

Abstract
Although the clinical outcomes of diffuse large B‐cell lymphoma (DLBCL) have improved in the immunochemotherapy era, approximately one‐third of patients develop intractable disease. To improve clinical outcomes for these patients, it is important to identify those with poor prognosis prior to initial treatment in order to select optimal therapies. Here, we investigated the clinical and biological significance of SPIB, an Ets family transcription factor linked to lymphomagenesis, in DLBCL. We classified 134 DLBCL patients into SPIB negative (n = 108) or SPIB positive (n = 26) groups by immunohistochemical staining. SPIB positive patients had a significantly worse treatment response and poor prognosis compared with SPIB negative patients. Multivariate analysis for patient survival indicated that SPIB expression was an independent poor prognostic factor for both progression free survival (PFS) and overall survival (OS) (PFS, hazard ratio [HR] 2.65, 95% confidence interval [CI] 1.31–5.33, P = 0.006; OS, HR 3.56, 95% CI 1.43–8.91, P = 0.007). Subsequent analyses of the roles of SPIB expression in DLBCL pathogenesis revealed that SPIB expression in lymphoma cells resulted in resistance to the BH3‐mimetic ABT‐263 and contributed to apoptosis resistance via the PI3K‐AKT pathway. The inhibition of AKT phosphorylation re‐sensitized SPIB expressing lymphoma cells to ABT‐263‐induced cell death. Together, our data indicate that SPIB expression is a clinically novel poor prognostic factor in DLBCL that contributes to treatment resistance, at least in part, through an anti‐apoptotic mechanism.
Keywords: Apoptosis, diffuse large B‐cell lymphoma, PI3K‐AKT pathway, prognostic factor, SPIB
The clinical outcomes of diffuse large B‐cell lymphoma (DLBCL) have improved in the rituximab era.1, 2, 3 However, the prognoses for intractable subtypes such as double hit lymphoma and EBV positive lymphoma remain unsatisfactory.4, 5, 6, 7 To improve the clinical outcomes for patients diagnosed with these poor prognostic subtypes, it is essential to accurately stratify patients prior to initial treatment to facilitate the selection of optimal therapies.
SPIB, a member of the Ets family of transcription factors, is located on chromosome 19q, and plays an important role in the differentiation of mature B‐cells into plasma cells, and the differentiation of plasmacytoid dendritic cells.8, 9, 10, 11, 12 Activation of IRF4 is an initial event in the plasmacytic differentiation of mature B‐cells, and leads to the upregulation of BLIMP1 and downregulation of BCL6. The upregulation of BLIMP1 induces the subsequent downregulation of SPIB, which is indispensable for normal differentiation. Therefore, the ectopic expression of SPIB during terminal B‐cell differentiation results in inhibition of plasma cell differentiation and contributes to the development of the activated B‐cell (ABC) type of DLBCL.9, 13, 14 Moreover, the amplification of chromosome 19q, on which SPIB is located, is frequently detected in DLBCL and primary central nervous system lymphoma.9, 15, 16, 17 A recent report also revealed that the expression of SPIB is elevated in BCL2 and MYC double positive lymphoma,6 which suggests that SPIB overexpression might be associated with this poor prognostic phenotype.
Here, we investigated the clinical and biological significance of deregulated SPIB expression in DLBCL pathogenesis and identified SPIB as a novel molecular target in intractable DLBCL subtypes.
Materials and Methods
Patients
A total of 134 patients diagnosed with DLBCL at Nagoya University Hospital between 2006 and 2013 were analyzed in this study. The study protocol for the experimental use of pathological specimens and patient information was approved by the institutional review board of Nagoya University Hospital and complied with all provisions of the Declaration of Helsinki and the Ethical Guidelines for Medical and Health Research Involving Human Subjects issued by the Ministry of Health, Labour and Welfare in Japan.
Drugs and cell lines
The following drugs used in this study were purchased: ABT‐263 (AdooQ BioScience, Irwin, CA, USA), LY294002 (Cayman Chemical Company, Ann Arbor, MI, USA), Ibrutinib and Idelalisib (Selleck Chemicals, Houston, TX, USA). 293T and SU‐DHL4 (a kind gift from Dr Kunihiko Takeyama, Dana Farber Cancer Institute, Boston, MA, USA) cells were cultured in DMEM (Sigma Aldrich, St Louis, MO, USA) and RPMI (Sigma Aldrich), respectively. Both culture media were supplemented with 10% FCS, 2 mM l‐glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin and 1 mM sodium pyruvate.
Immunohistochemistry
Formalin fixed, paraffin embedded tissues were evaluated by routine HE and immunohistochemical staining (IHC). For IHC of CD20, BCL2, BCL6, CD10, MUM1, CD5 and MYC, the following primary antibodies were used: mouse monoclonal anti‐human CD20 (clone L26) and BCL2 (clone 124) antibodies (Dako, Glostrup, Denmark), mouse monoclonal anti‐human BCL6 (clone LN22), CD10 (clone 56C6) and CD5 (clone 4C7) antibodies (Novocastra, Leica Biosystems, Newcastle Upon Tyne, UK), M‐17 antibody against MUM1 (sc‐6059, Santa Cruz Biotechnology, Dallas, TX, USA), and rabbit monoclonal anti‐human c‐MYC antibody (clone Y69; Abcam, Cambridge, UK). EBV‐encoded RNA expression was also routinely evaluated by in‐situ hybridization (EBER‐ISH). Anti‐SPIB mouse monoclonal antibody (clone 4G5, ab135238; Abcam) was used as the primary antibody targeting SPIB. Immunohistochemical detection of SPIB was performed according to the following procedure. After deparaffinization and rehydration of the sections using a microwave oven, antigen retrieval was performed in low pH Target Retrieval Solution (Dako) for 20 min at 98°C. The sections were subsequently incubated with primary antibody at 4°C overnight followed by the addition of biotin‐conjugated secondary antibody for 1 h at room temperature, and staining was activated by addition of the avidin–biotin complex (ABC). HRP activity was detected using 3, 3‐diaminobenzidine tetrahydrochloride (DAB). All pathological specimens were reviewed by two hematopathologists (S.S. and S.N.) according to the current WHO classification. The specimens were observed with an Olympus BX51 N‐34 microscope (Olympus, Tokyo, Japan), and the photos were taken with a Nikon DS‐Fil1 (Nikon, Tokyo, Japan) and BZ‐9000 (Keyence, Osaka, Japan).
SPIB expression vector and transfection/transduction procedures
To develop a SPIB expression vector, a FLAG tagged fragment of full length SPIB (FLAG‐SPIB) was sub‐cloned into the pCRII‐TOPO plasmid (Invitrogen in Thermo Fisher Scientific, Waltham, MA, USA). Subsequently, the vector was digested with EcoRI and transferred to the retrovirus vector MIGR1 (a kind gift from Dr W. S. Pear, University of Pennsylvania, Philadelphia, PA, USA), which constitutively expresses GFP, to generate MIGR1‐FLAG‐SPIB. The DNA sequence of FLAG‐SPIB cloned into MIGR1 was confirmed by sequence analysis. Transient transfection of the MIGR1 control vector (MOCK) and MIGR1‐FLAG‐SPIB into 293T cells was performed using the Effectene transfection reagent (Qiagen, Venlo, the Netherlands) according to the manufacturer's protocol. The stable transduction of SU‐DHL4 cells using MIGR1 vectors was performed using a retroviral infection system (Retro‐X Expression systems; Clontech in Takara Bio, Shiga, Japan). In brief, the MIGR1 vector and envelope vector (pVSV‐G) were co‐transfected into the GP2‐293 packaging cell line using the FuGENE6 transfection reagent (Promega, Fitchburg, WI, USA). Two to 4 days later, retrovirus was harvested from the culture supernatant and applied to SU‐DHL4 cell lines. SU‐DHL4 cells transduced with MIGR1 vector were sorted for GFP expression using a FACSAria II (Becton‐Dickinson [BD], Franklin Lakes, NJ, USA).
Cell proliferation assay
Cell proliferation was assessed by trypan blue‐exclusion tests and MTT assays. Cells (2.0 × 104 SU‐DHL4 cells) were placed in 96‐well plates and cultured for 96 h at 37°C in a 5% CO2 incubator. The number of live cells was counted every 24 h using trypan blue staining. For MTT assays, the same number of cells used for the trypan blue test were placed in 96‐well plates and cultured for 72 h. MTT labelling reagent (Roche Diagnostics, Mannheim, Germany) was then added to each well and incubated for 3 h. Fluorescence was evaluated at 490 nM with a GloMax Multi+ (Promega, Madison, WI, USA).
Cell cycle and cell death assessment
A hypotonic propidium iodide (PI) assay was used to evaluate the cell cycle, as described previously.18, 19, 20 In brief, cells were incubated for 48 h with various drugs, washed, and re‐suspended in PBS containing 0.2% Triton X‐100 and 50 μg/mL PI. Subsequently, cells were analyzed by flow cytometry (FACSCalibur [BD]). For cell death assays, MIGR1‐FLAG‐SPIB transduced cells were incubated in 96‐well plates with the appropriate drugs for 48 h. Cells were then stained with 5 μg/mL PI for 15 min in the dark and evaluated by flow cytometry.
Immunofluorescence staining
To evaluate SPIB expression in MIGR1‐FLAG‐SPIB transfected 293T and transduced SU‐DHL4 cells, we performed immunofluorescence staining using the same anti‐SPIB monoclonal antibody used for IHC. In brief, 293T cells were transfected with MIGR1‐FLAG‐SPIB using the Effectene Transfection Reagent according to the manufacturer's protocol (Qiagen) and placed in a chamber slide (Nunc in Thermo Fisher Scientific, Waltham, MA, USA). At 50–80% confluency, cells were fixed with 1:1 methanol/acetone fixative at −20°C. Slides were subsequently dried and blocked in PBS containing 1% BSA. The slide was incubated with anti‐SPIB primary antibody at 4°C overnight, and subsequently Alexa‐568 conjugated anti‐mouse IgG (H+L) secondary antibody for 1 h at room temperature; DAPI was then applied as a counterstain. SU‐DHL4 cells were applied to slides using the cytospin method and then stained in a similar way. Specimens were viewed and photographed with an Axio Imager M2 (Carl Zeiss, Oberkochen, Germany).
Immunoblotting
Cells were treated with the various drugs and then lysed as described previously.18, 21 To prevent post‐lysis changes in phosphorylation, 50 mM NaF and 1.68 mM Na3VO4 were added. After centrifugation (11700 g for 10 min), the protein concentrations of lysates were evaluated by the Bradford method, and sample buffer containing 5% 2‐mercaptoethanol was added. After boiling for 5 min, 20‐μg samples were separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membranes, and blocked with 5% skim milk in TBS‐tween buffer (50 mM Tris‐HCL [pH 7.4], 150 mM NaCl and 0.05% Tween 20). Immunoblotting was performed using primary antibodies appropriately diluted in TBS‐tween buffer containing 5% BSA and 0.05% sodium azide (Table S1). HRP conjugated secondary antibodies (GE Healthcare, Little Chalfont, UK) were added and activated with Immobilon Western Substrate (Millipore, Billerica, MA, USA). Images were obtained using a LAS 4000mini bio‐imager (Fujifilm, Tokyo, Japan) and analyzed with MultiGauge software (Fujifilm).
Statistical analysis
The statistical significance of in vitro experiments was evaluated by an unpaired t‐test using GraphPad PRISMv5 software (GraphPad Software, La Jolla, CA, USA). The distribution of clinical variables was evaluated by Fisher's exact test or Mann‐Whitney U test. Treatment response was evaluated according to the revised response criteria.22 Progression free survival (PFS) was calculated from the day of diagnosis to the day of progression, relapse or death. Overall survival (OS) was calculated from the day of diagnosis to the day of death by any cause. PFS and OS were analyzed with the log‐rank test, and results were determined by the Kaplan–Meier method. Univariate and multivariate Cox regression analyses were performed to evaluate the following parameters: age, sex, performance status (PS), lactate dehydrogenase (LDH), clinical stage, extranodal involvement, B symptoms, DLBCL cell of origin determined by IHC,23 leukocytosis, anemia and thrombocytopenia, in addition to the levels of sIL‐2R, total protein, albumin, creatinine, serum urine acid, ALP, total bilirubin, C‐reactive protein, IgG, IgA and IgM. Multivariate analyses for PFS and OS were undertaken using the forward–backward stepwise method. All probability values were two‐sided, with an overall significance level of 0.05. All statistical analyses of clinical subjects were analyzed with Stata 10SE software (Stata Corp, College Station, TX, USA)
Results
Immunohistochemical staining of SPIB
Of 134 patients, 26 (19%) were assessed as SPIB positive, and the remaining 108 patients (81%) were SPIB negative. SPIB staining was scored positive only when it occurred in the cytoplasm and/or nucleus of lymphoma cells and not in the surrounding vascular endothelial cells. Representative images of SPIB staining are shown in Figure 1. Examples of SPIB positive specimens include UPN#107, UPN#123 and UPN#145, and SPIB negative specimens include UPN#15 and UPN#72.
Figure 1.
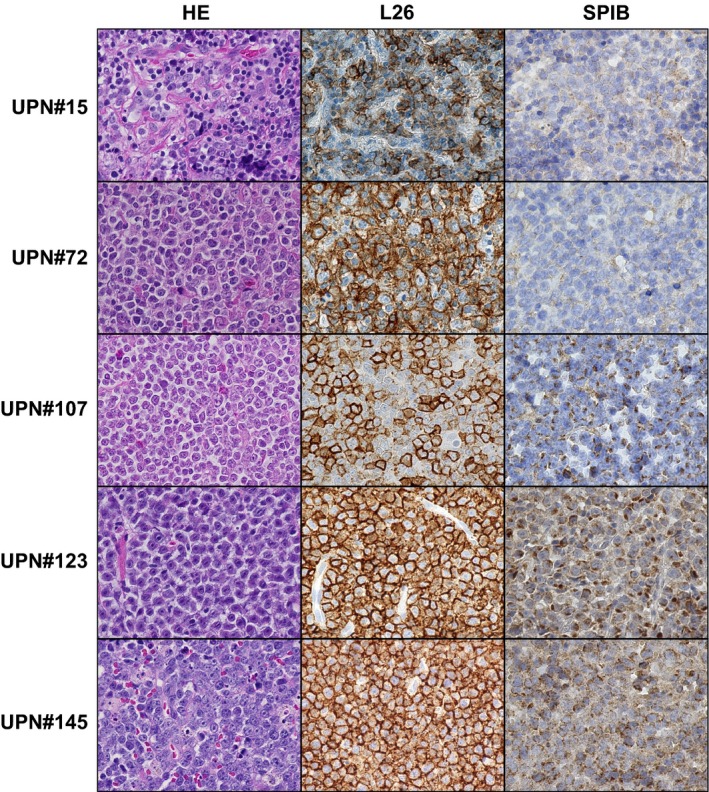
SPIB immunostaining of DLBCL specimens. Representative images of DLBCL stained by HE (left panels), anti‐CD20 antibody, L26 (center panels) and SPIB (right panels) are shown. CD20 positive tumor cells diffusely proliferate in the lymph node. Specimens UPN#15 and UPN#72 were assessed as SPIB negative, while UPN#107, UPN#123 and UPN#145 were assessed as SPIB positive (original magnification 400 × , Keyence BZ‐9000).
Clinical characteristics and outcomes
Patient characteristics according to SPIB expression are shown in Table 1. In all patients, the proportions of patients in the SPIB positive group who had extranodal involvement, higher international prognostic index score (IPI) and MYC positivity in IHC were significantly greater than those in the SPIB negative group. There was a significantly higher proportion of patients who were BCL6 positive by IHC in the SPIB negative group. The incidence of double positive lymphoma (MYC and BCL2 co‐expression by IHC)7, 24 did not significantly differ between the two groups. Other clinical variables including age, sex and DLBCL cell of origin determined by IHC were not significantly different. Ninety‐six (89%) and 25 patients (96%) in the SPIB negative and positive groups, respectively, received R‐CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine and prednisolone) or R‐CHOP‐like regimens as initial treatment. In patients who were evaluable for initial treatment, 96 of 104 patients (92%) in the SPIB negative group and 15 of 24 patients (63%) in the SPIB positive group achieved a treatment response (complete response [CR] + partial response [PR]) (P = 0.001). CR rates were 86% in the SPIB negative group and 50% in the SPIB positive group (P < 0.001). The rate of progressive disease during the initial treatment in the SPIB positive group was 38%, which was significantly higher than that in the SPIB negative group (P = 0.001). We subsequently assigned all patients to one of two groups according to the cell of origin. Of the 132 patients for whom the cell of origin was evaluable by IHC, 47 (36%) and 85 (64%) patients were classified into GCB type DLBCL and non‐GCB type DLBCL, respectively. Of the 47 patients, 12 (26%) with GCB type were assessed as SPIB positive, whereas 14 of the 85 patients (16%) with non‐GCB type were assessed as SPIB positive. In patients with GCB type, the proportion of patients in the SPIB positive group that were MYC positive by IHC was significantly higher than that in the SPIB negative group, whereas the proportion of patients in the SPIB negative group that were positive for BCL6 expression by IHC was significantly higher than that in the SPIB positive group, which was similar to the case in the total DLBCL population. CR rates were 85% in the SPIB negative group and 25% in the SPIB positive group (P < 0.001). In patients with the non‐GCB type, the proportions of patients with a higher IPI score and elevated sIL‐2R in the SPIB positive group were greater than those in the SPIB negative group. However, the CR rate did not differ between the two groups.
Table 1.
Patient characteristics
| Characteristic | All patients | GCB type | Non‐GCB type | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SPIB negative | SPIB positive | P | SPIB negative | SPIB positive | P | SPIB negative | SPIB positive | P | |||||||
| Number | % | Number | % | Number | % | Number | % | Number | % | Number | % | ||||
| Number of patients | 108 | 81 | 26 | 19 | 0.264 | 35 | 74 | 12 | 26 | 0.591 | 71 | 84 | 14 | 16 | 0.042 |
| Age at diagnosis, years | |||||||||||||||
| Median | 65 | 70 | 65 | 64 | 65 | 70 | |||||||||
| Range | 26–88 | 49–89 | 38–85 | 49–89 | 26–88 | 58–82 | |||||||||
| >60 | 79 | 73 | 19 | 73 | 1.000 | 27 | 77 | 6 | 50 | 0.140 | 50 | 70 | 13 | 93 | 0.102 |
| Sex, male | 72 | 67 | 19 | 73 | 0.643 | 21 | 60 | 11 | 92 | 0.071 | 49 | 69 | 8 | 57 | 0.535 |
| PS >1 | 22 | 20 | 5 | 19 | 1.000 | 7 | 20 | 2 | 17 | 1.000 | 14 | 20 | 3 | 21 | 1.000 |
| LDH >ULN | 61 | 56 | 20 | 77 | 0.074 | 21 | 60 | 11 | 92 | 0.071 | 38 | 54 | 9 | 64 | 0.563 |
| Clinical stage III or IV | 56 | 52 | 17 | 65 | 0.274 | 19 | 54 | 7 | 58 | 1.000 | 35 | 49 | 10 | 71 | 0.153 |
| Extranodal involvement >1 | 30 | 28 | 13 | 50 | 0.037 | 13 | 37 | 6 | 50 | 0.506 | 15 | 21 | 7 | 50 | 0.041 |
| Presence of “B” symptom | 24 | 22 | 8 | 31 | 0.442 | 6 | 17 | 5 | 42 | 0.118 | 18 | 25 | 3 | 21 | 1.000 |
| IPI score | 0.030 | 0.527 | 0.018 | ||||||||||||
| Low | 40 | 37 | 3 | 12 | 12 | 34 | 2 | 17 | 28 | 39 | 1 | 7 | |||
| Low–intermediate | 26 | 24 | 10 | 38 | 6 | 17 | 5 | 42 | 20 | 28 | 5 | 36 | |||
| High–intermediate | 15 | 14 | 4 | 15 | 7 | 20 | 2 | 17 | 8 | 11 | 2 | 14 | |||
| High | 27 | 25 | 9 | 35 | 10 | 29 | 3 | 25 | 15 | 21 | 6 | 43 | |||
| Leukocytosis | 12 | 11 | 3 | 3 | 1.000 | 3 | 9 | 1 | 3 | 1.000 | 9 | 13 | 2 | 14 | 1.000 |
| Anemia | 19 | 18 | 5 | 19 | 0.783 | 4 | 11 | 2 | 17 | 0.637 | 15 | 21 | 3 | 21 | 1.000 |
| Thrombocytopenia | 18 | 17 | 4 | 15 | 1.000 | 4 | 11 | 1 | 8 | 1.000 | 14 | 20 | 3 | 21 | 1.000 |
| Total protein level <6.5 g/dL | 23 | 21 | 9 | 35 | 0.199 | 8 | 23 | 4 | 33 | 0.471 | 14 | 20 | 5 | 36 | 0.289 |
| Albumin level <3.5 g/dL | 34 | 31 | 7 | 27 | 0.813 | 6 | 17 | 1 | 8 | 0.659 | 26 | 37 | 6 | 43 | 0.765 |
| Creatinine level >1.0 mg/dL | 17 | 16 | 8 | 31 | 0.094 | 3 | 9 | 6 | 50 | 0.005 | 14 | 20 | 2 | 14 | 1.000 |
| Urine acid level >8.0 mg/dL | 8 | 7 | 3 | 12 | 0.446 | 4 | 11 | 0 | 0 | 0.560 | 4 | 6 | 3 | 21 | 0.084 |
| ALP >400 U/L | 11 | 10 | 5 | 19 | 0.196 | 5 | 14 | 2 | 17 | 1.000 | 6 | 8 | 3 | 21 | 0.163 |
| T‐Bil >1.0 mg/dL | 9 | 8 | 3 | 12 | 0.701 | 3 | 9 | 0 | 0 | 0.560 | 6 | 8 | 3 | 21 | 0.163 |
| CRP >1.0 mg/dL | 38 | 35 | 13 | 50 | 0.182 | 11 | 31 | 5 | 42 | 0.725 | 26 | 37 | 8 | 57 | 0.232 |
| sIL‐2R ≥1000 U/mL | 50 | 46 | 17 | 65 | 0.125 | 14 | 40 | 6 | 50 | 0.737 | 34 | 48 | 11 | 79 | 0.043 |
| Immunoglobulin | |||||||||||||||
| IgG >1700 mg/dL | 23 | 21 | 2 | 8 | 0.160 | 7 | 20 | 0 | 0 | 0.166 | 15 | 21 | 2 | 14 | 0.725 |
| IgA >400 mg/dL | 17 | 16 | 1 | 4 | 0.197 | 3 | 9 | 0 | 0 | 0.560 | 13 | 19 | 1 | 7 | 0.447 |
| IgM >100 mg/dL | 29 | 27 | 4 | 15 | 0.312 | 4 | 11 | 1 | 8 | 1.000 | 25 | 36 | 3 | 21 | 0.367 |
| Immunophenotype | |||||||||||||||
| CD5 | 5 | 5 | 1 | 4 | 1.000 | 1 | 3 | 0 | 0 | 1.000 | 4 | 6 | 1 | 8 | 1.000 |
| CD10 | 29 | 27 | 11 | 42 | 0.157 | 29 | 83 | 11 | 92 | 0.659 | 0 | — | 0 | — | — |
| BCL‐2 | 56 | 53 | 18 | 69 | 0.186 | 14 | 40 | 9 | 75 | 0.049 | 42 | 59 | 9 | 64 | 0.775 |
| BCL‐6 | 86 | 83 | 16 | 62 | 0.031 | 33 | 97 | 8 | 67 | 0.013 | 53 | 76 | 8 | 57 | 0.192 |
| MYC | 30 | 28 | 13 | 50 | 0.037 | 9 | 26 | 8 | 67 | 0.016 | 21 | 30 | 5 | 36 | 0.753 |
| MUM‐1 | 85 | 80 | 22 | 85 | 0.783 | 17 | 50 | 8 | 67 | 0.502 | 68 | 96 | 14 | 100 | 1.000 |
| EBER | 16 | 16 | 2 | 8 | 0.526 | 1 | 3 | 0 | 0 | 1.000 | 13 | 19 | 2 | 14 | 1.000 |
| MYC/BCL2 | 20 | 19 | 8 | 31 | 0.190 | 5 | 14 | 5 | 42 | 0.096 | 15 | 21 | 3 | 21 | 1.000 |
| COO, GCB type | 35 | 33 | 12 | 46 | 0.255 | — | — | — | — | — | — | — | — | — | — |
| Initial treatment | 0.748 | 0.226 | 0.897 | ||||||||||||
| R‐CHOP based regimen | 95 | 88 | 24 | 92 | 32 | 91 | 10 | 83 | 61 | 86 | 14 | 100 | |||
| DA‐EPOCH‐R | 3 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 4 | 0 | 0 | |||
| CODOX/M‐IVAC | 1 | 1 | 0 | 0 | 1 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | |||
| CHOP based regimen | 1 | 1 | 1 | 4 | 0 | 0 | 1 | 8 | 1 | 1 | 0 | 0 | |||
| Irradiation therapy | 4 | 4 | 0 | 0 | 1 | 3 | 0 | 0 | 3 | 4 | 0 | 0 | |||
| Discontinuation of MTX | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | |||
| Prednisolone only | 1 | 1 | 0 | 0 | 1 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | |||
| Others | 1 | 1 | 1 | 4 | 0 | 0 | 1 | 8 | 1 | 1 | 0 | 0 | |||
| N/A | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | |||
| Anti‐tumor response | |||||||||||||||
| OR | 96 | 92 | 15 | 63 | 0.001 | 31 | 91 | 6 | 50 | 0.005 | 63 | 93 | 9 | 75 | 0.094 |
| CR | 89 | 86 | 12 | 50 | <0.001 | 29 | 85 | 3 | 25 | <0.001 | 59 | 87 | 9 | 75 | 0.376 |
| PR | 7 | 7 | 3 | 13 | 0.396 | 2 | 6 | 3 | 25 | 0.103 | 4 | 6 | 0 | 0 | 1.000 |
| PD | 8 | 8 | 9 | 38 | 0.001 | 3 | 9 | 6 | 50 | 0.005 | 5 | 7 | 3 | 25 | 0.094 |
| NE | 4 | 4 | 2 | 8 | — | 1 | 3 | 0 | 0 | — | 3 | 4 | 2 | 14 | — |
ALP, alkarine phosphatase; CODOX/M‐IVAC, cyclophosphamide, vincristine, doxorubicin, methotrexate, ifosfamide, etoposide and cytarabine; COO, cell of origin; CR, complete response; CRP, C‐reactive protein; DA‐EPOCH‐R, dose adjusted‐etoposide, prednisolone, vincristine, cyclophosphamide, doxorubicin and rituximab; GCB, germinal center B‐cell; R‐CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine and prednisolone; LDH, lactate dehydrogenase; MTX, methotrexate; NE, not evaluable; OR, overall response; PD, progressive disease; PR, partial response; PS, performance status; sIL‐2R, soluble interleukin‐2 receptor; T‐Bil, total bilirubin; ULN, upper limit of normal. Anemia: Hemoglobin <11 g/dL. Leukocytosis: White blood cell count ≥10 000/μL. Thrombocytopenia: Platelet count <16 × 10e4/μL.
With a median follow up of 39 months for surviving patients in all patients, PFS at 3 years was 38% in the SPIB positive group (95% confidence interval [CI], 18–58%) and 74% in the SPIB negative group (95% CI, 63–82%) (P < 0.001) (Fig. 2a). OS at 3 years was 59% in the SPIB positive group (95% CI, 32–78%) and 79% in the SPIB negative group (95% CI, 69–86%) (P = 0.024) (Fig. 2b). In patients with GCB type, PFS at 3 years was 17% in the SPIB positive group (95% confidence interval (CI), 1–48%) and 75% in the SPIB negative group (95% CI, 61–84%) with a median follow‐up duration of 39 months for surviving patients (P < 0.001) (Fig. 2c). OS at 3 years was 22% in the SPIB positive group (95% CI, 1–58%) and 85% in the SPIB negative group (95% CI, 68–94%) (P = 0.002) (Fig. 2d). In patients with non‐GCB type, PFS at 3 years was 56% in the SPIB positive group (95% confidence interval [CI], 23–79%) and 75% in the SPIB negative group (95% CI, 61–84%) with a median follow‐up duration of 40 months for surviving patients (P = 0.201) (Fig. 2e). OS at 3 years was 88% in the SPIB positive group (95% CI, 39–98%) and 77% in the SPIB negative group (95% CI, 64–86%) (P = 0.682) (Fig. 2f). Overall, the outcomes for PFS of patients with non‐GCB type DLBCL were significantly better than those with GCB type DLBCL (P = 0.013; Fig. S1)
Figure 2.
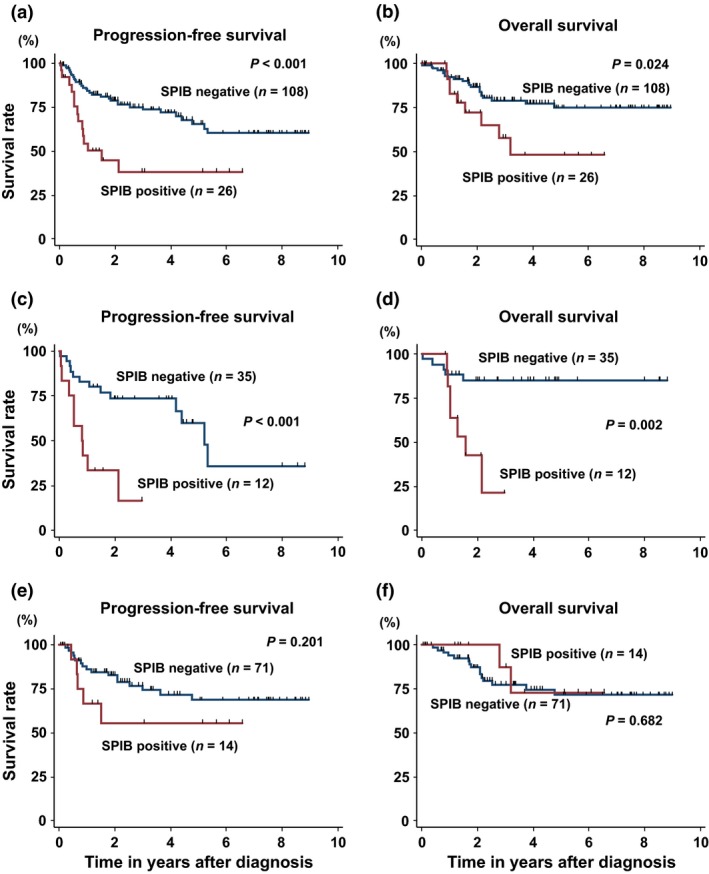
Patient survival and SPIB expression. Progression free survival (PFS) (a) and overall survival (OS) (b) according to SPIB expression in all patients (SPIB negative [n = 108] and SPIB positive [n = 26]). PFS (c) and OS (d) according to SPIB expression in patients with GCB type (SPIB negative [n = 35] and SPIB positive [n = 12]). PFS (e) and OS (f) according to SPIB expression in patients with non‐GCB type (SPIB negative [n = 71] and SPIB positive [n = 14]).
Prognostic factors
Prognostic factors for patient survival were evaluated by the Cox proportional hazard model. The univariate analyses for PFS and OS for all patients, and for patients with GCB type and non‐GCB type, are shown in Tables S2–S4, respectively. In all patients, multivariate analyses of prognostic factors revealed that SPIB positivity was an independent poor prognostic factor for PFS (HR 2.65, 95% CI 1.31–5.33, P = 0.006) and for OS (HR 3.56, 95% CI 1.43–8.91, P = 0.007). Intriguingly, male sex was a poor prognostic factor for both PFS and OS (PFS, HR 3.55, 95% CI 1.58–8.02, P = 0.002; OS, HR 10.5, 95% CI 2.36–46.2, P = 0.002). In patients with GCB type, SPIB was also an independent poor prognostic factor for PFS (HR 4.50, 95% CI 1.39–14.6, P = 0.012) and for OS (HR 18.9, 95% CI 3.59–99.5, P = 0.001). In contrast, SPIB was not identified as a poor prognostic factor in patients with non‐GCB type. Male sex was a poor prognostic factor for PFS and OS of non‐GCB type (PFS, HR 4.60, 95% CI 1.33–15.9, P = 0.016; OS, HR 10.2, 95% CI 1.31–78.8, P = 0.027) (Table 2).
Table 2.
Prognostic factors for PFS or OS
| Variable | All patients | GCB type | Non‐GCB type | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PFS | OS | PFS | OS | PFS | OS | ||||||||||||||
| HR | 95% CI | P | HR | 95% CI | P | HR | 95% CI | P | HR | 95% CI | P | HR | 95% CI | P | HR | 95% CI | P | ||
| Sex | Male | 3.55 | 1.58–8.02 | 0.002 | 10.5 | 2.36–46.2 | 0.002 | — | — | — | — | — | — | 4.60 | 1.33–15.9 | 0.016 | 10.2 | 1.31–78.8 | 0.027 |
| PS | >1 | 2.40 | 1.13–5.09 | 0.023 | 4.14 | 1.79–9.58 | 0.001 | — | — | — | 6.65 | 1.72–25.7 | 0.006 | — | — | — | 5.75 | 1.94–17.0 | 0.002 |
| LDH | >ULN | — | — | — | 3.09 | 1.21–7.89 | 0.018 | — | — | — | — | — | — | — | — | — | — | — | — |
| Clinical stage | >2 | — | — | — | — | — | — | 6.69 | 1.98—22.6 | 0.002 | — | — | — | — | — | — | — | — | — |
| Cell of origin | GCB type | 2.85 | 1.52–5.36 | 0.001 | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — |
| MYC/BCL2 | Positive | 2.02 | 1.03–3.99 | 0.041 | — | — | — | 4.66 | 1.37–15.9 | 0.014 | — | — | — | — | — | — | — | — | — |
| SPIB | Positive | 2.65 | 1.31–5.33 | 0.006 | 3.56 | 1.43–8.91 | 0.007 | 4.50 | 1.39–14.6 | 0.012 | 18.9 | 3.59–99.5 | 0.001 | — | — | — | — | — | — |
| sIL‐2R | ≥ 1000 U/mL | 2.70 | 1.33–5.45 | 0.006 | — | — | — | — | — | — | — | — | — | 5.50 | 1.82–16.6 | 0.002 | — | — | — |
| Serum urine acid | >8.0 mg/dL | — | — | — | — | — | — | 6.01 | 1.44–25.1 | 0.014 | 38.0 | 5.25–274.5 | <0.001 | — | — | — | — | — | — |
| ALP | >400 U/L | — | — | — | — | — | — | — | — | — | — | — | — | 9.84 | 2.81–34.4 | <0.001 | — | — | — |
| IgG | >1700 mg/dL | — | — | — | 2.73 | 1.15–6.50 | 0.023 | — | — | — | — | — | — | — | — | — | 4.40 | 1.62–12.0 | 0.004 |
CI, confidence interval; HR, hazard ratio; LDH, lactate dehydrogenase; OS, overall survival; PFS, progression free survival; PS, performance status; sIL‐2R, soluble interleukin‐2 receptor; ALP, alkarine phosphatase; ULN, upper limit of normal; non‐GCB type, non‐germinal center B‐cell type.
Development of an SPIB expression vector
The analysis of clinical outcomes suggested that SPIB expression was a poor prognostic factor in DLBCL, especially in GCB‐type DLBCL. To address this possibility in more detail and evaluate the biological significance of SPIB expression in DLBCL, we developed a retroviral SPIB expression vector, MIGR1‐FLAG‐SPIB (Fig. 3a). We confirmed the expression of Flag‐tagged SPIB by immunoblotting after transient transfection of 293T cells (Fig. 3b(i)). Next, we used the MIGR1‐FLAG‐SPIB retrovirus to transduce the SU‐DHL4 cell line, which has a relatively low endogenous expression of SPIB mRNA. Cells constitutively expressing GFP were purified by cell sorting, and the expression of Flag‐tagged SPIB was confirmed by immunoblotting (Fig. 3b(ii)). To confirm that the SPIB antibody used for IHC staining recognized SPIB, we assessed the immunofluorescence (IF) of MIGR1‐FLAG‐SPIB transfected 293T cells and retrovirally transduced SU‐DHL4 cells. As shown in Figure 3(c,d), 293T and SU‐DHL4 cells were clearly positive for SPIB expression by IF compared with the control 293T and SU‐DHL4 cells, which indicated that the antibody targeting SPIB appropriately recognized SPIB expression.
Figure 3.
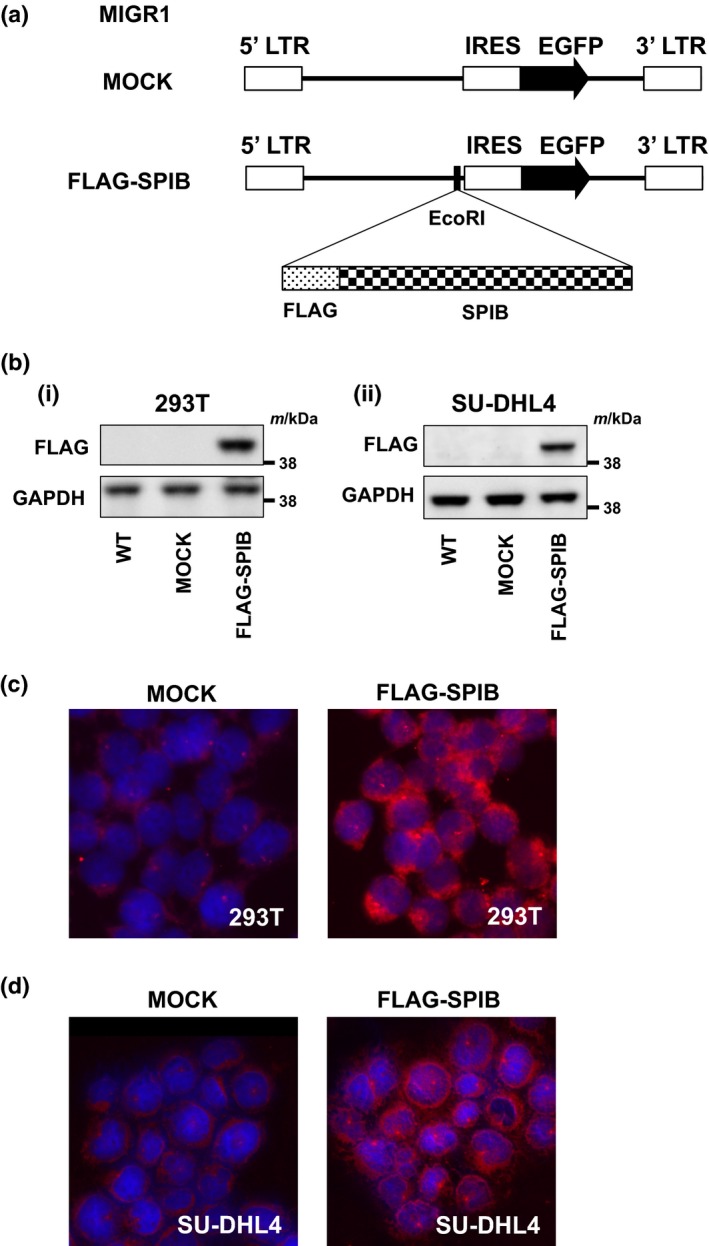
Development of a FLAG‐tagged SPIB expression vector. (a) Structure of the retroviral construct MIGR1. MIGR1‐FLAG‐SPIB was used for the retroviral transduction of the SU‐DHL4 cell line. (b) Immunoblotting analysis of MIGR1‐FLAG‐SPIB transiently transfected 293T cells and stably transduced SU‐DHL4 cells. Whole cell lysates were obtained 48 h after transfection of 293T cells with the MIGR1 construct indicated (i) or after retroviral transduction of SU‐DHL4 cells (ii). Immunoblotting for FLAG and glyceraldehyde phosphate dehydrogenase (GAPDH) as a loading control was performed. (c) Immunofluorescent analysis of SPIB expression in 293T cells. Cells were assessed 48 h after transfection of 293T cells with vector backbone (MOCK) (left) or FLAG‐SPIB (right). DAPI was used as a nuclear counterstain. (d) Immunofluorescent analysis of SPIB expression in SU‐DHL4 cells transduced with retroviral backbone (MOCK) (left) and FLAG‐SPIB (right). DAPI was used as a nuclear counterstain.
Effects of forced expression of SPIB on cell proliferation and the cell cycle
The proliferation of the MOCK and FLAG‐SPIB‐DHL4 cell lines cultured in 96‐well plates was evaluated every 24 h. The proliferation of MOCK cells was faster than that of FLAG‐SPIB‐DHL4 cells by the 96‐h time point (Fig. 4a). The cell cycle was not significantly different between the wild type (WT), MOCK and FLAG‐SPIB‐DHL4 cell lines (Fig. 4b).
Figure 4.
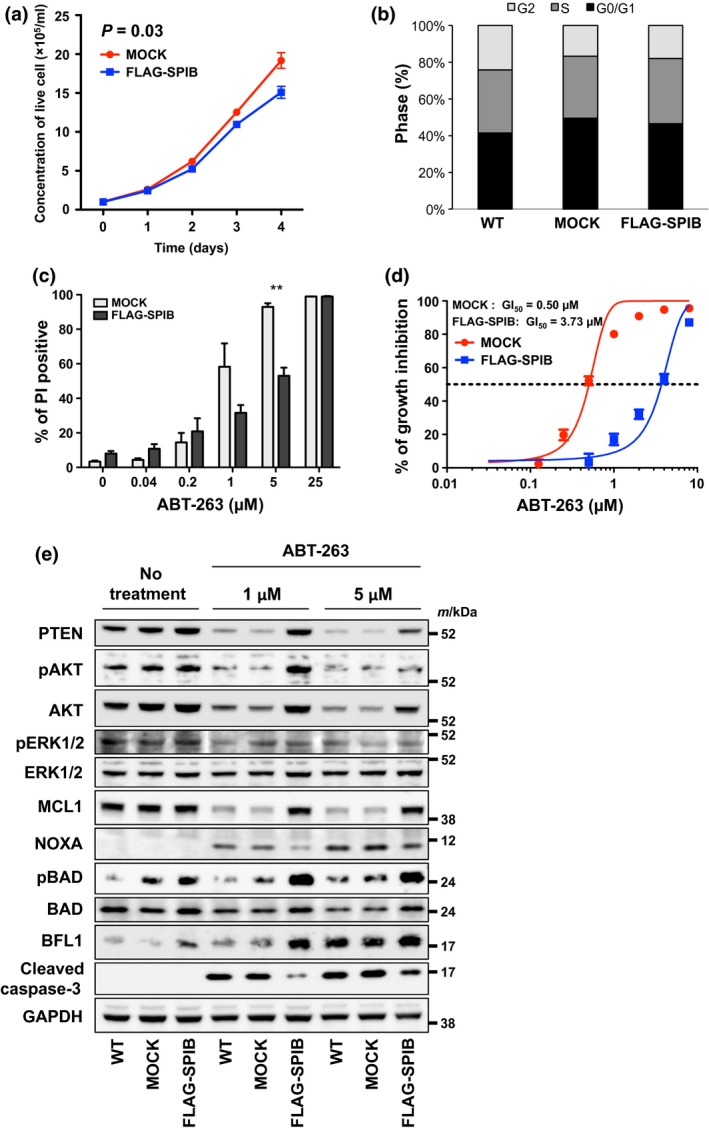
Forced expression of SPIB in SU‐DHL4 cells results in resistance to the BH3‐mimetic ABT‐263, via the AKT pathway. (a) The growth curves of SU‐DHL4 cells stably transduced with vector backbone (MOCK) (circles) and FLAG‐SPIB (squares) are shown. (b) Cell cycle analysis of control SU‐DHL4 cells transduced with the MIGR1 vector backbone. G0/G1 (black), S (grey) and G2 (pale grey) phases are indicated. (c) Sensitivity of stably transduced MOCK and FLAG‐SPIB SU‐DHL4 cells to ABT‐263. SU‐DHL4 cells transduced with MIGR1 were treated with various concentrations of ABT‐263 for 48 h, before analysis of cell death using flow cytometric propidium iodide (PI) assays. Asterisks indicate P‐values; **P < 0.01. Each point represents the mean value taken from three independent experiments with error bars indicating the SEM. (d) Inhibition of growth of stably transduced MOCK (red circles) and FLAG‐SPIB (blue squares) SU‐DHL4 cells by ABT‐263. Each point represents the mean value from three independent experiments with error bars indicating SEM. (e) Immunoblotting analysis of SU‐DHL4 cells stably transduced with the MIGR1 vector backbone. Whole cell lysates were obtained 6 h after treatment with 1 and 5 μM ABT‐263. Immunoblotting for PTEN, pAKT, AKT, pERK1/2, ERK1/2, MCL‐1, NOXA, pBAD, BAD, BFL1, Cleaved Caspase‐3 and GAPDH as a loading control were analyzed. All of the images except those of pERK1/2 and ERK1/2 were derived from the same membrane. The pERK1/2 and ERK1/2 images were taken from a separate membrane using the same cell lysate.
Forced SPIB expression and resistance to the BH3 mimetic, ABT‐263
As the treatment response was poor in the SPIB positive group, we next evaluated the effects of forced SPIB expression on susceptibilities to the standard anti‐cancer drugs used in the clinic. First, we analyzed the susceptibility of FLAG‐SPIB‐DHL4 cells to the chemotherapeutic drugs used in CHOP therapy, and found that these drugs killed the MOCK and FLAG‐SPIB‐DHL4 cells with equal efficiency (data not shown). Next, we evaluated their susceptibilities to molecular targeted drugs. In the presence of the BH3 mimetic, ABT‐263, the FLAG‐SPIB‐DHL4 cell line was more resistant to ABT‐263 mediated cell death than the MOCK cell line (Fig. 4c). The concentration of ABT‐263 required for 50% growth inhibition (GI50) in the FLAG‐SPIB‐DHL4 cell line (3.73 μM) was significantly higher than that required for the MOCK cell line (0.50 μM) (P = 0.015) (Fig. 4d). Subsequently, we analyzed the expression of various apoptotic proteins in the WT, MOCK and FLAG‐SPIB‐DHL4 cell lines in the presence or absence of ABT‐263. Compared to MOCK cells, in FLAG‐SPIB‐DHL4 cells treated with ABT‐263, the induction of cleaved caspase 3 was suppressed, and the expression of the anti‐apoptotic protein, MCL1, was maintained, which indicated that forced SPIB expression had an anti‐apoptotic effect (Fig. 4e). The expression of BCL‐xL, an ABT‐263 target, was similar to that of MCL1, while the expression of another ABT‐263 target, BCL2, did not differ among the WT, MOCK and FLAG‐SPIB‐DHL4 cell lines irrespective of the presence or absence of ABT‐263 (Fig. S2). In concert with the maintenance of MCL1 expression in the presence of ABT‐263 in FLAG‐SPIB‐DHL4 cells, the induction of pro‐apoptotic NOXA expression was suppressed. Next we analyzed the AKT and ERK cell survival pathways upstream of MCL1 and found that the expression of phosphorylated AKT (pAKT) was retained in FLAG‐SPIB‐DHL4 cells, consistent with AKT pathway activation. Subsequently, we also analyzed the expression of the pro‐apoptotic BAD protein that acts downstream from AKT. As anticipated, expression of phosphorylated BAD (pBAD) was retained in the FLAG‐SPIB‐DHL4 cell line. Moreover, the expression of the anti‐apoptotic protein, BFL1, was induced in the FLAG‐SPIB‐DHL4 cell line (Fig. 4e). These combined data indicated that SPIB expression had an anti‐apoptotic effect and acted via the AKT pathway.
Overcoming resistance to apoptosis through the inhibition of AKT phosphorylation
As SPIB expression is involved in anti‐apoptosis via the AKT pathway, we investigated whether SPIB induced anti‐apoptosis could be overcome through the inhibition of pAKT in the FLAG‐SPIB‐DHL4 cell line. In the presence of 5 μM ibrutinib, 25 μM idelalisib and 10 μM LY294002 without ABT‐263, limited cell death was observed. However, when these drugs at the same dosages were combined with >0.2 μM ABT‐263, cell death was clearly increased (Fig. 5a). In the reverse experiment, various doses of ibrutinib, idelalisib and LY294002 combined with 0.2 μM ABT‐263 increased cell death compared with cells treated with ibrutinib, idelalisib and LY294002 individually (Fig. 5b). To confirm that these combination effects were specifically associated with the inhibition of AKT, we investigated the phosphorylation status of AKT by immunoblotting in the presence or absence of ABT‐263 and ibrutinib, idelalisib and LY294002. As expected, phosphorylation of AKT and ERK decreased in the presence of high doses of ibrutinib, idelalisib and LY294002 individually or combined with ABT‐263. Consistent with the cell death induced by the combination of AKT inhibitors and ABT‐263, MCL1 was degraded in concert with the induction of NOXA expression and decreased expression of pBAD (Fig. 5c). Taken together these data indicate that inhibition of pAKT and pERK, upstream of MCL1, re‐sensitizes FLAG‐SPIB‐DHL4 cells to ABT‐263‐induced apoptosis. This effect also indicates that AKT is a potential therapeutic target in SPIB high expressing lymphoma cells.
Figure 5.
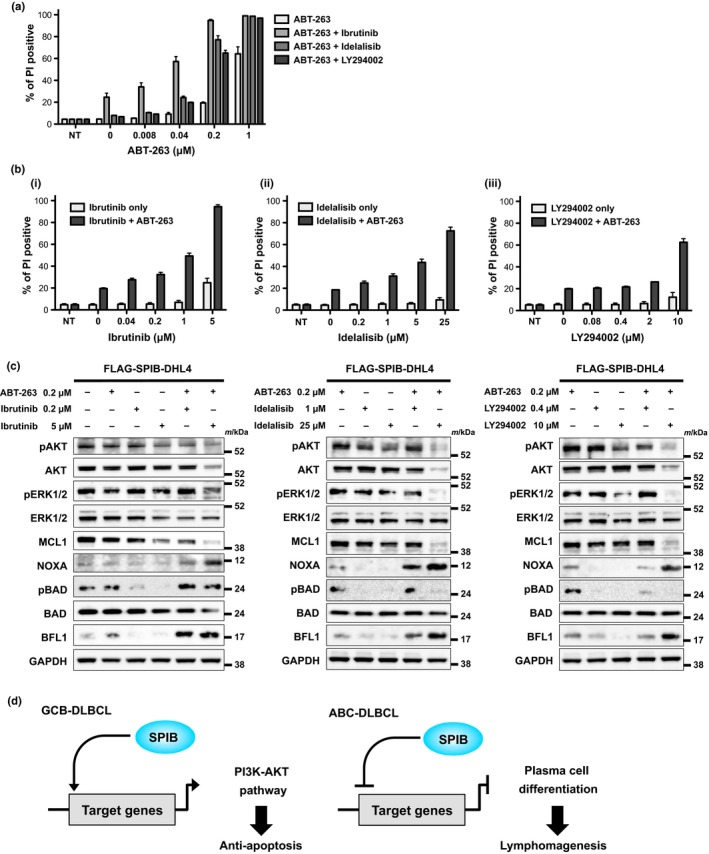
Resistance of stably transduced FLAG‐SPIB SU‐DHL4 cells to ABT‐263 is overcome by AKT inhibition. (a) FLAG‐SPIB SU‐DHL4 cells were treated with ABT‐263 combined with AKT inhibitors (ibrutinib, idelalisib and LY294002) for 48 h, before analysis of cell death as in Figure 4. Cell death observed using various concentrations of ABT‐263 with or without a constant dose of ibrutinib (5 μM), idelalisib (25 μM) and LY294002 (10 μM) is indicated. Each point represents the mean value taken from three independent experiments, with error bars indicating the SEM. (b) Cell death using various concentrations of ibrutinib (i), idelalisib (ii) and LY294002 (iii) with or without a constant dose of 0.2 μM ABT‐263 is indicated. Each point represents the mean value taken from three independent experiments, with the error bar indicating the SEM. (c) Immunoblotting analyses of FLAG‐SPIB SU‐DHL4 cells. Whole cell lysates were obtained 6 h after incubation with a constant dose of 0.2 μM ABT‐263 with or without various doses of ibrutinib (0.2 and 5 μM), idelalisib (1 and 25 μM) and LY294002 (0.4 and 10 μM). Immunoblotting for pAKT, AKT, pERK1/2, ERK1/2, MCL1, NOXA, pBAD, BAD, BFL1 and GAPDH as a loading control was performed. All of the images except those of pBAD and BAD in each panel were derived from the same membrane. The pBAD and BAD images were taken from a separate membrane using the same cell lysates. (d) The schema of the putative role of SPIB expression in the pathogenesis of GCB and ABC type DLBCL.
Discussion
In this analysis, we evaluated SPIB expression in DLBCL by routine immunohistochemical staining and demonstrated that SPIB expression was a novel poor prognostic factor and potential biomarker for DLBCL that is, at least in part, involved in apoptosis resistance via the PI3K‐AKT pathway.
We focused on 134 patients with DLBCL in a single institute. Overall, male sex, poor PS, elevated LDH, elevated IgG and SPIB expression were independent poor prognostic factors for OS, and male sex, poor PS, GCB type DLBCL, MYC/BCL2 co‐expression, SPIB expression and elevated sIL‐2R were poor prognostic factors for PFS. The fact that poor PS and MYC/BCL2 co‐expression were poor prognostic factors for survival is consistent with other studies.24, 25 However, the finding that male sex was the strongest poor prognostic factor for OS in all patients and in patients with non‐GCB type was unexpected and may reflect the potential difference in therapeutic efficacy of rituximab in men and women,26, 27 another as yet unidentified sex‐specific susceptibility factor, or a cohort‐specific statistical anomaly. These possibilities should be prospectively addressed in larger cohorts.
We detected SPIB expression by IHC in 19% of all patients, 26% of patients with GCB type and 16% of non‐GCB type, which is consistent with a previous report of chromosome 19q amplification in 15% of DLBCL patients.9 Moreover, patients with SPIB expression in all patients and in GCB type demonstrated poorer outcomes, whereas SPIB expression was not associated with poorer prognoses in patients with non‐GCB type, possibly due to the better outcomes of non‐GCB type DLBCL. In accordance with previous findings regarding the role of SPIB expression in lymphomagenesis in ABC type DLBCL, the present findings regarding the association of SPIB expression with clinical presentations and its anti‐apoptotic effect in GCB type DLBCL suggest that SPIB expression is linked to pathogenesis in both GCB and ABC type DLBCL. Intriguingly, a recent finding indicated that SPIB expression was associated with MYC/BCL2 double positive lymphoma,6 which is considered to be a poor prognostic phenotype;7, 24 however, SPIB was not linked to MYC/BCL2 co‐expression in our multivariate analysis. These combined findings might suggest that SPIB expression is linked to a poor prognostic phenotype through its anti‐apoptotic effect in GCB type DLBCL and to lymphomagenesis through the inhibition of plasma cell differentiation in ABC type DLBCL (Fig. 5d). Although we uncovered some aspects of the role of SPIB expression in DLBCL pathogenesis, SPIB may be involved in other processes linked to patient survival. Further investigation is warranted.
Forced expression of SPIB in tumor cells produced an anti‐apoptotic effect linked to the AKT‐BAD axis and MCL1 expression. Moreover, the inhibition of AKT phosphorylation overcame apoptosis resistance. These findings highlight a novel function of SPIB in the regulation of the AKT pathway. Considering that inhibition of AKT pathway signaling is an attractive molecular target, SPIB expression could be a useful surrogate marker for future combination therapy.
Although our data suggest that SPIB is a novel prognostic factor in DLBCL and the SPIB linked anti‐apoptotic effect might be a potential mechanism underlying the poor prognostic phenotype, it is important to acknowledge the limitations of our study. First, our cohort was derived from a single institute in a retrospective manner, raising the possibility that institutional and/or unrecognized bias might lead to an overestimation of the role of SPIB expression in DLBCL pathogenesis. Second, we did not detect resistance to anti‐chemotherapy drugs other than ABT‐263 in FLAG‐SPIB‐DHL4 cells. Nevertheless, our data contribute significantly to uncovering the role(s) of SPIB in DLBCL pathogenesis, including, importantly, an anti‐apoptotic function acting via the PI3K‐AKT pathway.
Disclosure Statement
H.K. has received research funding from FUJIFILM Corporation, Nippon Boehringer Ingelheim Co. Ltd., Chugai Pharmaceutical Co. Ltd., Kyowa‐Hakko Kirin Co. Ltd., Zenyaku Kogyo Company Ltd. and Dainippon Sumitomo Pharma Co. Ltd.
Supporting information
Fig. S1. progression free survival (PFS) and overall survival (OS) of all patients according to the cell of origin.
Fig. S2. The expression of apoptotic proteins in SU‐DHL4 cells transduced with MIGR1 vectors.
Table S1. Source of antibodies for immunoblotting.
Table S2. Prognostic factors for progression free survival (PFS) or overall survival (OS) in all patients.
Table S3. Prognostic factors for progression free survival (PFS) or overall survival (OS) in patients with GCB type.
Table S4. Prognostic factors for progression free survival (PFS) or overall survival (OS) in patients with non‐GCB type.
Acknowledgments
We would like to thank Mr Kuniyoshi Kitou, Ms Kazuko Matsuba, Ms Yuko Katayama and Mr Yoshiaki Inagaki (Nagoya University) for IHC work, Ms Yoko Matsuyama, Ms Chika Wakamatsu, Ms Manami Kira, Ms Rie Kojima, Ms Yukie Konishi, Ms Yuko Kojima and Ms Emi Kohno (Nagoya University) for assistance with laboratory work, Dr W. S. Pear (University of Pennsylvania) for providing the MIGR1 vector, and Dr Kunihiko Takeyama (Dana Farber Cancer Institute) for providing the SU‐DHL4 cell line. This work was supported by the Program to Disseminate Tenure Tracking System, MEXT, Japan, by a JSPS Grant‐in‐Aid for Young Scientists (B) 26860724 and by the Kanae Foundation for the Promotion of Medical Science grant to K.S., and by a JSPS Grant‐in‐Aid for Scientific Research (B) 25293218 grant to T.N.
Cancer Sci 107 (2016) 1270–1280
Funding Information
Program to Disseminate Tenure Tracking System, MEXT, Japan, JSPS Grant‐in‐Aid for Young Scientists (B) 26860724; Kanae Foundation for the Promotion of Medical Science; JSPS Grant‐in‐Aid for Scientific Research (B) 25293218; FUJIFILM Corporation, Nippon Boehringer Ingelheim Co. Ltd., Chugai Pharmaceutical Co. Ltd., Kyowa‐Hakko Kirin Co. Ltd., Zenyaku Kogyo Company Ltd.; Dainippon Sumitomo Pharma Co. Ltd.
References
- 1. Coiffier B, Thieblemont C, Van Den Neste E et al Long‐term outcome of patients in the LNH‐98.5 trial, the first randomized study comparing rituximab‐CHOP to standard CHOP chemotherapy in DLBCL patients: A study by the Groupe d'Etudes des Lymphomes de l'Adulte. Blood 2010; 116: 2040–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pfreundschuh M, Schubert J, Ziepert M et al Six versus eight cycles of bi‐weekly CHOP‐14 with or without rituximab in elderly patients with aggressive CD20+ B‐cell lymphomas: A randomised controlled trial (RICOVER‐60). Lancet Oncol 2008; 9: 105–16. [DOI] [PubMed] [Google Scholar]
- 3. Pfreundschuh M, Trumper L, Osterborg A et al CHOP‐like chemotherapy plus rituximab versus CHOP‐like chemotherapy alone in young patients with good‐prognosis diffuse large‐B‐cell lymphoma: A randomised controlled trial by the MabThera International Trial (MInT) Group. Lancet Oncol 2006; 7: 379–91. [DOI] [PubMed] [Google Scholar]
- 4. Oyama T, Yamamoto K, Asano N et al Age‐related EBV‐associated B‐cell lymphoproliferative disorders constitute a distinct clinicopathologic group: A study of 96 patients. Clin Cancer Res 2007; 13: 5124–32. [DOI] [PubMed] [Google Scholar]
- 5. Asano N, Yamamoto K, Tamaru J et al Age‐related Epstein–Barr virus (EBV)‐associated B‐cell lymphoproliferative disorders: Comparison with EBV‐positive classic Hodgkin lymphoma in elderly patients. Blood 2009; 113: 2629–36. [DOI] [PubMed] [Google Scholar]
- 6. Hu S, Xu‐Monette ZY, Tzankov A et al MYC/BCL2 protein coexpression contributes to the inferior survival of activated B‐cell subtype of diffuse large B‐cell lymphoma and demonstrates high‐risk gene expression signatures: A report from The International DLBCL Rituximab‐CHOP Consortium Program. Blood 2013; 121: 4021–31; quiz 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Green TM, Young KH, Visco C et al Immunohistochemical double‐hit score is a strong predictor of outcome in patients with diffuse large B‐cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 2012; 30: 3460–7. [DOI] [PubMed] [Google Scholar]
- 8. Rui L, Schmitz R, Ceribelli M, Staudt LM. Malignant pirates of the immune system. Nature Immunol 2011; 12: 933–40. [DOI] [PubMed] [Google Scholar]
- 9. Lenz G, Wright GW, Emre NC et al Molecular subtypes of diffuse large B‐cell lymphoma arise by distinct genetic pathways. Proc Nat Acad Sci U S A 2008; 105: 13520–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schmidlin H, Diehl SA, Nagasawa M et al Spi‐B inhibits human plasma cell differentiation by repressing BLIMP1 and XBP‐1 expression. Blood 2008; 112: 1804–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schotte R, Nagasawa M, Weijer K, Spits H, Blom B. The ETS transcription factor Spi‐B is required for human plasmacytoid dendritic cell development. J Exp Med 2004; 200: 1503–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sasaki I, Hoshino K, Sugiyama T et al Spi‐B is critical for plasmacytoid dendritic cell function and development. Blood 2012; 120: 4733–43. [DOI] [PubMed] [Google Scholar]
- 13. Pasqualucci L, Compagno M, Houldsworth J et al Inactivation of the PRDM1/BLIMP1 gene in diffuse large B cell lymphoma. J Exp Med 2006; 203: 311–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou Y, Liu X, Xu L et al Transcriptional repression of plasma cell differentiation is orchestrated by aberrant over‐expression of the ETS factor SPIB in Waldenstrom macroglobulinaemia. Br J Haematol 2014; 166: 677–89. [DOI] [PubMed] [Google Scholar]
- 15. Monti S, Chapuy B, Takeyama K et al Integrative analysis reveals an outcome‐associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B cell lymphoma. Cancer Cell 2012; 22: 359–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Novak AJ, Asmann YW, Maurer MJ et al Whole‐exome analysis reveals novel somatic genomic alterations associated with outcome in immunochemotherapy‐treated diffuse large B‐cell lymphoma. Blood Cancer J 2015; 5: e346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sung CO, Kim SC, Karnan S et al Genomic profiling combined with gene expression profiling in primary central nervous system lymphoma. Blood 2011; 117: 1291–300. [DOI] [PubMed] [Google Scholar]
- 18. Shimada K, Tomita A, Minami Y et al CML cells expressing the TEL/MDS1/EVI1 fusion are resistant to imatinib‐induced apoptosis through inhibition of BAD, but are resensitized with ABT‐737. Exp Hematology 2012; 40: 724–37.e2. [DOI] [PubMed] [Google Scholar]
- 19. Minami Y, Kiyoi H, Yamamoto Y et al Selective apoptosis of tandemly duplicated FLT3‐transformed leukemia cells by Hsp90 inhibitors. Leukemia 2002; 16: 1535–40. [DOI] [PubMed] [Google Scholar]
- 20. Cragg MS, Howatt WJ, Bloodworth L, Anderson VA, Morgan BP, Glennie MJ. Complement mediated cell death is associated with DNA fragmentation. Cell Death Diff 2000; 7: 48–58. [DOI] [PubMed] [Google Scholar]
- 21. Hiraga J, Tomita A, Sugimoto T et al Down‐regulation of CD20 expression in B‐cell lymphoma cells after treatment with rituximab‐containing combination chemotherapies: Its prevalence and clinical significance. Blood 2009; 113: 4885–93. [DOI] [PubMed] [Google Scholar]
- 22. Cheson BD, Pfistner B, Juweid ME et al Revised response criteria for malignant lymphoma. J Clin Oncol 2007; 25: 579–86. [DOI] [PubMed] [Google Scholar]
- 23. Hans CP, Weisenburger DD, Greiner TC et al Confirmation of the molecular classification of diffuse large B‐cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004; 103: 275–82. [DOI] [PubMed] [Google Scholar]
- 24. Sarkozy C, Traverse‐Glehen A, Coiffier B. Double‐hit and double‐protein‐expression lymphomas: aggressive and refractory lymphomas. Lancet Oncol 2015; 16: e555–67. [DOI] [PubMed] [Google Scholar]
- 25. Shipp MA, Harrington DP. A predictive model for aggressive non‐Hodgkin's lymphoma. The International Non‐Hodgkin's Lymphoma Prognostic Factors Project. N Engl J Med 1993; 329: 987–94. [DOI] [PubMed] [Google Scholar]
- 26. Witzens‐Harig M, Benner A, McClanahan F et al Rituximab maintenance improves survival in male patients with diffuse large B‐cell lymphoma. Results of the HD2002 prospective multicentre randomized phase III trial. Br J Haematol 2015; 171: 710–9. [DOI] [PubMed] [Google Scholar]
- 27. Pfreundschuh M, Muller C, Zeynalova S et al Suboptimal dosing of rituximab in male and female patients with DLBCL. Blood 2014; 123: 640–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. progression free survival (PFS) and overall survival (OS) of all patients according to the cell of origin.
Fig. S2. The expression of apoptotic proteins in SU‐DHL4 cells transduced with MIGR1 vectors.
Table S1. Source of antibodies for immunoblotting.
Table S2. Prognostic factors for progression free survival (PFS) or overall survival (OS) in all patients.
Table S3. Prognostic factors for progression free survival (PFS) or overall survival (OS) in patients with GCB type.
Table S4. Prognostic factors for progression free survival (PFS) or overall survival (OS) in patients with non‐GCB type.