

Abstract
Podoplanin (aggrus) is highly expressed in several types of cancers, including malignant pleural mesothelioma (MPM). Previously, we developed a rat anti‐human podoplanin mAb, NZ‐1, and a rat–human chimeric anti‐human podoplanin antibody, NZ‐8, derived from NZ‐1, which induced antibody‐dependent cellular cytotoxicity (ADCC) and complement‐dependent cytotoxicity against podoplanin‐positive MPM cell lines. In this study, we showed the antitumor effect of NZ‐1, NZ‐8, and NZ‐12, a novel rat–human chimeric anti‐human podoplanin antibody derived from NZ‐1, in an MPM orthotopic xenograft SCID mouse model. Treatment with NZ‐1 and rat NK (CD161a+) cells inhibited the growth of tumors and the production of pleural effusion in NCI‐H290/PDPN or NCI‐H226 orthotopic xenograft mouse models. NZ‐8 and human natural killer (NK) (CD56+) cells also inhibited tumor growth and pleural effusion in MPM orthotopic xenograft mice. Furthermore, NZ‐12 induced potent ADCC mediated by human MNC, compared with either NZ‐1 or NZ‐8. Antitumor effects were observed following treatment with NZ‐12 and human NK (CD56+) cells in MPM orthotopic xenograft mice. In addition, combined immunotherapy using the ADCC activity of NZ‐12 mediated by human NK (CD56+) cells with pemetrexed, led to enhanced antitumor effects in MPM orthotopic xenograft mice. These results strongly suggest that combination therapy with podoplanin‐targeting immunotherapy using both NZ‐12 and pemetrexed might provide an efficacious therapeutic strategy for the treatment of MPM.
Keywords: antibody‐dependent cellular cytotoxicity, mesothelioma, NZ‐12, orthotopic xenograft model, podoplanin
Expression of podoplanin (aggrus), a transmembrane sialomucin‐like glycoprotein, has been detected in various normal tissues, including kidney podocytes, endothelium of lymphatic vessels, and type I alveolar epithelium,1, 2, 3 as well as many types of cancers, including malignant brain tumor, oral cancers, esophageal cancers, squamous carcinoma, testicular seminomas, bladder cancers, fibrosarcomas, and malignant pleural mesothelioma (MPM).4, 5, 6, 7, 8, 9 Podoplanin, which binds to the platelet aggregation‐stimulating domain of PDPN–C‐type lectin‐like receptor 2 in platelets, induces platelet aggregation, resulting in cancer metastasis.4, 10 Furthermore, high expression of podoplanin in cancer‐associated fibroblasts is associated with severe malignancy and poor prognosis in cancer patients.11, 12, 13, 14 Therefore, it is expected that podoplanin will become a target for cancer diagnosis and therapies.
Malignant pleural mesothelioma, which is mainly caused by exposure to asbestos, develops in the pleural cavity with a high likelihood of malignancy.15 It is expected that the number of MPM patients will increase from 2030 to 2040 in Asia, and from 2010 to 2020 in Europe.16, 17 The standard therapy for MPM involves a combination of surgical operations, radiation therapy, and systemic chemotherapy. However, the prognosis of MPM is very poor as MPM is one of the most progressive cancers and frequently resists treatment.18, 19 Treatment with pemetrexed, which is the only validated chemotherapy drug for MPM, combined with cisplatin, is the standard chemotherapy used in MPM patients; however, this combination therapy often only prolongs progression‐free survival by approximately 2.8 months, compared with treatment without pemetrexed.20 Thus, development of novel therapies for MPM is warranted in order to improve the prognosis.
Immunotherapy using therapeutic antibodies against tumor‐associated antigens or antigenic peptides derived from pemetrexed is a novel therapy for the treatment of various cancers, including MPM.21, 22 Several therapeutic antibodies, including trastuzumab and rituximab, are already being used in clinical practice. Antibody‐dependent cellular cytotoxicity (ADCC) and complement‐dependent cytotoxicity (CDC) are critical mechanisms by which therapeutic antibodies provide their antitumor effects.23 Previously, we generated a rat anti‐human podoplanin mAb, NZ‐1,24, 25, 26 and a rat–human chimeric anti‐human podoplanin antibody, NZ‐8, derived from NZ‐1.9, 27 These anti‐podoplanin antibodies induce potent ADCC and CDC activity against podoplanin‐positive MPM cell lines in vitro, and had significant antitumor effects in an MPM s.c. transplantation SCID mouse model. However, whether the therapeutic effects of anti‐podoplanin antibodies in MPM orthotopic xenograft mice are similar to clinical presentation of MPM is still unknown. In the present study, we investigated whether anti‐human podoplanin antibodies NZ‐1, NZ‐8, and NZ‐12, a novel rat–human chimeric anti‐human podoplanin antibody derived from NZ‐1, can induce antitumor effects in an MPM orthotopic xenograft SCID mouse model. Furthermore, we evaluated the antitumor effects of combined treatment using anti‐human podoplanin antibody‐based immunotherapy, and pemetrexed.
Materials and Methods
Cell lines
In this study, we used four human MPM cell lines. NCI‐H290, ACC‐MESO‐1, and ACC‐MESO‐4 were provided by Dr. Yoshitaka Sekido (Division of Molecular Oncology, Aichi Cancer Center Research Institute, Nagoya, Japan).28 NCI‐H226 and Chinese hamster ovary (CHO) were purchased from ATCC (Rockville, MD, USA). NCI‐H290 cells were transfected with appropriate amounts of pcDNA3/human podoplanin or pcDNA3/mock plasmids, using Metafectene (Nippon Genetics, Tokyo, Japan), according to the manufacturer's instructions. Stable transfectants (NCI‐H290/PDPN and NCI‐H290/mock) were selected by culture in medium containing 0.5 mg/mL Geneticin (Invitrogen, Carlsbad, CA, USA). These cells were maintained in RPMI‐1640 medium supplemented with 10% FBS (CRPMI‐1640; Gibco, Grand Island, NY, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin (Meiji Seika Kaisha, Tokyo, Japan) in 5% CO2 at 37°C.
Antibodies and reagents
Rat anti‐human podoplanin mAb, NZ‐1, and rat–human chimeric anti‐human podoplanin antibody, NZ‐8, were developed as described previously.24, 27 For the generation of rat–human chimera anti‐human podoplanin (NZ‐12), the appropriate VH of a rat NZ‐1 antibody and CH of human IgG1 were subcloned into the pCAG‐Neo (Wako Pure Chemical Industries, Osaka, Japan), and VL of a rat NZ‐1 antibody and CL of human lambda chain were subcloned into pCAG‐Ble vectors (Wako Pure Chemical Industries). Antibody expression vectors were transfected into CHO cells using a Lipofectamine LTX kit (Life Technologies, Carlsbad, CA, USA). Stable transfectants of CHO/NZ‐12 were selected by cultivating the transfectants in medium containing 1 mg/mL Geneticin and 0.5 mg/mL Zeocin (Life Technologies). The CHO/NZ‐12 cells were cultivated in CHO‐S‐SFM II medium (Life Technologies). The media containing NZ‐12 were centrifuged, and the obtained supernatant was applied to a column of protein G‐Sepharose (Thermo Fisher Scientific, Rockford, IL, USA). After extensive washing with PBS, the fusion proteins were eluted using 0.1 M glycine and 0.15 M NaCl (pH 2.8), and then neutralized with 1 M Tris (pH 10.0). The antibodies were dialyzed against PBS. Expression and purity of the proteins were confirmed by SDS‐PAGE, using 5–20% gradient gels (Wako Pure Chemical Industries). Rat IgG (rIgG) was purchased from Southern Biotechnology (Birmingham, AL, USA). Human IgG (hIgG) was purchased from Cappel (Cochranville, PA, USA). Pemetrexed was obtained from Eli Lilly Japan (Kobe, Japan).
Animals
Five‐ to 6‐week‐old male SCID mice and 6‐week‐old male Wistar rats were obtained from CLEA Japan (Osaka, Japan) and maintained under specific pathogen‐free conditions throughout experiments. All animals were acclimatized for at least 1 week before experiments. All experiments were carried out in accordance with the guidelines of the Committee on Animal Care and Use of Tokushima University (Tokushima, Japan).
Flow cytometry
Expression of podoplanin was detected by flow cytometry, as described previously.9 Cells (5 × 105) were washed with PBS and stained with NZ‐1 (1 μg/mL) or rIgG (1 μg/mL). After 30 min of incubation, cells were washed with PBS and incubated for 30 min with FITC‐conjugated goat F(ab’)2 fragment anti‐rat IgG (H+L) antibody (Beckman Coulter, Fullerton, CA, USA). Cells were washed again and resuspended in PBS. A FACSCalibur flow cytometer with CellQuest software (BD Biosciences, Franklin Lakes, NJ, USA) was used for the analysis.
Preparation of effector cells
Effector cells were prepared as previously described.9, 29, 30 Rat splenocytes were harvested from Wistar rat spleens. Spleens were homogenized in RPMI‐1640 and centrifuged. To deplete red blood cells, the cell pellet was suspended in red blood cell lysis buffer (Sigma‐Aldrich, St. Louis, MO, USA). After washing and resuspension in CRPMI‐1640, splenocytes were used as effector cells. To separate rat natural killer (NK) cells from rat splenocytes, a magnetic cell‐sorting system was used. Splenocytes were incubated with FITC‐conjugated anti‐CD161a antibody (BD Biosciences), followed by anti‐FITC mAb‐coupled super‐paramagnetic microbeads (Miltenyi Biotec, Auburn, CA, USA). CD161a+ selection was carried out using an autoMACS (Miltenyi Biotec). Isolated CD161a+ cells yielding purity ≥90%, determined by flow cytometry, were used in experiments. Human peripheral blood mononuclear cells (MNCs) were obtained from leukocytes in lymphocyte separation medium (Litton Bionetics, Kensington, MD, USA). Leukocytes were separated from peripheral blood of healthy donors using a Kubota KR‐400 centrifuge with an RS‐6600 rotor (Kubota, Tokyo, Japan). CD56+ cells were purified from human MNCs, which were treated with CD56 microbeads (Miltenyi Biotec), and then cells were separated by autoMACS. The purity of CD56+ cells was ≥90%. The human study was approved by the ethics committee of University of Tokushima, and written informed consent was obtained from all donors.
Antibody‐dependent cellular cytotoxicity
Antibody‐dependent cellular cytotoxicity was determined using 51Cr release assays.9, 29, 31 Target cells were incubated with 51Cr‐sodium chromate (3.7 MBq) at 37°C for 1 h. After washing with CRPMI‐1640 three times, 51Cr‐labeled target cells were placed in triplicate in 96‐well plates. Effector cells and anti‐human podoplanin antibody or control IgG were added to the plates. After 6 h of incubation, 51Cr release of the supernatant from each well (100 μL) was measured using a gamma counter (PerkinElmer, Waltham, MA, USA). Percent of cytotoxicity was calculated using the following formula: % specific lysis = (E − S) / (M − S) × 100, where E is the release in the test sample, S is the spontaneous release, and M is the maximum release.
Complement‐dependent cytotoxicity
Complement‐dependent cytotoxicity was evaluated by 51Cr release assay, as described previously.9, 32 Target cells were incubated with 51Cr‐sodium chromate (3.7 MBq) for 1 h at 37°C. Following this, cells were washed in CRPMI‐1640. The 51Cr‐labeled cells were incubated with baby rabbit complement (dilution of 1:4) (Cedarlane, Burlington, VT, Canada) and NZ‐12 (1 μg/mL) or control hIgG (1 μg/mL) for 6 h in 96‐well plates. After incubation, the supernatant, including 51Cr, was measured using a gamma counter. Percent cytotoxicity was calculated as described above.
Animal experiments
SCID mice were injected into the thoracic cavity with NCI‐H290/PDPN (1.0 × 106 cells) or NCI‐H226 (1.0 × 106 cells) on day 0. Intrathoracic administration or i.p. injection of anti‐human podoplanin antibody or control IgG began on day 0, and continued twice a week for 2–3 weeks. Rat CD161a+ cells (1.0 × 106 cells), human CD56+ cells (1.0 × 106 cells), or control normal saline were injected into the thoracic cavity from day 3, and continued weekly for 2–3 weeks. SCID mice of the pemetrexed combination group were treated with pemetrexed (100 mg/kg, i.p.) on days 4, 5, 6, 11, 12, and 13, as described previously.33 Three weeks (NCI‐H290/PDPN) or 9 weeks (NCI‐H226) after tumor cell inoculation, the mice were killed, thoracic tumors were weighed, and the volume of pleural effusion was measured using a 1‐mL syringe.
Statistical analyses
The statistical significance of differences in in vitro and in vivo data was analyzed using standard Student's t‐test and one‐way anova. P‐values < 0.05 were considered significant in all experiments.
Results
Rat anti‐human podoplanin antibody NZ‐1 induces ADCC against podoplanin‐transfected MPM cell lines
First, we confirmed that NZ‐1 induces ADCC against NCI‐H290/PDPN in target cells. NCI‐H290, one of the podoplanin‐negative MPM cell lines, develops thoracic tumors and pleural effusion in an orthotropic xenograft SCID mouse model.34 We generated NCI‐H290/PDPN to transfect pcDNA3/podoplanin into NCI‐H290. As shown in Fig. 1(a), podoplanin expression in NCI‐H290/PDPN was determined by flow cytometry. Using NCI‐H290/PDPN as target cells, we could detect ADCC induced by NZ‐1 with rat splenocytes (Fig. 1b). Figure 1(c) shows that effector/target cell‐dependent and dose‐dependent ADCC activities against NCI‐H290/PDPN were also induced by NZ‐1. Moreover, NZ‐1 showed significant ADCC activity against NCI‐H290/PDPN and NCI‐H226 when rat NK cells were used as target cells (Fig. 1d).
Figure 1.
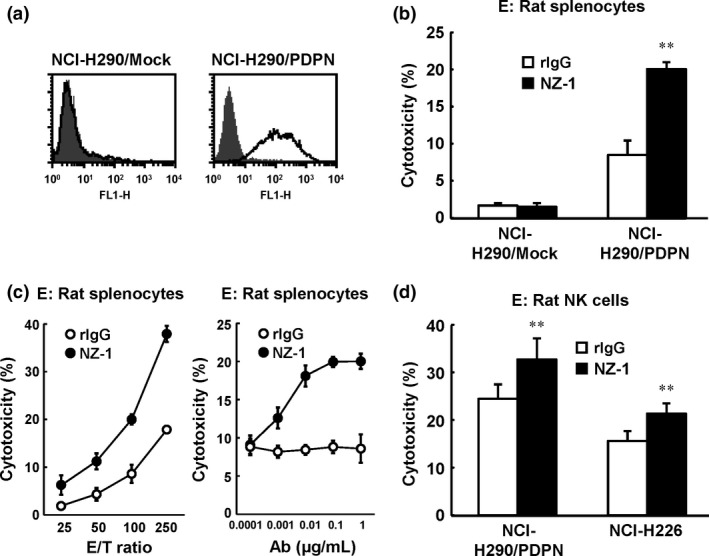
Antibody‐dependent cellular cytotoxicity (ADCC) activity of rat anti–human podoplanin antibody NZ‐1 against NCI‐H290/PDPN in vitro. (a) Expression of podoplanin was detected by FACS analysis. (b) ADCC activity of NZ‐1 using rat splenocytes against NCI‐H290/PDPN or NCI‐H290/Mock was evaluated by 6‐h 51Cr release assay in the presence of antibody (1 μg/mL; effector/target [E/T] ratio 100). (c) E/T ratio‐dependent and antibody dose‐dependent effects of ADCC against NCI‐H290/PDPN mediated by NZ‐1 with rat splenocytes are shown by 51Cr release assay. (d) Rat natural killer (NK) (CD161a+) cells were isolated from rat splenocytes. ADCC activity of NZ‐1 (1 μg/mL) mediated by NK (CD161a+) cells was evaluated by 6‐h 51Cr release assay at an E/T ratio of 10. **P < 0.01 versus control (values represent mean ± SE). rIgG, rat IgG.
Antitumor activity of NZ‐1 in MPM orthotopic xenograft model
Previously, we reported that injection of both NZ‐1 and rat NK cells inhibited the growth of podoplanin‐positive MPM cells inoculated s.c. in SCID mice.9 To determine whether NZ‐1 also induced an antitumor effect in an orthotopic xenograft model, we first evaluated the effects of local administration of NZ‐1 in NCI‐H290/PDPN orthotopic xenograft mice. For 2 weeks, NZ‐1 or control IgG was injected into the thoracic cavity twice a week, and rat NK (CD161a+) cells or PBS was injected into the thoracic cavity once a week. Three weeks after tumor cell inoculation, tumor growth and production of pleural effusion was almost entirely inhibited in both the NZ‐1 and rat NK cells treatment groups (Fig. 2). In contrast, treatment with NZ‐1 alone did not produce an antitumor effect in our model. Next, we investigated whether systemic treatment of NZ‐1 induced antitumor effects in the orthotopic xenograft SCID mouse model. For 3 weeks, NZ‐1 or rIgG was injected i.p. twice a week, and rat NK cells or PBS were injected once a week into the thoracic cavity. In both NCI‐H290/PDPN‐ and NCI‐H226‐inoculated mice, systemic administration of NZ‐1 with rat NK cells significantly inhibited growth of intrathoracic tumors and production of pleural effusion, compared with either NZ‐1 or rat NK cell treatment alone (Fig. 3).
Figure 2.
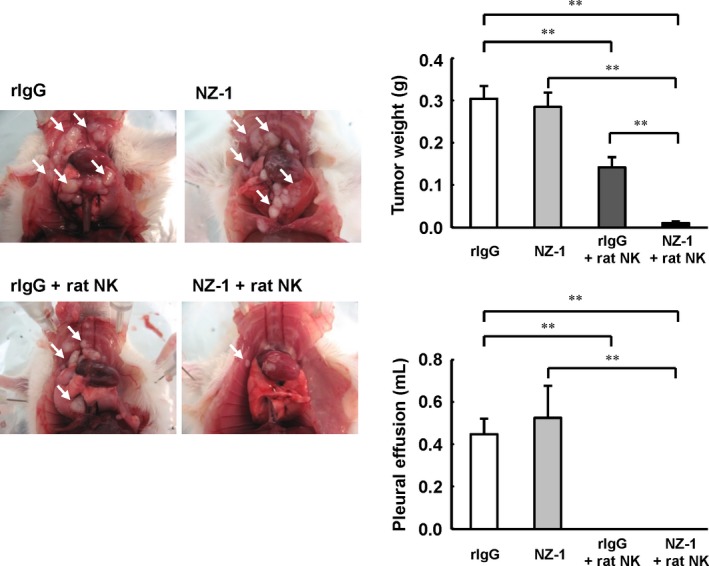
Antitumor effects of NZ‐1 injected into the thoracic cavity in an NCI‐H290/PDPN malignant pleural mesothelioma orthotopic xenograft model. SCID mice (n = 5) were injected into the thoracic cavity with 1.0 × 106 NCI‐H290/PDPN cells. NZ‐1 (100 μg) or rat IgG (rIgG; 100 μg) intrathoracic injection began on day 0, and continued twice a week for 2 weeks. Rat NK (CD161a+) cells (1.0 × 106 cells) or control normal saline intrathoracic injections continued weekly for 2 weeks. Three weeks after tumor cell inoculation, the mice were killed, and the weight of thoracic tumors (white arrows) and volume of pleural effusion were measured. **P < 0.01 (values represent mean ± SE).
Figure 3.
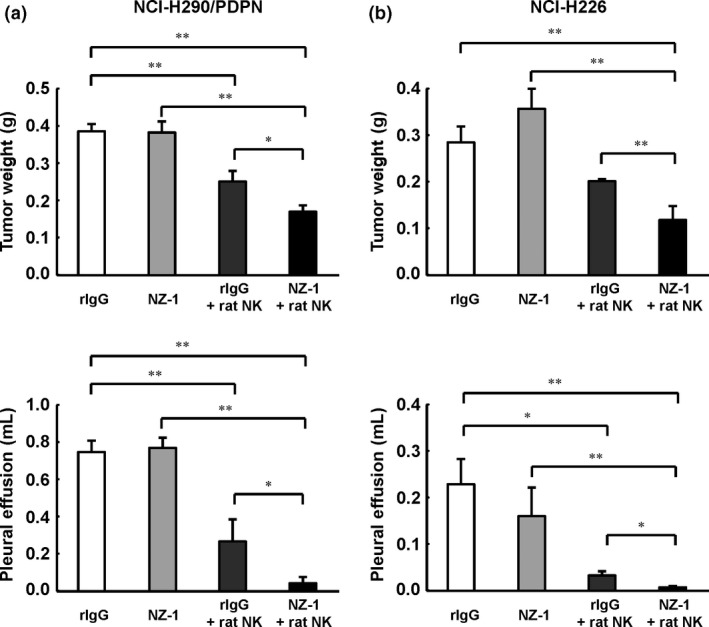
Antitumor effects of NZ‐1 i.p. injection in a malignant pleural mesothelioma orthotopic xenograft model. SCID mice (n = 5) were injected into the thoracic cavity with NCI‐H290/PDPN (a) or NCI‐H226 (b) (1.0 × 106 cells). Intraperitoneal injection of NZ‐1 (100 μg) or rat IgG (rIgG; 100 μg) began on day 0, and continued twice a week for 3 weeks. Intrathoracic injection of rat CD161a+ cells (1.0 × 106 cells) or control normal saline began on day 3, and continued weekly for 3 weeks. Three weeks (NCI‐H290/PDPN) or 9 weeks (NCI‐H226) after tumor cell inoculation, mice were killed. *P < 0.05, **P < 0.01 (values represent mean ± SE).
Antitumor activity of NZ‐8 in MPM orthotopic xenograft model
We previously reported that NZ‐8 induced ADCC activity mediated by human NK cells.9 To evaluate the antitumor effects of NZ‐8 combined with human NK cells in an MPM orthotopic xenograft model, we used the NCI‐H290/PDPN orthotopic xenograft mouse model. As shown in Figure 4(a), ADCC activity against NCI‐H290/PDPN was observed following treatment with NZ‐8 and human MNC. Injection of NZ‐8 (i.p.) twice a week and human NK (CD56+) cells injected into the thoracic cavity weekly for 2 weeks significantly inhibited tumor weight and pleural effusion production, compared with NZ‐8 or human NK cells alone (Fig. 4b).
Figure 4.
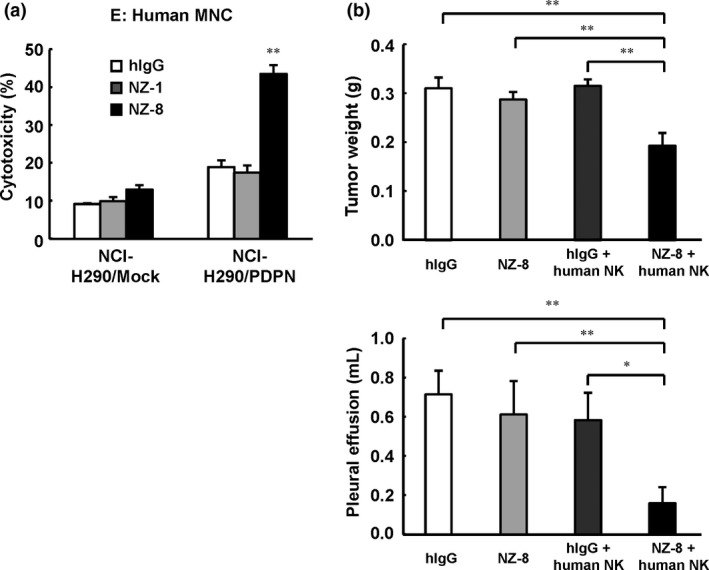
Antitumor activity of rat–human chimeric anti‐human podoplanin antibody NZ‐8 in an NCI‐H290/PDPN malignant pleural mesothelioma orthotopic xenograft model. (a) Antibody‐dependent cellular cytotoxic activity against NCI‐H290/PDPN was determined with 6‐h 51Cr release assay (effector/target ratio 100) in the presence of human IgG (hIgG; 1 μg/mL), NZ‐1 (1 μg/mL), or NZ‐8 (1 μg/mL), with human peripheral blood mononuclear cells (MNC). (b) NCI‐H290/PDPN (1.0 × 106 cells) was inoculated into the thoracic cavity on day 0 (n = 5). NZ‐8 (100 μg) or hIgG (100 μg) injection (i.p.) also began on day 0, and continued twice a week for 2 weeks. Human natural killer (NK) (CD56+) cells (1.0 × 106 cells) or control normal saline were injected into the thoracic cavity from day 3, and continued weekly for 2 weeks. Mice were killed 3 weeks after tumor cell inoculation. *P < 0.05, **P < 0.01 (values represent mean ± SE).
In vitro and in vivo antitumor effects of NZ‐12
Given that NZ‐1 and NZ‐8 induced antitumor effects in MPM in an orthotopic xenograft model, we generated a novel rat–human chimeric anti‐human podoplanin antibody, NZ‐12, derived from NZ‐1, in order to establish a more potent target therapy for podoplanin. As shown in Figure 5(a), NZ‐12 induced a significant level of ADCC, mediated by human MNC, against podoplanin‐positive MPM cells. The ADCC activity induced by NZ‐12 was significantly higher than that of NZ1 or NZ‐8. NZ‐12 also induced CDC activity against podoplanin‐positive MPM cells (Fig. 5b). Moreover, ADCC activity of NZ‐12 was mediated by human NK (CD56+) cells (Fig. 5c). In the NCI‐H290/PDPN orthotopic xenograft SCID mouse model, tumor weight and production of pleural effusion was significantly inhibited by 2 weeks of injections of NZ‐12 (i.p.), given twice a week, co‐administered with injections of human NK (CD56+) cells into the thoracic cavity, given once a week (Fig. 5d,e). By contrast, treatment with NZ‐12 alone did not inhibit tumor growth.
Figure 5.
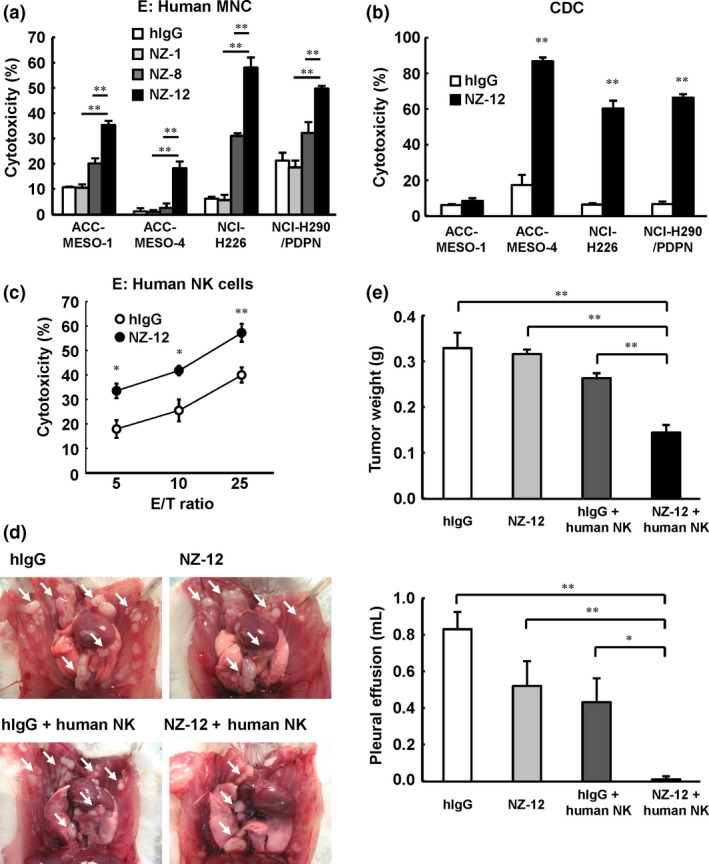
Antitumor effects of novel rat–human chimeric anti‐human podoplanin antibody NZ‐12 in vitro and in vivo. (a) Antibody‐dependent cellular cytotoxicity induced by human peripheral blood mononuclear cells (MNC) against malignant pleural mesothelioma cell lines ACC‐MESO‐1, ACC‐MESO‐4, NCI‐H226, and NCI‐H290/PDPN, was evaluated by 6‐h 51Cr release assay (effector/target ratio 100) in the presence of human IgG (hIgG; 1 μg/mL), NZ‐1 (1 μg/mL), NZ‐8 (1 μg/mL), and NZ‐12 (1 μg/mL), with human MNC. (b) Complement‐dependent cytotoxic activity was indicated by 6‐h 51Cr release assay in the presence of NZ‐12 (1 μg/mL) or hIgG with baby rabbit complement (1:4 dilution). (c) Human natural killer (NK) (CD56+) cells were isolated from human MNC. Antibody‐dependent cellular cytotoxic activity of NZ‐12 (1 μg/mL) mediated by human NK (CD56+) cells was evaluated by 6‐h 51Cr release assay at an effector/target ratio of 5, 10, and 25. (d, e) SCID mice (n = 5) were injected with NCI‐H290/PDPN (1.0 × 106 cells) into the thoracic cavity. NZ‐12 (100 μg) or hIgG (100 μg) injection (i.p.) was continued twice a week for 2 weeks. Human NK (CD56+) cells (1.0 × 105 cells) were injected into the thoracic cavity weekly for 2 weeks. Three weeks after tumor cell inoculation, the mice were killed and the weight of thoracic tumors (white arrows) and volume of pleural effusion were measured. *P < 0.05, **P < 0.01 (values represent mean ± SE).
Effect of combination treatment of NZ‐12 and pemetrexed
Our result suggested that combined treatment with NZ‐12 and human NK cells inhibited tumor growth in an MPM orthotropic model. Additionally, we further revealed the efficacy of NZ‐12 against MPM using combined treatment of NZ‐12 and pemetrexed in an MPM SCID mouse model. As shown in Figure 6(a), expression of podoplanin in NCI‐H290/PDPN was unchanged following 72 h of incubation with pemetrexed (0.1 μM). In contrast, the same treatment condition of pemetrexed inhibited the proliferation of NCI‐H290/PDPN in vitro (data not shown). Furthermore, treatment with pemetrexed in target cells did not inhibit ADCC activity induced by NZ‐12 with human MNC (Fig. 6b). Using the orthotopic xenograft SCID mouse model, injection of NZ‐12 (i.p.), human NK (CD56+) cells injected into thoracic cavity, and pemetrexed (i.p.) significantly reduced intrathoracic tumor growth and production of pleural effusion, compared with the immunotherapy of NZ‐12 (NZ‐12 with human NK cells) or pemetrexed alone (Fig. 6c).
Figure 6.
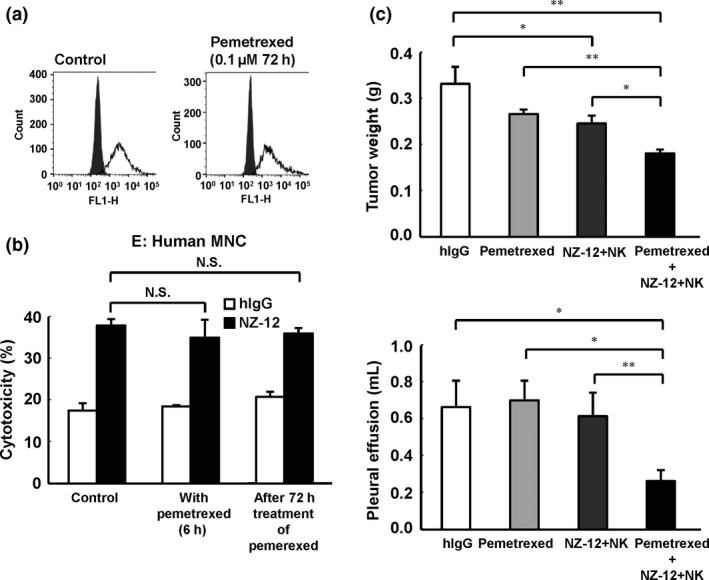
Combinatory effects of treatment for malignant pleural mesothelioma with NZ‐12‐based immunotherapy and pemetrexed in vivo. (a) NCI‐H290/PDPN was incubated with pemetrexed (0.1 μM). After 72 h of incubation, expression of podoplanin was evaluated by FACS analysis. (b) Antibody‐dependent cellular cytotoxic activity of NZ‐12 (1 μg/mL) against NCI‐H290/PDPN mediated by human peripheral blood mononuclear cells (MNC) was evaluated by 6‐h 51Cr release assay (effector/target ratio 100) in the presence or absence of pemetrexed (0.1 μM). NCI‐H290/PDPN treated with pemetrexed (0.1 μM) for 72 h was also used in target cells. (c) SCID mice (n = 5) were injected with NCI‐H290/PDPN (1.0 × 106 cells) into the thoracic cavity. NZ‐12 (100 μg) or human IgG (hIgG; 100 μg) was injected i.p. twice a week for 2 weeks. Human natural killer (NK) (CD56+) cells (1.0 × 105 cells) or normal saline was injected into the thoracic cavity weekly for 2 weeks. Pemetrexed (100 mg/kg, i.p.) was given on days 4, 5, 6, 11, 12, and 13. *P < 0.05, **P < 0.01 (values represent mean ± SE). N.S., not significant.
Discussion
In the present study, we have shown that anti‐human podoplanin antibodies NZ‐1, NZ‐8, and NZ‐12 possess therapeutic antitumor effects in an MPM orthotopic xenograft SCID mouse model. Furthermore, NZ‐12 induced more ADCC activity than NZ‐1 or NZ‐8. In addition, we have shown that combination treatment of NZ‐12 and pemetrexed produced significantly greater antitumor activity than each single therapy. These findings suggested that NZ‐12 is an effective antibody against MPM.
We previously established a human MPM orthotopic xenograft SCID mouse model, in which MPM cells were inoculated into the thoracic cavity.33, 35 Several weeks after inoculation, the mice died due to increased size of thoracic tumors and production of bloody pleural effusion inside the thoracic cavity. As this disease state is similar to that of human MPM patients, this model could be used to evaluate the efficacy of therapeutic treatments of MPM.34, 36 Although our previous study only showed the antitumor effects of an anti‐human podoplanin antibody given with NK cells in an MPM s.c. xenograft model,9 in this study, administration of anti‐human podoplanin antibodies NZ‐1, NZ‐8, and NZ‐12 with NK cells reduced growth of thoracic tumors and production of pleural effusion in an MPM orthotopic xenograft model. By contrast, no antitumor effects were observed following injection of the anti‐human podoplanin antibody alone in this experimental condition. Treatment of NK cells with control IgG partly inhibited the tumor growth because of antitumor activity of NK cells alone. Production of pleural effusion closely depends on the tumor size above a certain level in MPM orthotopic xenograft model. Therefore, pleural effusion was completely inhibited in Figure 2. However, anti‐human podoplanin antibody with NK cells significantly inhibited tumor weight compared with NK cells alone. These results indicate that the antitumor effects of anti‐human podoplanin antibodies in the MPM orthotopic xenograft mouse model are mediated by NK cell‐mediated ADCC. Furthermore, cancer progression was reduced not only by intrathoracic injection of the anti‐human podoplanin antibody, but also by i.p. injection. As the drug injection into the thoracic cavity produced several side‐effects, including pneumothorax, inflammation, and infection, systemic administration is preferable in clinical practice. Therefore, in the current study, it appears that systemic treatment with anti‐human podoplanin antibodies would be beneficial in podoplanin‐positive MPM patients.
We have shown that NZ‐12, a novel rat–human chimeric anti‐human podoplanin antibody generated from NZ‐1, with human NK cells induced antitumor effects against MPM in vitro and in vivo. The ADCC activity of NZ‐12 mediated by human NK cells was much higher than that of NZ‐1 or NZ‐8. In our previous study, NZ‐1 could only induce ADCC activity by rat NK cells because NZ‐1 was a rat anti‐human podoplanin antibody.9 However, NZ‐8, another rat‐human chimeric anti‐human podoplanin antibody generated from NZ‐1, could induce ADCC activity by human NK cells as effector cells. The reason why ADCC activity of NZ‐12 was significantly higher than that of NZ‐8 is still unknown. However, it may be related to differences in the development method of NZ‐12 and NZ‐8. In development of the human chimeric antibody, the kappa chain was generally used as the antibody light chain, and NZ‐8 was also established using the kappa chain. In contrast, because NZ‐1 has a lambda chain of the rat antibody, NZ‐12 was also generated using the human lambda chain. Therefore, it might be considered that the potent ADCC activity of NZ‐12, compared with NZ‐8, was caused by differences in the binding affinity of NZ‐12 against podoplanin due to the development method. In addition, CDC activity in ACC‐MESO‐1 and ADCC activity in ACC‐MESO‐4 were relatively lower compared to other cell lines. We previously showed that ACC‐MESO‐1, ACC‐MESO‐4, and NCI‐H226 express high levels of podoplanin.(9) Therefore, the expression level of podoplanin was probably not related. We speculated that the sensitivity of several cell lines against the cytotoxic mechanisms induced by NK cells or complements were related to differences in ADCC and CDC activity.
Pemetrexed combined with cisplatin is the standard chemotherapy used in the treatment of MPM patients. However, this combined treatment often only prolongs survival in patients by approximately 2.8 months, compared with cisplatin treatment alone.20 Therefore, development of novel therapeutic strategies in combination with pemetrexed are needed to improve therapeutic treatments for MPM. In this study, prolonged treatment with pemetrexed did not change the expression level of podoplanin in MPM cells, suggesting that ADCC activity of NZ‐12 against MPM was not inhibited by pemetrexed in vitro. In addition, the combined immunotherapy of NZ‐12, mediated by human NK cells, with pemetrexed injection enhanced antitumor effects by both induction of ADCC activity and antifolate action in an MPM orthotopic xenograft model. Although many anticancer chemotherapy drugs suppress immune function due to myelosuppression, several chemotherapy drugs, including gemcitabine, combine with the effects of immunotherapy to modulate immune function.37, 38 Pemetrexed, which is an antifolate antimetabolite agent, is likely to induce myelosuppression. Nevertheless, it is reported that antifolate agents enhanced the production of interleukin‐2 and interleukin‐12.39 The detailed mechanism is still unclear, but our results suggest that treatment with both NZ‐12‐mediated immunotherapy and pemetrexed induces combined effects against intrathoracic MPM. These are likely additive effects, as pemetrexed had no effect on the expression of podoplanin or ADCC activity of NZ‐12 mediated by human NK cells.
In conclusion, we found that anti‐human podoplanin antibodies possess ADCC activity‐induced antitumor effects in an MPM orthotopic xenograft model. Furthermore, combined treatment of NZ‐12, which induced potent ADCC activity mediated by human NK cells, and pemetrexed produced enhanced antitumor effects in an MPM orthotopic xenograft model. These findings suggest that combination therapy using both NZ‐12 podoplanin‐targeting immunotherapy and pemetrexed could provide a promising therapeutic strategy in the treatment of MPM.
Disclosure Statement
Yasuhiko Nishioka received research funding from Eli Lilly Japan. The other authors have no conflicts of interest.
Acknowledgments
We thank Tomoko Oka for excellent technical assistance. This work was supported in part by: KAKENHI (24390210, 25460189, 26440019, and 25462242) for Scientific Research (C) (to Y.N., S.A., M.K.K., and Y.K.) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan; a research program for development of intelligent Tokushima artificial exosome (iTEX) from Tokushima University (to S.A.); the Platform for Drug Discovery, Informatics, and Structural Life Science from the Japan Agency for Medical Research and Development (AMED) (to Y.K.); the Basic Science and Platform Technology Program for Innovative Biological Medicine from AMED (to Y.K.); and by the Regional Innovation Strategy Support Program from MEXT, Japan (to Y.K.).
Cancer Sci 107 (2016) 1198–1205
Funding Information
Ministry of Education, Culture, Sports, Science and Technology of Japan KAKENHI (24390210, 25460189, 26440019, and 25462242); Tokushima University; Japan Agency for Medical Research and Development.
References
- 1. Breiteneder‐Geleff S, Matsui K, Soleiman A et al Podoplanin, novel 43‐kd membrane protein of glomerular epithelial cells, is down‐regulated in puromycin nephrosis. Am J Pathol 1997; 151: 1141–52. [PMC free article] [PubMed] [Google Scholar]
- 2. Breiteneder‐Geleff S, Soleiman A, Kowalski H et al Angiosarcomas express mixed endothelial phenotypes of blood and lymphatic capillaries: podoplanin as a specific marker for lymphatic endothelium. Am J Pathol 1999; 154: 385–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vanderbilt JN, Allen L, Gonzalez RF et al Directed expression of transgenes to alveolar type I cells in the mouse. Am J Respir Cell Mol Biol 2008; 39: 253–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kato Y, Fujita N, Kunita A et al Molecular identification of Aggrus/T1alpha as a platelet aggregation‐inducing factor expressed in colorectal tumors. J Biol Chem 2003; 278: 51599–605. [DOI] [PubMed] [Google Scholar]
- 5. Kato Y, Sasagawa I, Kaneko M, Osawa M, Fujita N, Tsuruo T. Aggrus: a diagnostic marker that distinguishes seminoma from embryonal carcinoma in testicular germ cell tumors. Oncogene 2004; 23: 8552–6. [DOI] [PubMed] [Google Scholar]
- 6. Kato Y, Kaneko M, Sata M, Fujita N, Tsuruo T, Osawa M. Enhanced expression of Aggrus (T1alpha/podoplanin), a platelet‐aggregation‐inducing factor in lung squamous cell carcinoma. Tumour Biol 2005; 26: 195–200. [DOI] [PubMed] [Google Scholar]
- 7. Mishima K, Kato Y, Kaneko MK, Nishikawa R, Hirose T, Matsutani M. Increased expression of podoplanin in malignant astrocytic tumors as a novel molecular marker of malignant progression. Acta Neuropathol 2006; 111: 483–8. [DOI] [PubMed] [Google Scholar]
- 8. Kato Y, Vaidyanathan G, Kaneko MK et al Evaluation of anti‐podoplanin rat monoclonal antibody NZ‐1 for targeting malignant gliomas. Nucl Med Biol 2010; 37: 785–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abe S, Morita Y, Kaneko MK et al A novel targeting therapy of malignant mesothelioma using anti‐podoplanin antibody. J Immunol 2013; 190: 6239–49. [DOI] [PubMed] [Google Scholar]
- 10. Kaneko MK, Kato Y, Kitano T, Osawa M. Conservation of a platelet activating domain of Aggrus/podoplanin as a platelet aggregation‐inducing factor. Gene 2006; 378: 52–7. [DOI] [PubMed] [Google Scholar]
- 11. Kawase A, Ishii G, Nagai K et al Podoplanin expression by cancer associated fibroblasts predicts poor prognosis of lung adenocarcinoma. Int J Cancer 2008; 123: 1053–9. [DOI] [PubMed] [Google Scholar]
- 12. Hoshino A, Ishii G, Ito T et al Podoplanin‐positive fibroblasts enhance lung adenocarcinoma tumor formation: podoplanin in fibroblast functions for tumor progression. Cancer Res 2011; 71: 4769–79. [DOI] [PubMed] [Google Scholar]
- 13. Schoppmann SF, Jesch B, Riegler MF et al Podoplanin expressing cancer associated fibroblasts are associated with unfavourable prognosis in adenocarcinoma of the esophagus. Clin Exp Metastasis 2013; 30: 441–6. [DOI] [PubMed] [Google Scholar]
- 14. Shindo K, Aishima S, Ohuchida K et al Podoplanin expression in cancer‐associated fibroblasts enhances tumor progression of invasive ductal carcinoma of the pancreas. Mol Cancer 2013; 12: 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Robinson BW, Lake RA. Advances in malignant mesothelioma. N Engl J Med 2005; 353: 1591–603. [DOI] [PubMed] [Google Scholar]
- 16. Pelucchi C, Malvezzi M, La Vecchia C, Levi F, Decarli A, Negri E. The Mesothelioma epidemic in Western Europe: an update. Br J Cancer 2004; 90: 1022–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Murayama T, Takahashi K, Natori Y, Kurumatani N. Estimation of future mortality from pleural malignant mesothelioma in Japan based on an age‐cohort model. Am J Ind Med 2006; 49: 1–7. [DOI] [PubMed] [Google Scholar]
- 18. Ray M, Kindler HL. Malignant pleural mesothelioma: an update on biomarkers and treatment. Chest 2009; 136: 888–96. [DOI] [PubMed] [Google Scholar]
- 19. Scherpereel A, Astoul P, Baas P et al Guidelines of the European Respiratory Society and the European Society of Thoracic Surgeons for the management of malignant pleural mesothelioma. Eur Respir J 2010; 35: 479–95. [DOI] [PubMed] [Google Scholar]
- 20. Vogelzang NJ, Rusthoven JJ, Symanowski J et al Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol 2003; 21: 2636–44. [DOI] [PubMed] [Google Scholar]
- 21. Li Q, Wang W, Machino Y et al Therapeutic activity of glycoengineered anti‐GM2 antibodies against malignant pleural mesothelioma. Cancer Sci 2015; 106: 102–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hassan R, Cohen SJ, Phillips M et al Phase I clinical trial of the chimeric anti‐mesothelin monoclonal antibody MORAb‐009 in patients with mesothelin‐expressing cancers. Clin Cancer Res 2010; 16: 6132–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jiang XR, Song A, Bergelson S et al Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat Rev Drug Discov 2011; 10: 101–11. [DOI] [PubMed] [Google Scholar]
- 24. Kato Y, Kaneko MK, Kuno A et al Inhibition of tumor cell‐induced platelet aggregation using a novel anti‐podoplanin antibody reacting with its platelet‐aggregation‐stimulating domain. Biochem Biophys Res Commun 2006; 349: 1301–7. [DOI] [PubMed] [Google Scholar]
- 25. Ogasawara S, Kaneko MK, Price JE, Kato Y. Characterization of anti‐podoplanin monoclonal antibodies: critical epitopes for neutralizing the interaction between podoplanin and CLEC‐2. Hybridoma (Larchmt) 2008; 27: 259–67. [DOI] [PubMed] [Google Scholar]
- 26. Kato Y, Kaneko MK, Kunita A et al Molecular analysis of the pathophysiological binding of the platelet aggregation‐inducing factor podoplanin to the C‐type lectin‐like receptor CLEC‐2. Cancer Sci 2008; 99: 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kaneko MK, Kunita A, Abe S et al Chimeric anti‐podoplanin antibody suppresses tumor metastasis through neutralization and antibody‐dependent cellular cytotoxicity. Cancer Sci 2012; 103: 1913–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kawaguchi K, Murakami H, Taniguchi T et al Combined inhibition of MET and EGFR suppresses proliferation of malignant mesothelioma cells. Carcinogenesis 2009; 30: 1097–105. [DOI] [PubMed] [Google Scholar]
- 29. Wang W, Nishioka Y, Ozaki S et al HM1.24 (CD317) is a novel target against lung cancer for immunotherapy using anti‐HM1.24 antibody. Cancer Immunol Immunother 2009; 58: 967–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kishuku M, Nishioka Y, Abe S et al Expression of soluble vascular endothelial growth factor receptor‐1 in human monocyte‐derived mature dendritic cells contributes to their antiangiogenic property. J Immunol 2009; 183: 8176–85. [DOI] [PubMed] [Google Scholar]
- 31. Kato Y, Kunita A, Abe S et al The chimeric antibody chLpMab‐7 targeting human podoplanin suppresses pulmonary metastasis via ADCC and CDC rather than via its neutralizing activity. Oncotarget 2015; 6: 36003–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang W, Nishioka Y, Ozaki S et al Chimeric and humanized anti‐HM1.24 antibodies mediate antibody‐dependent cellular cytotoxicity against lung cancer cells. Lung Cancer 2009; 63: 23–31. [DOI] [PubMed] [Google Scholar]
- 33. Li Q, Yano S, Ogino H et al The therapeutic efficacy of anti vascular endothelial growth factor antibody, bevacizumab, and pemetrexed against orthotopically implanted human pleural mesothelioma cells in severe combined immunodeficient mice. Clin Cancer Res 2007; 13: 5918–25. [DOI] [PubMed] [Google Scholar]
- 34. Ikuta K, Yano S, Trung VT et al E7080, a multi‐tyrosine kinase inhibitor, suppresses the progression of malignant pleural mesothelioma with different proangiogenic cytokine production profiles. Clin Cancer Res 2009; 15: 7229–37. [DOI] [PubMed] [Google Scholar]
- 35. Nakataki E, Yano S, Matsumori Y et al Novel orthotopic implantation model of human malignant pleural mesothelioma (EHMES‐10 cells) highly expressing vascular endothelial growth factor and its receptor. Cancer Sci 2006; 97: 183–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Van TT, Hanibuchi M, Kakiuchi S et al The therapeutic efficacy of S‐1 against orthotopically implanted human pleural mesothelioma cells in severe combined immunodeficient mice. Cancer Chemother Pharmacol 2011; 68: 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shevchenko I, Karakhanova S, Soltek S et al Low‐dose gemcitabine depletes regulatory T cells and improves survival in the orthotopic Panc02 model of pancreatic cancer. Int J Cancer 2013; 133: 98–107. [DOI] [PubMed] [Google Scholar]
- 38. Tongu M, Harashima N, Monma H et al Metronomic chemotherapy with low‐dose cyclophosphamide plus gemcitabine can induce anti‐tumor T cell immunity in vivo. Cancer Immunol Immunother 2013; 62: 383–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shurin GV, Tourkova IL, Kaneno R, Shurin MR. Chemotherapeutic agents in noncytotoxic concentrations increase antigen presentation by dendritic cells via an IL‐12‐dependent mechanism. J Immunol 2009; 183: 137–44. [DOI] [PMC free article] [PubMed] [Google Scholar]