

Abstract
The novel human gene family encoding neuronal leucine rich repeat (NLRR) proteins were identified as prognostic markers from our previous screening of primary neuroblastoma (NB) cDNA libraries. Of the NLRR gene family members, NLRR1 and NLRR3 are associated with the regulation of cellular proliferation and differentiation, respectively. However, the functional regulation and clinical significance of NLRR2 in NB remain unclear. Here, we evaluated the differential expression of NLRR2, where high expressions of NLRR2 were significantly associated with a poor prognosis of NB (P = 0.0009), in 78 NBs. Enforced expression of NLRR2 in NB cells enhanced cellular proliferation and induced resistance to retinoic acid (RA)‐mediated cell growth inhibition. In contrast, knockdown of NLRR2 exhibited growth inhibition effects and enhanced RA‐induced cell differentiation in NB cells. After RA treatment, NLRR2 expression was increased and correlated with the upregulation of c‐Jun, a member of the activator protein‐1 (AP‐1) family in NB cells. Moreover, the expressions of NLRR2 and c‐Jun were suppressed by treatment with a JNK inhibitor, which ameliorated the promoter activity of the NLRR2 gene while knockdown of c‐Jun reduced NLRR2 expression. We then searched AP‐1 binding consensus in the NLRR2 promoter region and confirmed c‐Jun recruitment at a consensus. Conclusively, NLRR2 must be an inducible gene regulated by the JNK pathway to enhance cell survival and inhibit NB cell differentiation. Therefore, NLRR2 should have an important role in NB aggressiveness and be a potential therapeutic target for the treatment of RA resistant and aggressive NB.
Keywords: c‐Jun, differentiation, JNK, neuroblastoma, NLRR2
Neuroblastoma (NB) is the most common form of malignancy in early childhood and is derived from the sympathoadrenal lineage of neural crest progenitor cells.1 NB exerts heterogeneous clinical behavior. Some NB regress spontaneously while others progress to highly metastatic tumors with a poor prognosis despite intensive multimodal therapy.2 However, the pathogenesis of NB is poorly understood as very few gene defects have been identified in this often‐lethal tumor. Frequently detected genetic alterations of NB are limited to MYCN amplification and ALK activation.3, 4, 5 Hence, aberrant gene expression patterns have been shown to be important to identify mechanisms behind the clinical outcome of NB. For example, high expression of the neurotrophic receptor TrkA has been identified as a prognostic factor for a favorable outcome because it inhibits tumor growth and angiogenesis, whereas TrkB and its ligand, brain‐derived neurotrophic factor, are associated with aggressive NB with MYCN amplification that enhances tumor growth and angiogenesis.4, 6, 7, 8 Some factors related to NB pathogenesis are involved in proliferation, differentiation and metastasis. However, little is known about their biological function with regards to tumor growth aggressiveness and this has negatively impacted on the development of therapeutic drugs. Therefore, studies on the molecular mechanisms of NB are required to develop new therapeutic strategies to treat aggressive NB.
Retinoic acid is an important metabolite that functions as a morphogen in the developing nervous system in vivo and induces the differentiation of neuronal cells in vitro.9 It has been implicated in human NB because RA treatment inhibits NB proliferation, differentiates into neuronal‐like cells, and reduces cell migration and invasiveness.10, 11, 12, 13 These properties have been used as a basis for the clinical application of RA, one of the limited drugs currently used to treat this type of cancer.10, 11, 12, 13, 14 It has become standard practice to treat high‐risk NB patients after marrow or stem cell transplantation.15 However, despite multiple clinical efforts, the prognosis remains poor for this enigmatic disease because of the high rate of resistance and metastasis. Human NB cells also often express multidrug resistance genes during differentiation by RA treatment.16, 17 Therefore, to overcome drug resistance in the aggressive NB, it is important to identify factors that contribute to RA resistant and RA‐mediated NB cell differentiation. RA binds to RAR and activates a signal transduction pathway that involves multiple cytoplasmic signaling molecules such as JNK and ERK. The JNK pathway has been proposed to induce a signaling cascade during RA‐mediated differentiation of NB as well as non‐NB.18, 19, 20 The downstream component of JNK/SAP MAPK is activator protein‐1 (AP‐1), which is a homodimeric or heterodimeric complex consisting of c‐Jun, c‐fos, Maf and ATF2. Based on the cellular context, the composition of the AP‐1 dimeric complex determines its functions in the regulation of differentiation, proliferation and apoptosis.19, 20, 21, 22, 23 However, the direct molecular mediator in transmitting RA signal to regulate neuritis extension in NB has remained unknown. Here, we report that NLRR2 acts as an inhibitor for the RA‐mediated differentiation of NB, and, therefore, contributes to NB aggressiveness.
NLRR2 is a member of the NLRR gene family that encode a glycosylated transmembrane protein with a leucine rich repeat (LRR) domain containing 11 or 12 LRR, an immunoglobulin c2‐type domain, and a type III fibronectin domain in its extracellular domain. Like other NLRR family proteins, NLRR2 has highly conserved amino acid sequences in the extracellular domain, which includes substrate/chemical binding sites and four potential glycosylation sites. NLRR2 also possesses ww interacting domains in the short intracellular region, which mediate protein–protein interactions and might provide a basis of signaling events for NLRR function in tumorigenesis.24, 25, 26 We previously reported that NLRR1 enhances epidermal growth factor (EGF)‐mediated MYCN induction in NB, resulting in the acceleration of tumor growth in vivo, and that a high expression of NLRR1 mRNA levels is associated with a poor prognosis of NB. In contrast, low NLRR3 expression levels were correlated with a poor prognosis in NB patients.27, 28, 29 However, very little is known about the role of NLRR2 in tumor progression, except it has been reported to be amplified and overexpressed in malignant gliomas.30 The current study reveals that RA functions as a negative feedback regulator through the upregulation of NLRR2 during RA‐mediated differentiation in NB. NLRR2 might be a useful pharmacological indicator to predict RA efficiency in NB treatment and should be considered as a therapeutic target for RA‐resistant aggressive NB.
Materials and Methods
Cell culture and agents
Human NB‐derived TGW, SMS‐SAN and non‐NB HeLa cells were collected from The Children's Hospital of Philadelphia cell line bank (Philadelphia, PA, USA), and SK‐N‐BE NB cells were collected from the European Collection of Cell Cultures (Wiltshire, UK) cell bank. NB cells were maintained in RPMI 1640 medium (Wako, Osaka, Japan), supplemented with 10% heat‐inactivated FBS (Invitrogen, CA, USA), 50 μg/mL penicillin and 50 μg/mL streptomycin (Invitrogen). HeLa cells were maintained in DMEM medium (Wako) with the same supplements. All cells were cultured in a humidified chamber provided with 5% CO2 at 37°C. RA and cisplatin (CDDP) were purchased from Sigma‐Aldrich (St. Louis, MO, USA).
siRNA‐mediated knockdown
A mixture of two sets of siRNA sense and antisense sequences (NLRR2: siNLRR2‐1, 5′‐CUACAGGAACUCUAUCUCATT‐3′ [sense] and 5′‐UGAGAUAGAGUUCCUGUAGTT‐3′ [antisense]; siNLRR2‐2, 5′‐CCAACUUGGAGAUACUCAUTT‐3′ [sense] and 5′‐AUGAGUAUCUCCAAGUUGGTT‐3′ [antisense]) was used to target human NLRR2 (Takara, Shiga, Japan). c‐Jun siRNA was purchased from Cell Signaling Technology (#6203; Boston, MA, USA) and Santa Cruz Biotechnology (sc‐29223; Dallas, TX, USA). Control non‐targeting siRNA was purchased from Thermo Fisher Scientific (Waltham, MA, USA). NB cells were transfected with siRNA by forward‐transfection according to the manufacturer's protocol using Lipofectamine RNAiMAX reagent (Invitrogen). We used siRNA (concentration 50 nM) for siNLRR2 and 100 nM for sic‐Jun because these concentrations worked well in a preliminary study (Fig. S1).
In vivo tumorigenicity assays
SK‐N‐BE cells at a density of 1 × 107 were inoculated s.c. into 7‐week‐old female SCID mice. One week after inoculation, when the tumors had an average volume of 70 ± 30 mm3, a mixture of 1 nmol of control or a mixture of two sets of NLRR2 siRNA and 200 μL of atelocollagen (Koken, Tokyo, Japan) was injected to the site of the tumor to evaluate the growth inhibition effect. Animal experiments were performed in compliance with the regulations for animal experiments of IACUC (IACUC approved # 15‐4).
Statistical analysis
Results were shown as the mean ± SD. Student's t‐test was used to compare the differences in means between two groups. The difference between groups was evaluated by repeated measures ANOVA followed by Bonferroni post‐test. P < 0.05 was considered statistically significant.
More detailed descriptions of the material and methods are described in Appendix S1.
Results
Expression of NLRR2 is associated with the poor prognosis of neuroblastoma and enhances oncogenic transformation in vitro and in vivo
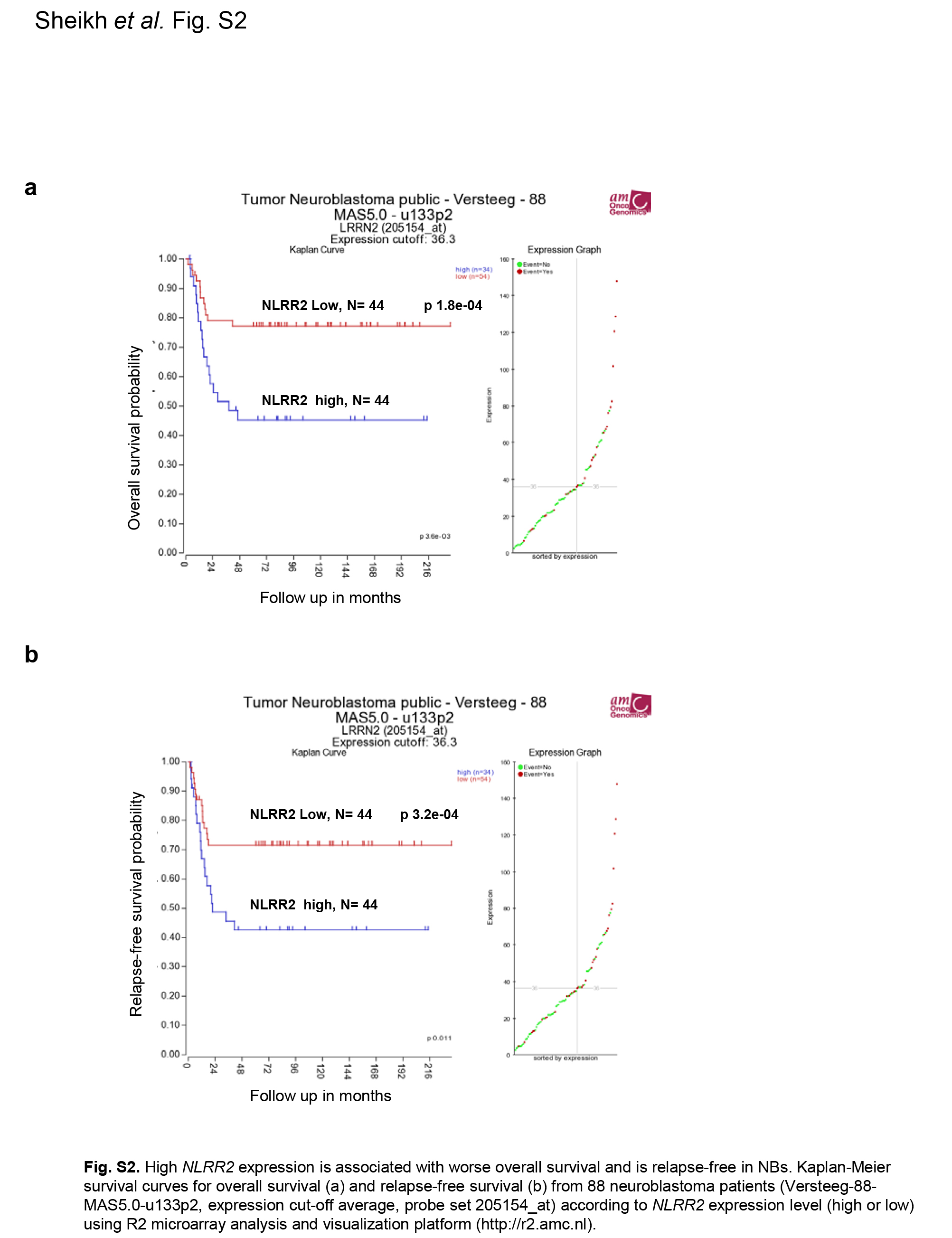
Previously, we identified NLRR family genes which are differentially expressed between favorable and unfavorable NB from the screening of nearly 2000 genes using our unique NB cDNA libraries. Human NLRR2 is a highly expressed gene in NB‐derived cell lines26 and may be used to define clinical relevancies of NB. We performed gene expression analysis by real‐time PCR using NB cDNA from 78 patients. Kaplan–Meier survival curves indicated that NB with high NLRR2 expression significantly associated with a poor clinical prognosis (P < 0.001) (Fig. 1a). A Kaplan–Meier survival analysis using a public R2 microarray dataset from 88 NB patients (http://r2.amc.nl) also revealed that high expression of NLRR2 was associated with worse overall survival and relapse‐free NB outcome (Fig. S2) To investigate the oncogenic effect of NLRR2 in NB, we stably or transiently expressed NLRR2 in SK‐N‐BE cells that resulted in significant increase of proliferation compared with the mock control cells (Fig. 1b,c). Overexpression of NLRR2 also resulted in a significant increase of cell proliferation in SMS‐SAN cells (Fig. 1d). In addition, the downregulation of NLRR2 by siRNA‐mediated knockdown significantly (P < 0.01) reduced the cell growth in SK‐N‐BE and SMS‐SAN cells (Fig. 1e,f). To confirm the function of NLRR2 in vivo, we locally treated mice bearing tumors derived from SK‐N‐BE cells with atelocollagen complexed with siNLRR2 (Fig. 1g). Compared with the control siRNA group, siNLRR2 treatment significantly reduced the tumor growth of SK‐N‐BE xenograft tumors (Fig. 1h,i and j). These data suggest that the function of NLRR2 is involved in tumorigenesis and is associated with a poor prognosis of NB.
Figure 1.
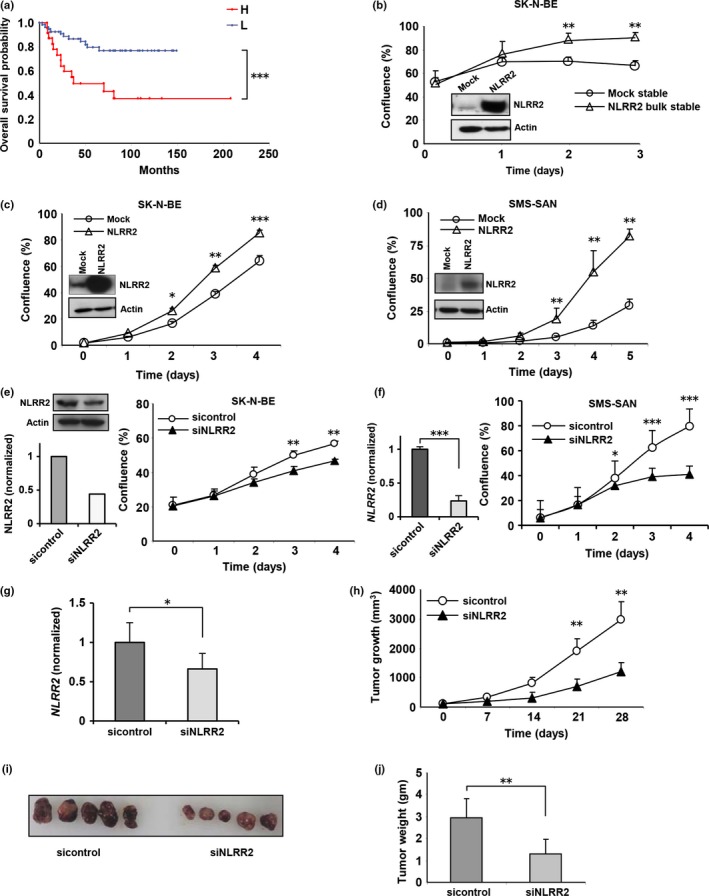
NLRR2 is correlated with poor survival outcome in human neuroblastoma (NB) and regulates tumor cell growth in vitro and in vivo. (a) Kaplan–Meier survival curves. The average of the expression level was used as a threshold to divide the tumors with low (n = 55, blue line) from high (n = 23, red line) expression. High NLRR2 mRNA expression was significantly correlated with poor survival outcome (***P < 0.001). (b) Mock and NLRR2 bulk stable cells were generated in SK‐N‐BE cells and NLRR2 expressions were confirmed by western blotting (left panel). Stable expression of NLRR2 exhibited significant (**P < 0.01) enhancement of cell growth (right panel). (c, d) NLRR2 was transiently overexpressed in SK‐N‐BE and SMS‐SAN cells and confirmed by western blotting. Overexpression of NLRR2 significantly (*P < 0.05) enhanced cell growth in SK‐N‐BE (c) and SMS‐SAN (d) cells. (e, f) In vitro knockdown of NLRR2 reduced cell growth in SK‐N‐BE (e) and SMS‐SAN (f) cells. (g, h) SCID mice were subcutaneously inoculated with 1 × 107 SK‐N‐BE cells. Two weeks after inoculation, when the tumors had an average volume of 70 ± 30 mm3, 1 nmol of control siRNA or a mixture of two siNLRR2 siRNA with 200 μL of atelocollagen was injected to the tumor mass to evaluate the growth inhibition effect. In vivo knockdown efficiency was measured by quantitative PCR (g). siNLRR2 treatment significantly (**P < 0.01) reduced the tumor growth compared with the control siRNA‐treated group (h). (i, j) Four weeks after treatment, the tumors were removed (i). The tumor weight was significantly (**P < 0.01) reduced in the siNLRR2‐treated group compared with the control siRNA group (j).
NLRR2 inhibits differentiation in neuroblastoma cells
We investigated whether or not NLRR2 has a function in cell growth and differentiation processes of NB. The transfection of siNLRR2 had no effect on the expression levels of other family genes, NLRR1 and NLRR3 (Fig. 2a). After transfection of siNLRR2, the growth of SK‐N‐BE cells was significantly repressed by RA treatment (Fig. 2b). Interestingly, the differentiation data revealed that NLRR2‐knockdown cells were more susceptible to RA‐mediated differentiation (Fig. 2c,d). The enhanced cell differentiation by NLRR2 knockdown was confirmed by higher levels of GAP43 and NF‐M,31, 32, 33 neuronal differentiation markers (Fig. 2e). These data suggest that NLRR2 knockdown retards cell growth and enhances cell differentiation induced by RA treatment.
Figure 2.
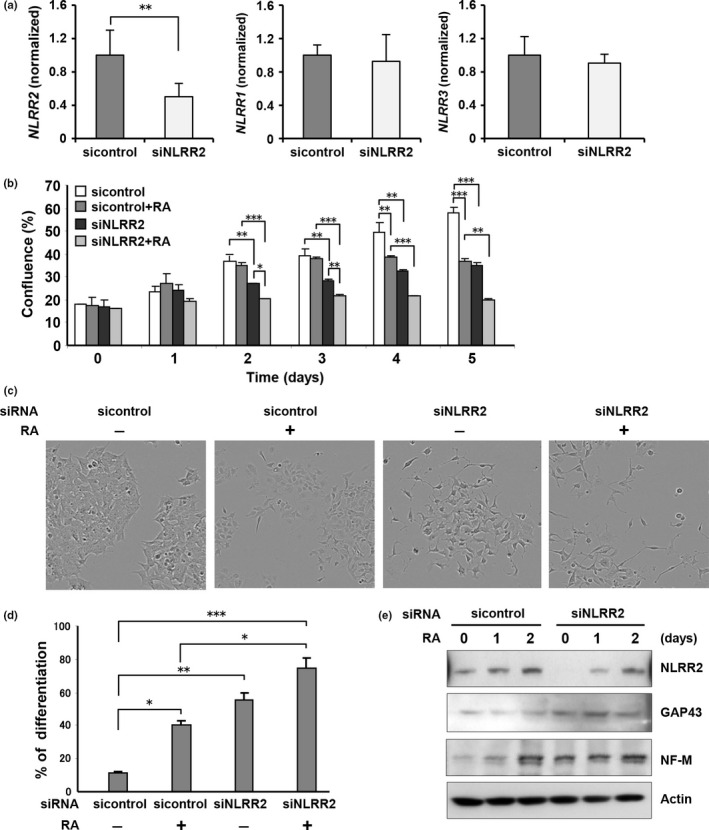
NLRR2 knockdown cells are susceptible to retinoic acid (RA)‐mediated inhibition of cell survival and differentiation. (a, b) SK‐N‐BE cells were transiently transfected with control siRNA or siNLRR2 and cell growth was measured using a real‐time cell imaging system. Knockdown of NLRR2 expression, as well as the effect of siNLRR2 on other NLRR family members were confirmed by quantitative PCR (a). The inhibition of cell growth by RA treatment was significantly increased in NLRR2 knockdown cells (P < 0.01). (c, d) SK‐N‐BE cells were transiently transfected with control siRNA or siNLRR2 followed by RA (2.5 μM) treatment for 3 days. The percentage of differentiated cells per field was counted (5 fields per well, 5 wells in each group). Data are presented as the mean ± SD. (e) SK‐N‐BE cells were transiently transfected with control siRNA or siNLRR2 followed by RA treatment for the indicated time period. GAP43, and neurofilament medium (NF‐M), markers for the neuronal differentiation, and NLRR2 expressions were examined by western blot analysis. NLRR2 knockdown cells with RA treatment showed a higher expression of GAP43 and NF‐M.
c‐Jun is important for the regulation of NLRR2 expression
We next examined NLRR2 expression during RA‐induced differentiation (Fig. S3) of NB cells. Interestingly, mRNA and protein levels of NLRR2 expression were elevated in RA‐treated NB cells (Fig. 3a,b). Consistent with previous reports showing the RA‐mediated induction of c‐Jun,18, 19, 34 c‐Jun expression in NB cells was also increased upon RA treatment, whereas expressions of other AP‐1 members, such as c‐fos and ATF‐2, were reduced (Fig. S4). To investigate whether c‐Jun was important for the regulation of NLRR2 expression, NB cells were transiently transfected with c‐Jun siRNA, resulting in reduced expressions of NLRR2 mRNA (Figs 3c,d and S1) and protein (Fig. 3e). Because RA treatment failed to rescue the NLRR2 expression in c‐Jun knockdown cells, these data demonstrated that c‐Jun functions by regulating RA‐induced NLRR2 expression.
Figure 3.

c‐Jun is important for regulating NLRR2 expression induced by retinoic acid (RA). (a, b) NLRR2 and c‐Jun were upregulated during RA‐mediated differentiation at the mRNA (a) and protein (b) levels in SK‐N‐BE, TGW and SMS‐SAN cells. Neuroblastoma (NB) cells were treated with RA (1.0 μM for TGW cells, 2.5 μM for SK‐N‐BE and SMS‐SAN cells) for the indicated time periods. NLRR2 and c‐Jun expressions were determined by RT‐PCR and western blot analyses. (c–e) SK‐N‐BE cells were transfected with control or c‐Jun siRNA (c and e, Santa Cruz; d, Cell Signaling Technology). Forty‐eight hours after transfection, expressions of c‐Jun and NLRR2 were measured by quantitative PCR (c, d). The data were shown as the mean ± SD. c‐Jun knockdown cells showed a reduced expression of NLRR2 upon RA treatment (e).
Retinoic acid enhances the recruitment of c‐Jun to the NLRR2 promoter
To clarify the transcriptional regulation of the NLRR2 gene, we generated luciferase reporter constructs containing −790 bp to +110 bp fragments of the NLRR2 gene where +1 represents the transcriptional initiation site. We performed a promoter assay in HeLa cells because they have high transfection efficiency. RA‐treatment also induced NLRR2 and c‐Jun expression in HeLa cells (Fig. S5). The promoter activity determined by luciferase reporter analysis was increased in response to RA treatment (Fig. 4a). The study using a series of deletion mutants of the promoter constructs showed that the luciferase activity was highest in between the −790 and −560 region, which contains a c‐Jun binding site of TPA‐responsive element (TRE), TGACAAA. We confirmed similar luciferase activity data in SK‐N‐BE cells (Fig. 4b) using a −790 to +110 promoter construct including an AP‐1 binding site. Next, we performed a ChIP assay to determine whether c‐Jun directly binds to the promoter region of NLRR2. As shown in Figure 4c, the recruitment of c‐Jun to the NLRR2 promoter was increased by RA treatment, indicating that NLRR2 is transcriptionally regulated by c‐Jun.
Figure 4.
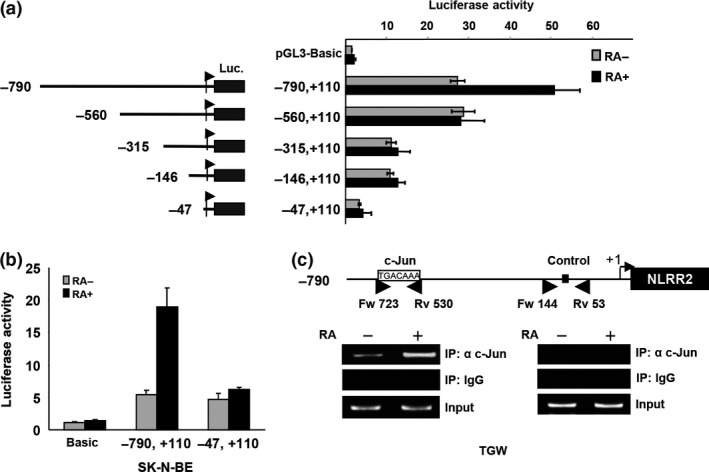
NLRR2 transcription is enhanced by retinoic acid (RA) through the recruitment of c‐Jun onto the promoter of NLRR2 while JNK inhibition suppresses the promoter activity. (a) Luciferase reporter constructs −790, +110; −560, +110; −315, +110; −147, +110 and −47, +110 (left panel) with renilla luciferase vector were introduced to HeLa cells and the promoter activity was measured by dual luciferase assay. RA treatment enhanced the NLRR2 promoter activity (−790, +110) (right panel). (b) RA treatment enhanced NLRR2 promoter activity in SK‐N‐BE cells. SK‐N‐BE cells were transfected with NLRR2 core luciferase reporter constructs (−790, +110) with renilla luciferase vector followed by RA treatment for 36 h and promoter activity was measured. (c) RA‐mediated c‐Jun recruitment to the NLRR2 promoter region containing AP‐1 consensus sequence was confirmed by ChIP assay.
NLRR2 expression is regulated by the JNK pathway
NLRR2 promoter activity was suppressed in SK‐N‐BE cells following treatment with a JNK inhibitor (Fig. 5a). To confirm that NLRR2 is regulated through the JNK pathway, we treated TGW and SK‐N‐BE cells with a JNK inhibitor to examine the expression of NLRR2. As expected, JNK inhibition reduced the expression of NLRR2 and c‐Jun at both the mRNA and protein levels (Fig. 5b,c), although NLRR2 induction upon RA treatment was marginally reduced by JNK inhibition (Fig. S6). To confirm the function of the JNK pathway in the regulation of NLRR2 expression, we used sorbitol, which induces JNK activation.35, 36 The phosphorylation of JNK and c‐Jun as well as the expression of NLRR2 were induced by sorbitol treatment in SK‐N‐BE cells and co‐treatment with a JNK inhibitor ameliorated the induction of NLRR2 expression (Fig. 5d,e). Thus, the JNK pathway is particularly important for regulating NLRR2 expression in NB cells.
Figure 5.
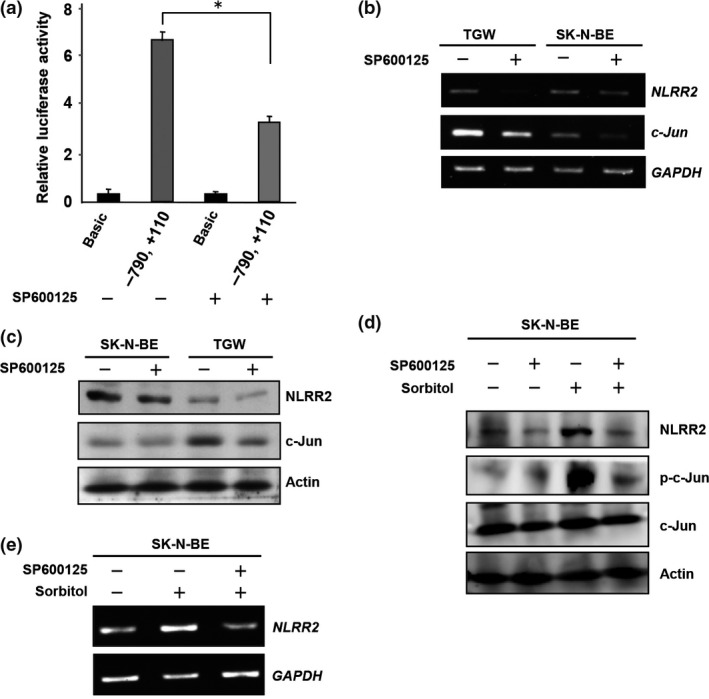
The JNK pathway is important for regulating NLRR2 expression. (a) JNK inhibition reduced NLRR2 promoter activity in SK‐N‐BE cells. SK‐N‐BE cells were transfected with the (−790, +110) promoter construct and a Renilla luciferase vector. At 24 h after the transfection, cells were treated with SP600125 (50 μM) for 24 h and luciferase assay was performed. (b, c) TGW and SK‐N‐BE cells were treated with SP600125 for 24 h and the expression of c‐Jun and NLRR2 were analyzed by western blotting and RT‐PCR analyses. JNK inhibition by SP600125 suppressed the expression of NLRR2 and c‐Jun at mRNA (b) and protein levels (c) in TGW and SK‐N‐BE cells. (d, e) JNK activation by sorbitol treatment (300 μM) in SK‐N‐BE cells induced NLRR2 expression, which was blocked by SP600125 pretreatment. Total JNK, p‐JNK, total c‐Jun and p‐c‐Jun were examined by western blot analysis (d) and NLRR2 mRNA expression was determined by RT‐PCR (e).
NLRR2 exhibits survival to retinoic acid and other stress‐mediating agents in neuroblastoma cells
Because the JNK pathway was induced by not only RA but the other agents such as sorbitol, we next treated NB cells with a DNA‐damaging agent that causes cellular stress. The treatment of cisplatin (CDDP) showed a clear induction of NLRR2 expression in SMS‐SAN cells and the smaller induction was observed in SK‐N‐BE cells (Figs 6a and S7). To examine whether NLRR2 expression was associated with cell survival against cellular stress, NLRR2‐stably expressing cells were treated with RA and cell viability was examined. As shown in Figure 6b, NLRR2‐stable cells were significantly resistant to RA treatment compared with the mock stable cell control. In contrast, NLRR2 knockdown cells showed significant susceptibility to CDDP treatment (Fig. S8). These data suggest that the cellular stress responses and NLRR2 induction vary more or less depending on the types of agents and cell lines. Next, we checked the morphology of the NLRR2 stable and mock cells upon RA treatment to study the differentiation function of NLRR2. The data showed that the stable expression of NLRR2 significantly (P < 0.01) inhibited RA‐induced differentiation compared with controls (Fig. S9). Therefore, after the induction of NLRR2 by cellular stress‐mediating agents, NLRR2, in turn, reduced cytotoxicity of NB cells.
Figure 6.
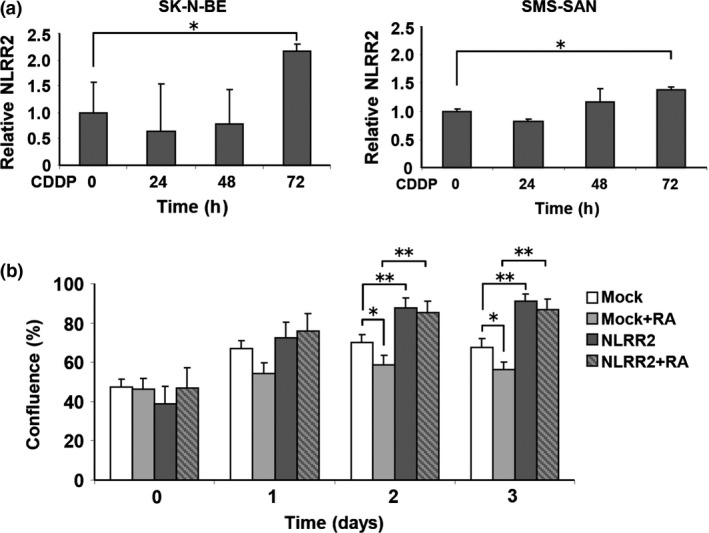
NLRR2 expression is induced by CDDP and retinoic acid (RA) and contributes to RA resistance in neuroblastoma cells. (a) SK‐N‐BE and SMS‐SAN cells were treated with CDDP (10 μM) for the indicated time periods. The expressions of NLRR2 mRNA were measured by quantitative PCR. NLRR2 expression was induced by CDDP. (b) SK‐N‐BE mock and NLRR2 bulk stable cells were treated with RA and cell survival was analyzed using a real‐time cell imaging system. NLRR2 bulk stable cells showed significant resistance to RA‐mediated cell growth inhibition.
JNK inhibition in neuroblastoma cells overexpressing NLRR2 rescues resistance to retinoic acid and induces differentiation
To further confirm the functional regulation of NLRR2 through JNK pathway, RA treatment was given to NLRR2 overexpressing cells with or without prior JNK inhibitor treatment. Data demonstrated that JNK inhibition in NLRR2 overexpressing cells rescued the resistance phenotype (Fig. 7a) and also induced significant differentiation to RA treatment (Fig. 7b,c). The expression of NLRR2, phospho‐c‐Jun and c‐Jun were suppressed by JNK inhibition (Fig. 7d) in NLRR2 overexpressing NB cells.
Figure 7.
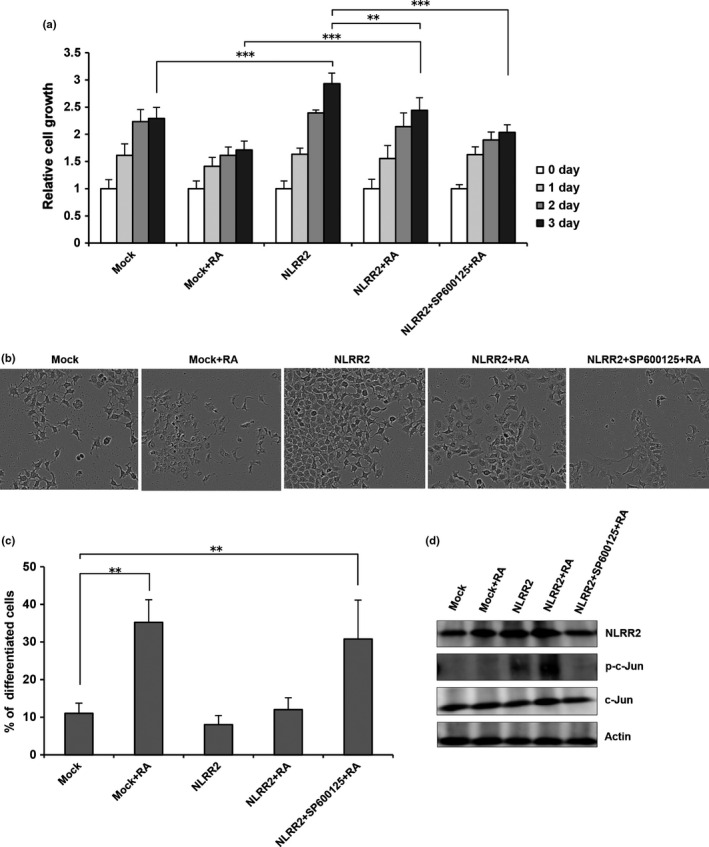
JNK inhibition in the cells overexpressing NLRR2 rescued the resistance phenotype to retinoic acid (RA) treatment and induced cell differentiation. SK‐N‐BE cells were enforced to express NLRR2 and treated with or without SP600125 followed by RA treatment. Cell growth (a), differentiation phenotype (b) and percent (%) of differentiated cells (c) were analyzed. The levels of p‐c‐Jun, NLRR2 and c‐Jun were determined by western blot analysis (d).
Discussion
Our results clearly demonstrate the clinical relevance of NLRR2 expression and its functional role in RA‐induced cell differentiation. NLRR2 enhances the cell survival of RA‐treated NB cells, while other family members, NLRR1 and NLRR3, are associated with cell proliferation and differentiation.28, 29 In addition, mouse NLRR3 was reported to modulate MAPK signaling through the EGFR pathway,35 indicating that each NLRR family protein might have functional relevance in cell signaling events. The present study suggests that the high expression of NLRR2 is significantly associated with the poor prognosis of NB, consistent with data analyzed using an R2 platform suggesting that NLRR2 serves as a prognostic marker of NB. Of note, a similar observation was reported for malignant gliomas.30 The local treatment of siNLRR2 in a mouse xenograft model of NB significantly reduced tumor growth, suggesting that the low expression of NLRR2 solely restricts tumor progression. Further study may clarify the clinical significance of NLRR2 expression in tumorigenesis.
Our in vitro experiments that modulated the expression of NLRR2 in NB cells indicated the potential molecular mechanisms of how NLRR2 might be involved in the poor clinical outcome of NB (Fig. 8). NLRR2‐overexpressing cells showed the enhanced cell proliferation and significant resistance phenotype to RA treatment. In contrast, NLRR2 knockdown cells were more susceptible to RA‐mediated cell growth inhibition and exhibited significantly increased numbers of differentiated cells compared with control cells. Even a 50% reduction of NLRR2 expression by siNLRR2 was enough to show these significant results. These data suggest that the function of NLRR2 is to inhibit the differentiation process and support the cell survival of RA‐treated cells.
Figure 8.
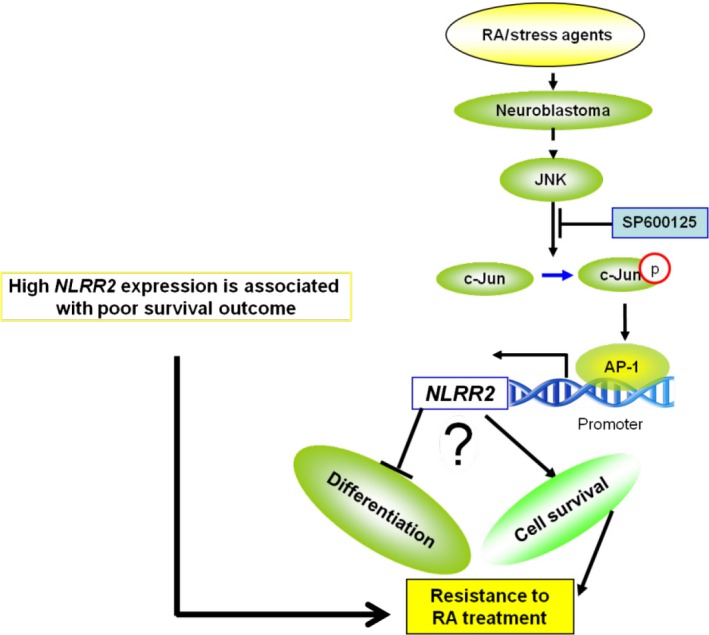
Schematic diagram of how NLRR2 contributes to poor prognosis of neuroblastoma (NB). Retinoic acid (RA)‐mediated JNK activation in NB cells induces NLRR2 expression that, in turn, ameliorates the differentiation and enhances cell survival.
Retinoic acid is a potent differentiation inducer of NB cells and causes a marked suppression of MYCN expression.29, 36, 37 However, our previous observation has demonstrated that NLRR2 expression is not correlated with MYCN expression (data not shown), while NLRR1 positively regulates cellular proliferation through MYCN induction and NLRR3 is negatively regulated by MYCN to facilitate differentiation of NB cells.28, 29 In the present study, we found that c‐Jun plays a key role in regulating NLRR2 gene expression in RA‐treated NB cells. RA‐treated NB cells exhibited the co‐induction of NLRR2 and c‐Jun. The activity of JNK appears to be important for the induction of NLRR2, which is consistent with previous reports regarding the induction and activation of c‐Jun downstream of the JNK pathway after RA treatment.19, 20 It is noteworthy that a DNA damaging agent, CDDP, also induced NLRR2 in NB cells. The significant survival difference observed in the clinical subsets with low and high expressions of NLRR2 may be explained by the stress‐induced upregulation of NLRR2, which, in turn, contributes to the drug resistance of NB cells. An anti‐apoptotic role of activated c‐Jun/AP‐1 transcription factor was reported to protect human tumor cells from DNA‐damaging agents.38 Thus, the effects observed in this study suggest that activated AP‐1 complexes exert a favorable influence on cell survival that counters diverse external stresses induced by drugs.
Although the JNK and p38MAPK pathways can potentially synergize to induce AP‐1 transcriptional activity and share c‐Jun as a common substrate,39 we found a limited effect of p38MAPK inhibition on RA‐induced differentiation as well as NLRR2 expression in NB cells (data not shown). There is considerable evidence that neurite outgrowth stimulated by RA is partially dependent on JNK activity.18 c‐Jun/AP‐1 was reported to be involved in neuronal differentiation and protection from apoptosis in NB cells.40 However, the relevant functions and target genes of AP‐1 in neuronal differentiation have not been elucidated yet. In particular, it is unclear whether AP‐1 in NB cells facilitates cell death or, conversely, is part of a protective response against differentiation or apoptosis. Our experimental evidence suggests that c‐Jun/AP‐1 in RA‐treated NB cells plays a protective role by upregulating the expression of NLRR2. Future studies might explain why the prognosis of NB treated with RA remains poor, with a high rate of resistance and metastasis.16, 17
The present study indicates the clinical relevance of NLRR2 and a regulatory mechanism of NLRR2 transcription in RA‐treated NB cells. The expression of NLRR2 is transcriptionally regulated by the JNK‐c‐Jun axis and enhances cell survival in drug‐treated NB cells. Therefore, NLRR2 might be a key target molecule for the development of new therapeutic strategies to overcome RA resistance in aggressive NLRR2‐expressing NB.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting information
Appendix S1. Supplementary materials and methods.
Fig. S1. siNLRR2 (20 or 50 nM) and sic‐Jun (50‒200 nM) reduced the expression of NLRR2 and c‐Jun in SK‐N‐BE cells, respectively.
{kind=link}
Fig. S2. High NLRR2 expression is associated with worse survival in neuroblastoma (NB).
{kind=link}
Fig. S3. Retinoic acid (RA) induced TGW cell differentiation in a dose dependent manner.
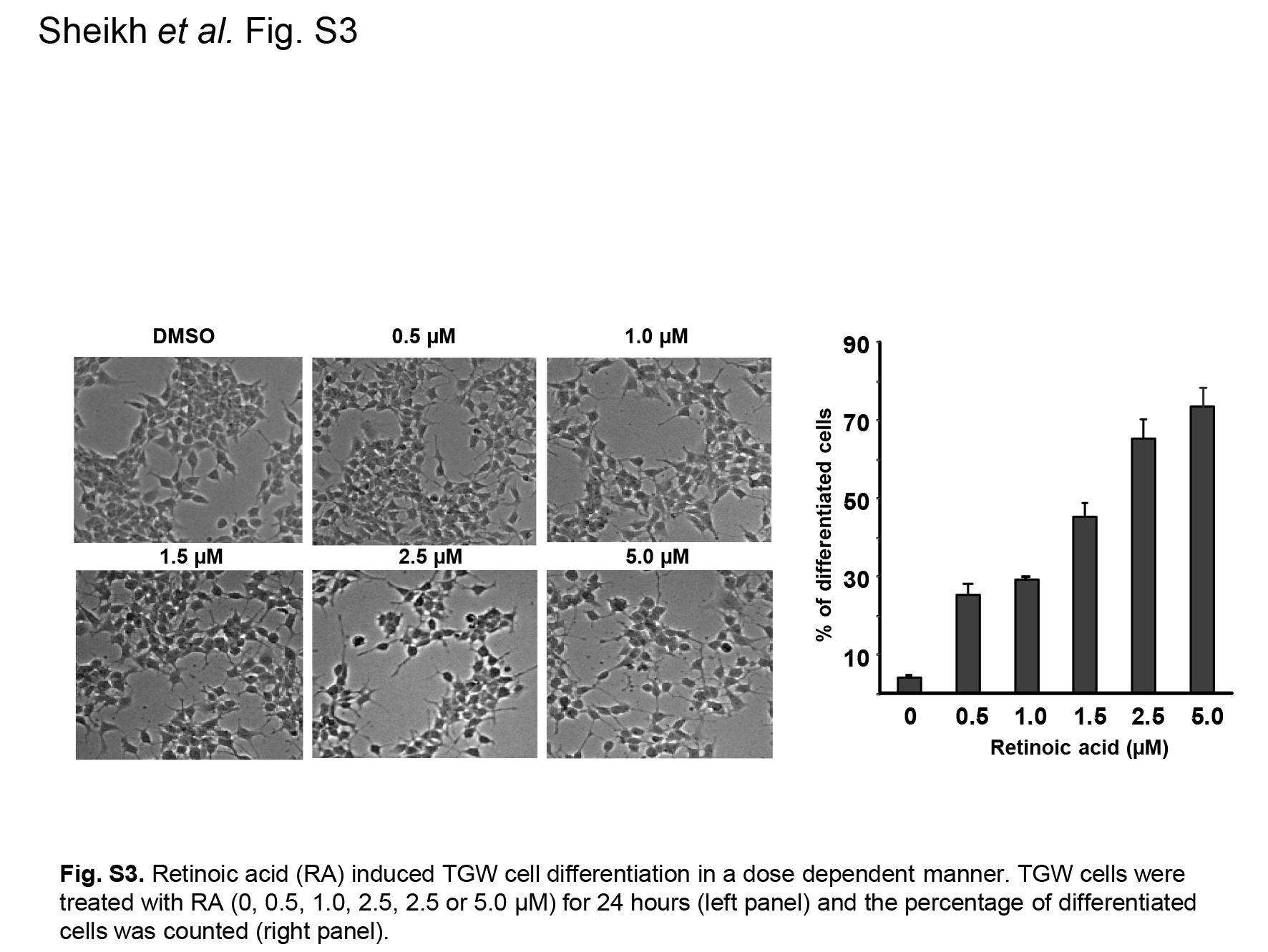{kind=link}
Fig. S4. c‐Fos and ATF‐2 expression was suppressed by retinoic acid (RA) in neuroblastoma (NB) cells.
{kind=link}
Fig. S5. Retinoic acid (RA) induced NLRR2 (a) and c‐Jun (b) expression in HeLa cells.
{kind=link}
Fig. S6. Retinoic acid (RA)‐treatment induced NLRR2 that was marginally reduced by JNK inhibition.
{kind=link}
Fig. S7. Cisplatin (CDDP) treatment induced NLRR2 expression in neuroblastoma (NB) cells.
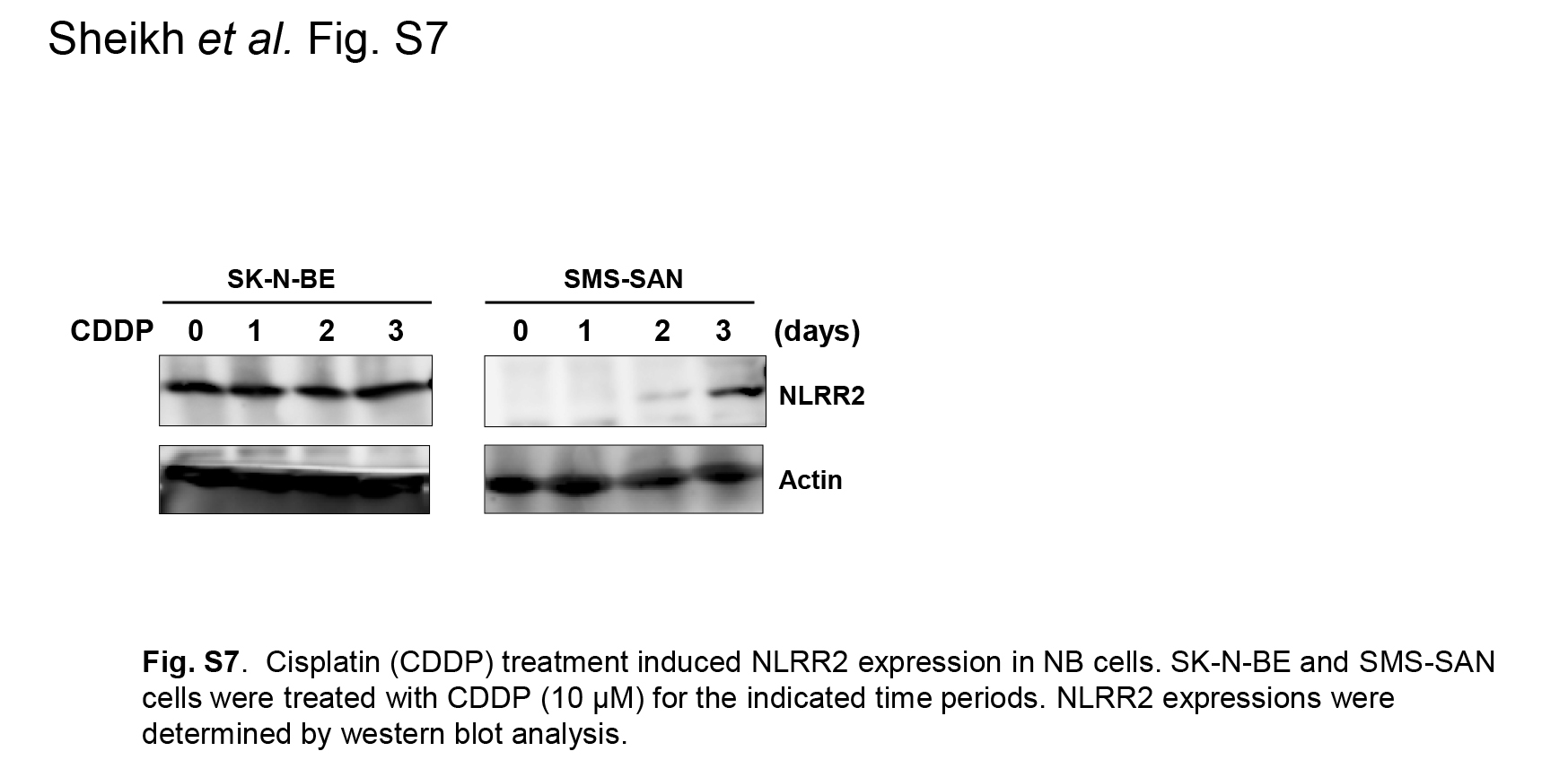{kind=link}
Fig. S8. NLRR2 knockdown cells are susceptible to cisplatin (CDDP) treatment.
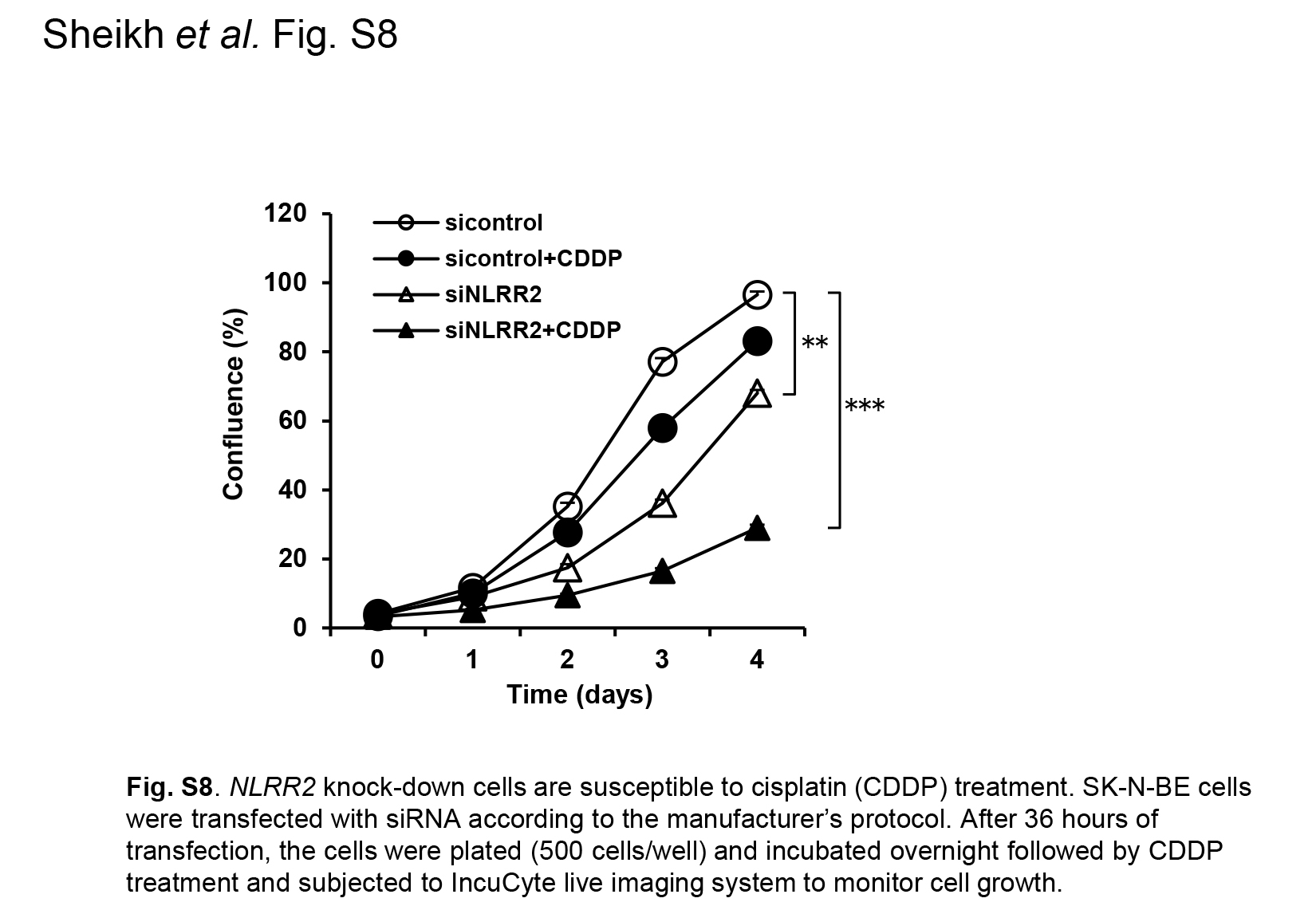{kind=link}
Fig. S9. Exogenous expression of NLRR2 inhibited retinoic acid (RA)‐induced differentiation of neuroblastoma (NB) cells.
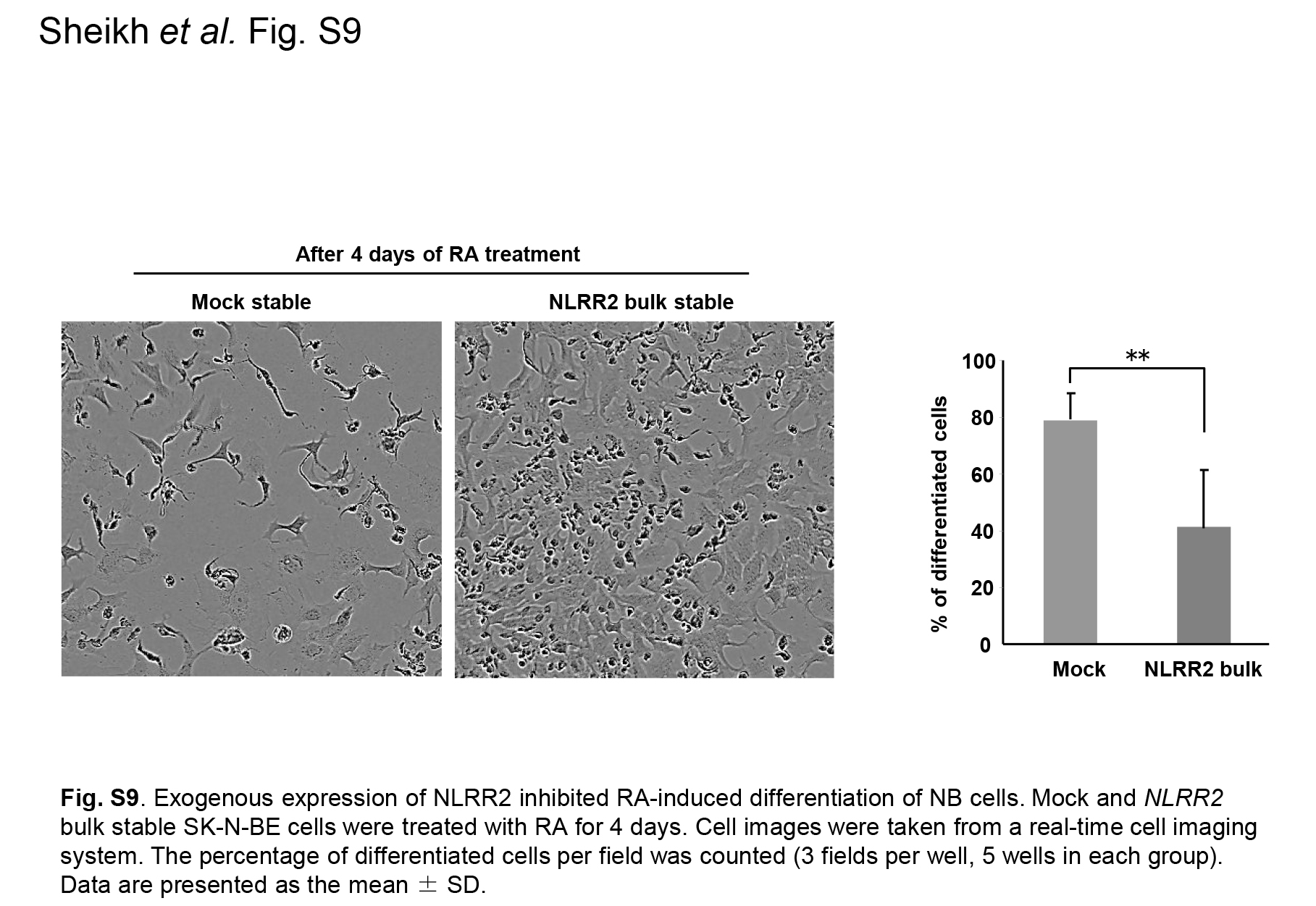{kind=link}
Acknowledgments
This work was supported by JSPS KAKENHI Grant Number 19890276 to A.T., and Ministry of Education, Culture, Sports, Science and Technology KAKENHI Grant Number 22791016 and 25830092 to A.T. The authors would like to thank, Drs Y. Nakamura and N. Koshikawa for their excellent advice and to Drs T. Ozaki, M. Ohira, Y. Suenaga, J. Akter, and R. Islam (Chiba Cancer Center Research Institute) for their fruitful scientific discussions.
Cancer Sci 107 (2016) 1223–1232
Funding Information This work was supported by JSPS KAKENHI Grant Number 19890276 to A.T., and MEXT KAKENHI Grant Numbers 22791016 and 25830092 to A.T.
References
- 1. Chang HH, Lee H, Hu MK et al Notch1 expression predicts an unfavorable prognosis and serves as a therapeutic target of patients with neuroblastoma. Clin Cancer Res 2010; 16: 4411–20. [DOI] [PubMed] [Google Scholar]
- 2. Brodeur GM, Nakagawara A. Molecular basis of clinical heterogeneity in Neuroblastoma. Am J Pediatr Hematol Oncol 1992; 14: 111–6. [DOI] [PubMed] [Google Scholar]
- 3. John M, Maris MD. Recent advances in neuroblastoma. N Engl J Med 2010; 362: 2202–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nakagawara A, Arima M, Azar CG, Scavarda NJ, Brodeur GM. Inverse relationship between trk expression and N‐myc amplification in human neuroblastomas. Cancer Res 1992; 52: 1364–8. [PubMed] [Google Scholar]
- 5. Chen Y, Takita J, Choi YL et al Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008; 16: 971–4. [DOI] [PubMed] [Google Scholar]
- 6. Nakagawara A, Arima‐Nakagawara M, Scavarda NJ, Cantor AB, Brodeur GM. Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N Engl J Med 1993; 328: 847–54. [DOI] [PubMed] [Google Scholar]
- 7. Nakagawara A, Azar CG, Scavarda NJ, Brodeur GM. Expression and function of TRK‐B and BDNF in human neuroblastomas. Mol Cell Biol 1994; 14: 759–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nakagawara A, Arima‐Nakagawara M, Azar CG, Scavarda NJ, Brodeur GM. Clinical significance of expression of neurotrophic factors and their receptors in neuroblastoma. Prog Clin Biol Res 1994; 385: 155–61. [PubMed] [Google Scholar]
- 9. Duester G. Retinoic acid synthesis and signaling during early embryogenesis. Cell 2008; 134: 921–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miloso M, Villa D, Crimi M et al Retinoic acid‐induced neuritogenesis of human neuroblastoma SH‐SY5Y cells is ERK independent and PKC dependent. J Neurosci Res 2004; 75: 241–52. [DOI] [PubMed] [Google Scholar]
- 11. Sidell N, Altman A, Haussler MR, Seeger RC. Effects of retinoic acid (RA) on the growth and phenotypic expression of several human Neuroblastoma cell lines. Exp Cell Res 1983; 148: 21–30. [DOI] [PubMed] [Google Scholar]
- 12. Messi E, Florian MC, Caccia C, Zanisi M, Maggi R. Retinoic acid reduces human neuroblastoma cell migration and invasiveness: effects on DCX, LIS1, neurofilaments‐68 and vimentin expression. BMC Cancer 2008; 8: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McBurney MW, Jones‐Villeneuve EM, Edwards MK, Anderson PJ. Control of muscle and neuronal differentiation in a cultured embryonal carcinoma cell line. Nature 1982; 299: 165–7. [DOI] [PubMed] [Google Scholar]
- 14. Tang XH, Gudas LJ. Retinoids, retinoic acid receptors, and cancer. Annu Rev Pathol 2011; 6: 345–64. [DOI] [PubMed] [Google Scholar]
- 15. Huang S, Laoukili J, Epping MT et al ZNF423 is critically required for retinoic acid‐induced differentiation and is a marker of neuroblastoma outcome. Cancer Cell 2009; 15: 328–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tallman MS, Andersen JW, Schiffer CA et al All‐trans retinoic acid in acute promyelocytic leukemia: long‐term outcome and prognostic factor analysis from the North American Intergroup protocol. Blood 2002; 100: 4298–302. [DOI] [PubMed] [Google Scholar]
- 17. Bates SE, Mickley LA, Chen YN et al Expression of a drug resistance gene in human neuroblastoma cell lines: modulation by retinoic acid‐induced differentiation. Mol Cell Biol 1989; 9: 4337–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yu YM, Han PL, Lee JK. JNK pathway is required for retinoic acid‐induced neurite outgrowth of human neuroblastoma, SH‐SY5Y. NeuroReport 2003; 14: 941–5. [DOI] [PubMed] [Google Scholar]
- 19. Kanungo J, Potapova I, Malbon CC, Wang HY. MEKK4 mediates differentiation in response to retinoic acid via activation of c‐Jun N‐terminal kinase in rat embryonal carcinoma P19 cells. J Biol Chem 2000; 275: 24032–9. [DOI] [PubMed] [Google Scholar]
- 20. Leppä S, Saffrich R, Ansorge W, Bohmann D. Differential regulation of c‐Jun by ERK and JNK during PC12 cell differentiation. EMBO J 1998; 17: 4404–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karin M. The regulation of AP‐1 activity by mitogen‐activated protein kinases. J Biol Chem 1995; 270: 16483–6. [DOI] [PubMed] [Google Scholar]
- 22. Ameyar M, Wisniewska M, Weitzman JB. A role for AP‐1 in apoptosis: the case for and against. Biochimie 2003; 85: 747–52. [DOI] [PubMed] [Google Scholar]
- 23. Raivich G, Behrens A. Role of the AP‐1 transcription factor c‐Jun in developing, adult and injured brain. Prog Neurobiol 2006; 78: 347–63. [DOI] [PubMed] [Google Scholar]
- 24. Ohira M, Morohashi A, Inuzuka H et al Expression profiling and characterization of 4200 genes cloned from primary neuroblastomas: identification of 305 genes differentially expressed between favorable and unfavorable subsets. Oncogene 2003; 22: 5525–36. [DOI] [PubMed] [Google Scholar]
- 25. Haines BP, Gupta R, Jones CM et al The NLRR gene family and mouse development: modified differential display PCR identifies NLRR‐1 as a gene expressed in early somitic myoblasts. Dev Biol 2005; 281: 145–59. [DOI] [PubMed] [Google Scholar]
- 26. Hamano S, Ohira M, Isogai E, Nakada K, Nakagawara A. Identification of novel human neuronal leucine‐rich repeat (hNLRR) family genes and inverse association of expression of Nbla10449/hNLRR‐1 and Nbla10677/hNLRR‐3 with the prognosis of primary neuroblastomas. Int J Oncol 2004; 24: 1457–66. [PubMed] [Google Scholar]
- 27. Hossain MS, Ozaki T, Wang H et al N‐MYC promotes cell proliferation through a direct transactivation of neuronal leucine‐rich repeat protein‐1 (NLRR1) gene in Neuroblastoma. Oncogene 2008; 27: 6075–82. [DOI] [PubMed] [Google Scholar]
- 28. Hossain S, Takatori A, Nakamura Y, Suenaga Y, Kamijo T, Nakagawara A. NLRR1 enhances EGF‐mediated MYCN induction in neuroblastoma and accelerates tumor growth in vivo. Cancer Res 2012; 72: 4587–96. [DOI] [PubMed] [Google Scholar]
- 29. Akter J, Takatori A, Hossain MS et al NLRR3 Orphan receptor gene is negatively regulated by MYCN and Miz‐1, and its downregulation is associated with unfavorable outcome in neuroblastoma. Clin Cancer Res 2011; 17: 6681–92. [DOI] [PubMed] [Google Scholar]
- 30. Almeida A, Zhu XX, Vogt N et al GAC1, a new member of the leucine‐rich repeat superfamily on chromosome band 1q32.1, is amplified and overexpressed in malignant gliomas. Oncogene 1998; 16: 2997–3002. [DOI] [PubMed] [Google Scholar]
- 31. Zhao JC, Zhang LX, Zhang Y, Shen YF. The differential regulation of Gap43 gene in the neuronal differentiation of P19 cells. J Cell Physiol 2012; 227: 2645–53. [DOI] [PubMed] [Google Scholar]
- 32. Benowitz LI, Routtenberg A. GAP‐43: an intrinsic determinant of neuronal development and plasticity. Trends Neurosci 1997; 20: 84–91. [DOI] [PubMed] [Google Scholar]
- 33. Aarts LH, Schotman P, Verhaagen J, Schrama LH, Gispen WH. The role of the neural growth associated protein B‐50/GAP‐43 in morphogenesis. Adv Exp Med Biol 1998; 446: 85–106. [DOI] [PubMed] [Google Scholar]
- 34. Kitabayashi I, Kawakami Z, Chiu R et al Transcriptional regulation of the c‐jun gene by retinoic acid and E1A during differentiation of F9 cells. EMBO J 1992; 11: 167–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fukamachi K, Matsuoka Y, Ohno H, Hamaguchi T, Tsuda H. Neuronal leucine‐rich repeat protein‐3 amplifies MAPK activation by epidermal growth factor through a carboxyl‐terminal region containing endocytosis motifs. J Biol Chem 2002; 277: 43549–52. [DOI] [PubMed] [Google Scholar]
- 36. Thiele CJ, Reynolds CP, Israel MA. Decreased expression of N‐myc precedes retinoic acid‐induced morphological differentiation of human neuroblastoma. Nature 1985; 313: 404–6. [DOI] [PubMed] [Google Scholar]
- 37. Reynolds CP, Kane DJ, Einhorn PA et al Response of neuroblastoma to retinoic acid in vitro and in vivo. Prog Clin Biol Res 1991; 366: 203–11. [PubMed] [Google Scholar]
- 38. Potapova O, Basu S, Mercola D, Nikki J, Holbrook NJ. Protective role for c‐Jun in the cellular response to DNA damage. J Biol Chem 2001; 276: 28546–53. [DOI] [PubMed] [Google Scholar]
- 39. Wagner EF, Angel RN. Signal integration by JNK and p38MAPK pathways in cancer development. Nat Rev Cancer 2009; 9: 537–49. [DOI] [PubMed] [Google Scholar]
- 40. Feng Z, Li L, Ng PY, Porter AG. Neuronal differentiation and protection from nitric oxide‐induced apoptosis require c‐Jun‐dependent expression of NCAM140. Mol Cell Biol 2002; 22: 5357–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supplementary materials and methods.
Fig. S1. siNLRR2 (20 or 50 nM) and sic‐Jun (50‒200 nM) reduced the expression of NLRR2 and c‐Jun in SK‐N‐BE cells, respectively.
Fig. S2. High NLRR2 expression is associated with worse survival in neuroblastoma (NB).
Fig. S3. Retinoic acid (RA) induced TGW cell differentiation in a dose dependent manner.
Fig. S4. c‐Fos and ATF‐2 expression was suppressed by retinoic acid (RA) in neuroblastoma (NB) cells.
Fig. S5. Retinoic acid (RA) induced NLRR2 (a) and c‐Jun (b) expression in HeLa cells.
Fig. S6. Retinoic acid (RA)‐treatment induced NLRR2 that was marginally reduced by JNK inhibition.
Fig. S7. Cisplatin (CDDP) treatment induced NLRR2 expression in neuroblastoma (NB) cells.
Fig. S8. NLRR2 knockdown cells are susceptible to cisplatin (CDDP) treatment.
Fig. S9. Exogenous expression of NLRR2 inhibited retinoic acid (RA)‐induced differentiation of neuroblastoma (NB) cells.