment. Blood 2011; 118: 1600–9. [DOI] [PubMed] [Google Scholar]
- 36. Watanabe K, Terakura S, Martens AC et al Target antigen density governs the efficacy of anti‐cd20‐cd28‐cd3 zeta chimeric antigen receptor‐modified effector CD8+ T Cells. J Immunol 2015; 194: 911–20. [DOI] [PubMed] [Google Scholar]
- 37. Kiyoi H, Towatari M, Yokota S et al Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia 1998; 12: 1333–7. [DOI] [PubMed] [Google Scholar]
- 38. Kiyoi H, Naoe T, Yokota S et al Internal tandem duplication of FLT3 associated with leukocytosis in acute promyelocytic leukemia. Leukemia Study Group of the Ministry of Health and Welfare (Kohseisho). Leukemia 1997; 11: 1447–52. [DOI] [PubMed] [Google Scholar]
- 39. Board RE, Williams VS, Knight L et al Isolation and extraction of circulating tumor DNA from patients with small cell lung cancer. Ann N Y Acad Sci 2008; 1137: 98–107. [DOI] [PubMed] [Google Scholar]
- 40. Morgan SR, Whiteley J, Donald E et al Comparison of KRAS mutation assessment in tumor DNA and circulating free DNA in plasma and serum samples. Clin Med Insights Pathol 2012; 5: 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Elshimali YI, Khaddour H, Sarkissyan M, Wu Y, Vadgama JV. The clinical utilization of circulating cell free DNA (CCFDNA) in blood of cancer patients. Int J Mol Sci 2013; 14: 18925–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Confirmation of the fragmentation pattern of plasma‐/serum‐PBcfDNA in agarose gel electrophoresis.
Figure S2. The difference of PBcfDNA concentration in peripheral blood samples harvested at the different time points.
Figure S3. Detection of genetic mutations in PBcfDNA.
Figure S4. Detection of IDH2 mutation using plasma‐/serum‐PBcfDNA in UPN #7.
Table S1. Patients’ characteristics.
Table S2. PCR primers.