

Abstract
miR-155 is an oncogenic microRNA (miR) that is often over-expressed in cancer and is associated with poor prognosis. miR-155 can target several DNA repair factors including RAD51, MLH1, and MSH6, and its over-expression results in an increased mutation frequency in vitro, although the mechanism has yet to be fully understood. Here, we demonstrate that over-expression of miR-155 drives an increased mutation frequency both in vitro and in vivo, promoting genomic instability by affecting multiple DNA repair pathways. miR-155 over-expression causes a decrease in homologous recombination, but yields a concurrent increase in the error-prone non-homologous end-joining (NHEJ) pathway. Despite repressing established targets MLH1 and MSH6, the identified mutation pattern upon miR-155 over-expression does not resemble that of a mismatch repair-deficient background. Further investigation revealed that all four subunits of polymerase delta, a high-fidelity DNA replication and repair polymerase, are down-regulated at the mRNA level in the context of miR-155 over-expression. FOXO3a, a transcription factor and known target of miR-155, has one or more putative binding site(s) in the promoter of all four polymerase delta subunits. Finally, suppression of FOXO3a by miR-155 or by siRNA knockdown is sufficient to repress the expression of the catalytic subunit of polymerase delta, POLD1, at the protein level, indicating that FOXO3a contributes to the regulation of polymerase delta levels.
Keywords: microRNA, DNA repair, mutagenesis, NHEJ, polymerase delta
Introduction
microRNAs have been described as master regulators of the genome. As such, their expression is tightly controlled in normal tissues. microRNAs bind to mRNA transcripts of target genes and promote translational repression. Cancer cells often hijack these translational regulators by modifying their expression to promote cell survival and tumorigenesis. Understanding the mechanism and function behind misregulated microRNAs will provide insight into the potential adjuvant manipulation of these RNA mediators to improve targeted cancer therapies.
It has been shown that several factors can influence the regulation of microRNA expression, most notably aspects of the tumor microenvironment, such as hypoxia and inflammation. These environmental changes employ transcription factors to aberrantly modulate gene expression and promote cell survival. Over-expression of several microRNAs has been observed in response to hypoxia, ultimately resulting in facilitation of hypoxic hallmarks such as therapy resistance, genomic instability, and the metabolic shift to glycolysis. One such miRNA, miR-155, is induced by hypoxia and subsequently promotes resistance to radiation therapy [1, 2].
miR-155 has been described as an oncomiR, an oncogenic microRNA, in part due to its association with poor prognosis in a variety of cancer types [3–5]. miR-155 over-expression was initially identified in B-cell lymphoma, but has since been associated with several other malignancies including lung, breast, and colon cancer [4–8]. Recently, it was demonstrated that miR-155 over-expression alone was sufficient to drive lymphoma in a mouse model [9]. Supporting its designation as an oncomiR, miR-155 has many tumor suppressor gene targets, including pro-apoptotic factors FOXO3a and TP53INP1 and negative regulators of proliferation SOCS1 and SHIP1 [10–13]. Further evidence suggests that miR-155 is also responsible for targeting several genes essential for maintaining genomic integrity including DNA repair factors MLH1 and RAD51 [14, 15]. The essential nature of these and many other miR-155 targets emphasizes the importance of tight miR-155 regulation in maintaining cellular homeostasis and inhibiting tumorigenesis.
DNA repair is fundamental to the maintenance of genomic integrity. Although both single and double-strand breaks (DSBs) are disruptive, DSBs are more toxic to a cell due to their propensity for genomic instability and cell death. DSB repair consists of two main pathways, non-homologous end-joining (NHEJ) and homologous recombination (HR). Although these pathways work in concert with one another, NHEJ is the more frequently used yet more error-prone of the two pathways. Inherently, HR occurs only during S-phase and early G2-phase due to the necessity of a homologous template. In cancer, DSB repair is a priority, regardless of fidelity, to assure cell survival. As such, an even more mutagenic version of end joining, microhomology-mediated end joining (MMEJ), is often favored by tumors. MMEJ utilizes small nearby regions of homology to repair a break, always resulting in deletions. Although many mechanisms are likely involved in determining which DSB repair pathway to use, microRNAs may play an indirect role in regulating the activity of one pathway over the other to promote cell survival. miR-155 has been shown to be involved in the regulation of DSB repair factor, RAD51 [14], which identifies it as an important candidate for studying the role of miRNAs in modulating NHEJ and HR activity. In the context of miR-155 over-expression, the balance between HR and error-prone NHEJ has yet to be examined.
In addition to DSB repair, miR-155 also plays a role in other repair pathways. Mismatch repair deficiency due to miR-155 over-expression results in microsatellite instability in vitro [15]. Additionally, it has been shown that inflammation-induced expression of miR-155 leads to an increase in mutation frequency in vitro as measured by HPRT [16]. Although this evidence is compelling, this phenomenon has yet to be explored in vivo and the mechanism behind the observed increase in mutation is not fully understood. We set out to determine whether miR-155 over-expression in vivo results in genomic instability using a mouse model system. Additionally, we hypothesized that other mechanisms beyond deficiency in mismatch repair might be responsible for the effect of miR-155 over-expression on genomic instability.
We report here that over-expression of miR-155 is sufficient to increase mutation frequency both in cells in culture and in vivo in a mouse model system. Upon over-expression of miR-155, we also find increased NHEJ activity and decreased HR activity, a pattern that is in keeping with an increase in mutation frequency based on the relative fidelity of these pathways. In addition to observing suppression of RAD51 and MLH1 in our system, we also analyzed global changes in mRNA levels by microarray in the presence of miR-155 over-expression to determine what other DNA repair mechanisms might be affected. We found that miR-155 over-expression leads to transcriptional repression of all four subunits of polymerase delta, a high-fidelity DNA repair polymerase. Interestingly, an established miR-155 target, FOXO3a, is a transcription factor with putative binding sites in the promoters of each of the four polymerase delta subunits, and we show that knocking-down FOXO3a leads to a suppression of POLD1 expression at the protein level. Taken together, the results suggest that miR-155 down-regulates polymerase delta by targeting the transcription factor FOXO3a, thereby inhibiting the transcription of the four polymerase delta subunits, resulting in an increased susceptibility to mutation by suppressing a high-fidelity polymerase and favoring error prone translesion synthesis.
Materials & Methods
Cells
AV16 mouse epithelial cells harboring two mutation reporter transgenes, λsupFG1 and cII, were derived as previously described in a C127 cell background [17]. BICKO and 155KI MEFs were derived from mice at embryonic day 13. AV16 cells were cultured in DMEM medium + 10% FBS + 0.8 mg/mL G418; BICKO mouse embryonic fibroblast (MEF) cells were cultured in DMEM medium + 15% FBS + 1X Pen/Strep. The xrs6 Chinese hamster ovary cells are deficient in Ku80 due to splice site mutation, and the xrs6+Ku80 cells are derived from stable transfection of hamster wild-type XRCC5 cDNA [18]. Both xrs6 and xrs6+Ku80 cells were cultured in Ham’s F12 medium + 10% FBS + 1X Pen/Strep. DLD1 and DLD1 BRCA2 −/− (Horizon Discovery, UK; HD 105-007) were cultured in McCoys 5A medium + 10%FBS + 1X Pen/Strep. BRCA2 deficient PEO1 and BRCA2 proficient PEO1 C4-2 were gifts from T. Taniguchi [19] and were cultured in DMEM medium +10% FBS + 1X Pen/Strep. U20S EJ-DR cells, which have been previously described [20], contain the DR-GFP chromosomal reporter for HR and the EJ-RFP chromosomal reporter for mutagenic end-joining and were cultured in DMEM medium + 10% tetracycline-free FBS and 1X Pen/Strep. The U2OS DR-GFP cells have been previously described [21] and were cultured in DMEM medium + 15% FBS + 1X Pen/Strep. AV16, BICKO MEFs, and 155KI MEFs are all fibroblasts directly isolated from transgenic mice. Presence of the expected transgene or knock in genotype was confirmed by PCR of genomic DNA. All other cell lines were obtained from Horizon Discovery (validation, SNP 6.0, and Sanger sequencing) or ATCC (validation, COI profiling, and STR profiling).
In vitro microRNA over-expression
Human shMIMIC miRNA lentiviral particles expressing pre-miR-155 (155) or a non-targeting control (NTC) along with GFP were purchased from GE Dharmacon (Lafayette, CO, USA; originally VSH5841-10120825, miRIDIAN and HMR5872; now VSH6185-202567165, SMARTchoice and S-005000-01). Briefly, AV16 cells were transduced with lentiviral particles at 20 MOI for 48 hours and pools were selected using puromycin after GFP expression appeared. Over-expression was confirmed by qRT-PCR.
Animals
miR-155 knock-in (NesCre8; miR-155LSLtTA) mice (generously provided by Frank Slack, Yale University, New Haven, CT, USA) were generated in a C57B6/129 hybrid background as previously described [9]. Briefly, pre-miR-155 was cloned into the ROSA26 locus behind a lox-stop-lox sequence along with a tTA, Tet-responsive element. This construct was used to generate a transgenic mouse model. Subsequently, these mice were crossed with NesCre8 FVB/NJ mice to initiate miR-155 expression in the lymph and nervous system tissues, where Nestin-driven Cre is expressed [9]. Breeding animals were kept on a doxycycline food diet to inhibit miR-155 expression in these tissues as it has been shown to induce a lymphoproliferative disease [9]. NesCre8; miR-155LSLtTA animals were crossed into another transgenic mouse line carrying the mutation reporter genes λsupFG1 and cII as previously described [22, 23]. The resulting animals (155KI) were genotyped for NesCre8, miR-155LSLtTA, and λsupFG1 as previously described [9, 24]. 155KI and BICKO animals [25] (provided by Frank Slack, Yale University, New Haven, CT, USA) were utilized to generate mouse embryonic fibroblasts (MEFs).
Western blot
AV16 or BICKO cells were collected at 80% confluence by rinsing with phosphate buffered saline (PBS) and scraping on ice. Protein was extracted using AZ lysis buffer (50 mM Tris pH 8, 250 mM NaCl, 1% Igepal, 0.1% SDS, 5 mM EDTA, 10 mM Na4P2O7, 10 mM NaF) plus 1X protease inhibitor cocktail. 50 μg total protein was loaded and size fractionated on a 4–15% SDS-polyacrylamide gel and transferred to a nitrocellulose membrane. Antibodies used: MLH1, RAD51, POLD1, FOXO3a, BRCA2, XRCC4, Vinculin, β-actin, Tubulin. Antibodies were as follows: mouse monoclonal anti-α-tubulin (Sigma-Aldrich; St. Louis, MO, USA; 1:10 000; B-5-1-2); rabbit monoclonal anti-FOXO3a (Cell Signaling Technology; Beverly, MA, USA; 1:1 000; 2497); rabbit polyclonal anti-RAD51 (Santa Cruz Biotechnology; Dallas, TX, USA; 1:500; sc-8349); mouse monoclonal anti-MLH1 (BD Biosciences; San Jose, CA, USA; 1:500; 554073); rabbit polyclonal anti-POLD1 (Santa Cruz Biotechnology; 1:200; sc-10784); mouse monoclonal anti-β-actin (Santa Cruz Biotechnology; 1:1 000; sc-47778); rabbit polyclonal anti-BRCA2 (Santa Cruz Biotechnology; 1:1 000; sc8326); mouse monoclonal anti-XRCC4 (BD Transduction Laboratories, 1:1 000; 611506); mouse monoclonal anti-Vinculin (Abcam, 1:5 000, ab18058). Primary antibodies were incubated for 2–3 hours at room temperature or overnight at 4°C. Secondary goat-anti-mouse or goat-anti-rabbit antibodies (Thermo Fisher Scientific/Pierce; Rockford, IL, USA) were used at a 1:5 000 dilution for 1 hour at room temperature. Primary and secondary antibodies were prepared in 5% milk. TBST washes were performed after primary incubation and after secondary incubation. Membranes were developed in SuperSignal® West Pico Chemiluminescent Substrate (Thermo Fisher Scientific). Western blot quantification was performed using ImageJ software (NIH; Bethesda, MD, USA), comparing the intensity of the bands of interest with the intensity of loading controls. Values are reported as a fold change in intensity compared to the control sample, which is normalized to 1.
qRT-PCR
RNA was extracted from tissue culture or from organs of NesCr8; miR-155LSLtTA mice using the mirVana miRNA Isolation kit (Applied Biosystems/Life Technologies; Grand Island, NY, USA). Frozen organs were ground to a powder in liquid nitrogen and then lysed according to the manufacturer’s protocol. cDNA was synthesized from total RNA using the Taqman miRNA High-Capacity cDNA Reverse Transcription kit (Applied Biosystems/Life Technologies) with primers specific to hsa-miR-155, mmu-miR-155, and RNU6B or snoRNA202, endogenous controls for human or mouse, respectively. qRT-PCR was performed according to the manufacturer’s protocol using the Taqman microRNA PCR system (Applied Biosystems/Life Technologies) as previously described [26]. Briefly, cDNA was combined with Taqman Universal PCR Master Mix and Taqman probes specific for hsa-miR-155, mmu-miR-155, hsa- miR-128, and RNU6B or snoRNA202 (Applied Biosystems/Life Technologies). qRT-PCR was performed in 96-well optical plates. For analysis of POLD1, POLD2, POLD3 and POLD4 mRNA levels, RNA was extracted using the RNeasy kit (Qiagen) with on column DNAse treatment. cDNA synthesis was carried out using the High Capacity cDNA reverse transcriptase kit (Applied Biosystems/Life Technologies). cDNA was combined with primers and Power SYBR Green PCR Master mix (Applied Biosystems/Life Technologies). Plates were spun down prior to analysis. miRNA Ct values were normalized to RNU6B or snoRNA202 Ct values and relative expression was calculated using the −ΔΔCt method and reported with standard error for technical triplicate samples.
λsupFG1 shuttle vector rescue and mutagenesis assay
High molecular weight DNA was isolated from organs or cells of interest and screened for mutation frequency as previously described [24]. Briefly, DNA was isolated from spleens and brains of miR-155/Nestin-Cre knock-in animals using Phenol: Chloroform extraction. Isolated DNA was packaged into λ in vitro bacterial packaging extracts to rescue phage vectors. Packaging reactions were subsequently plated on LB Agar or TB Agar plates and incubated overnight at 37°C, or 24°C, respectively for λsupFG1 or cII mutation analysis. Mutants were either screened by color (clear = mutants, blue = wild-type) or growth at 24°C for λsupFG1 or cII mutation reporter genes, respectively. Mutation frequencies were determined based on the number of mutants identified per total number of plaques. A total of 50 000 plaques were screened per sample or animal. Statistical significance was determined using unpaired t-test (GraphPad Prism; La Jolla, CA, USA).
Mutant sequencing
Mutant plaques were isolated from LB Agar plates, PCR amplified using λsupFG1 primers [23], purified, and sent to Yale’s Keck Facility for sequencing. Mutation spectrum was determined based on the mutants identified by sequencing.
HR and NHEJ luciferase assays
The HR luciferase reporter, modeled after a similar assay described by the Samson lab [27], was constructed by cloning an inactivating I-SceI recognition site into the BstBI site 56 amino acids into the firefly luciferase gene in the gWIZ.Luciferase vector (Gelantis), and cloning a promoterless copy of the firefly luciferase open reading frame 700 base pairs downstream in reverse orientation as a donor template for homologous recombination. A DSB in the firefly luciferase gene was induced by I-SceI digestion and confirmed by electrophoresis. Linearized plasmid was transfected into cells to measure HR as a function of luciferase activity (firefly luciferase activity can only be restored by HR, which removes the inactivating I-SceI site). Renilla luciferase activity, expressed from pCMV-RL (Promega) transfected at a 1:20 ratio, was used as a transfection efficiency control. 48 hours after transfection, luciferase activity from the HR reporter was normalized to gWIZ.Luciferase to calculate HR activity as percent reactivation.
The NHEJ luciferase assay is well characterized [28–32]. Briefly, a HindIII-mediated DSB is induced between the promoter and the coding region of the firefly luciferase gene of pGL3 (Promega) and confirmed by electrophoresis. Linearized plasmid is transfected and repair of this double strand break by NHEJ restores firefly luciferase activity. NHEJ activity is determined by firefly luciferase activity measured 24 hours after transfection and normalized to a Renilla luciferase transfection control. Percent reactivation is determined by normalization of luciferase activity to the undamaged pGL3-Control vector. All luciferase reporter assays were performed in 12-well format by seeding 5×104 cells per well 24 hours before transfection and transfecting 1μg of reporter or positive control vector and 50ng Renilla luciferase vector per well. Luciferase activity was measured using the Dual Luciferase Reporter Assay System (Promega) for all samples. Statistical significance was determined by biological triplicate.
Chromosomal GFP DSB reporter assays
U2OS DR-GFP reporter cell lines were transfected in triplicate with pre-miR-control (Ambion), pre-miR-155 (Ambion), or siRNA (BRCA2: Ambion s2085; RAD51: GE Dharmacon ON-TARGETplus SMART pool; XRCC4: GE:Dharmacon ON-TARGETplus SMART pool) to a final concentration of 20nM using an Amaxa Nucleofector (Lonza). SCE-I expression plasmid (4 ug) was transfected into 1×106 cells per replicate along with a second dose of siRNA to a final concentration of 20nM 72 hours before analysis. Cells were analyzed for GFP expression by flow cytometry and data was analyzed using the FlowJo software (Tree Star Inc.).
Mutagenic end-joining reporter assay
The Mutagenic end-joining assay in the U2OS DR-EJ cells is well characterized and reporter assays were carried out as previously described [20, 33]. Briefly, U2OS DR-EJ cells were transfected to a final concentration of 20nm with pre-miR-control (Ambion) or pre-miR-155 (Ambion) using the Amaxa Nucleofector (Lonza) in triplicate. 72 hours after transfection, cells were exposed to media containing the Shield1 and Triamcinolone Acetonide (TA) ligands at concentrations of 1 μM and 100 nM, respectively, for 24 hours to induce a DSB in the RFP reporter. 72 hours after the induction of the DSB, the percentage of RFP positive cells was analyzed by flow cytometry, and data was analyzed using the FlowJo software (Tree Star Inc.). The data was normalized to the non-targeting pre-miR-control.
MMEJ host cell reactivation assays
Extrachromosomal MMEJ assays using pEJ2 [34] have been previously described [35]. Briefly, pEJ2 was linearized with I-SceI digestion and transfected into 5×104 cells seeded 24 hours before transfection at a ratio of 1ug per well. In parallel, cells were transfected using the same conditions with a positive control GFP expression vector pMAXGFP (Lonza) and a transfection efficiency control pDS-Red-Express-N1 (Clontech Laboratories). Fluorescence was quantified by flow cytometry 48 hours after transfection, and data were analyzed using FlowJo software (Tree Star Inc.) For each sample, GFP signal was normalized to RFP transfection efficiency and then normalized to the positive control to calculate percent reactivation. These cells were transfected in triplicate, and significance was calculated using an unpaired t-test.
Microarray
RNA was isolated and purified from AV16 NTC or 155 or from BICKO +/+ or −/− cells in biological and technical triplicate using the RNeasy and QIAshredder kits (Qiagen; Venlo, Limburg, Netherlands). RNA was run on an Illumina (San Diego, CA, USA) MouseWG- 6 V2 BeadChip Microarray Kit by the Yale Center for Genome Analysis (YCGA; New Haven, CT, USA). Analysis was performed to determine genes with expression changes greater than 20% with a p-value of less than 0.01, which were depicted in a heat map.
siRNA knockdown
AV16 or U2OS-DR-GFP cells were seeded to 70% confluence. FOXO3a siRNA (GE Dharmacon; siGENOME mouse FOXO3 (56484) SMARTpool siRNA) was transfected using Dharmafect 1 (GE Dharmacon) at 100 nM final concentration according to the manufacturer’s protocol. BRCA2, RAD51, and XRCC4 siRNA (BRCA2: Ambion s2085; RAD51: GE:Dharmacon ON-TARGETplus SMART pool; XRCC4: GE:Dharmacon ON-TARGETplus SMART pool) were transfected to a final concentration of 20 nM according using the Amaxa Nucleofector (Lonza) to the manufacturer’s protocol. Cells were collected at several time points following transfection and processed for Western blot.
Comet assay
AV16 NTC, 155 BICKO +/+, or −/− cells were collected in the absence of treatment and examined by Comet assay (Trevigen; Gaithersburg, MD, USA) per manufacturer’s protocol, as previously described [36]. Comet slides were electrophoresed for 45 minutes and imaged on an EVOS® FL cell imaging microscope (Life Technologies). Comets were analyzed using CometScore™ software (TriTek Corp.; Summerduck, VA, USA), and at least 100 cells were scored for each sample. Data are presented as a mean of tail moment with standard error.
Results
miR-155 over-expression in vitro leads to an increase in mutation frequency
miR-155 over-expression has been shown to cause an increase in mutation frequency in vitro [16], but this phenomenon has yet to be examined in vivo. We decided to use a transgenic mouse reporter gene to assess the in vivo effect of miR-155 on mutagenesis in an animal model. To establish feasibility, we first tested for a detectable effect of miR-155 over-expression in cells in culture using the same reporter system as in the transgenic mice. For this, we generated a stable mouse cell line over-expressing miR-155 in a previously derived mouse epithelial cell line, AV16, carrying both cII and λsupFG1 mutation reporter transgenes [17]. Cells were infected with an shMIMIC lentiviral construct expressing pre-miR-155 (155) or a non-targeting control (NTC). Cells incorporating the construct were selected and pooled by treatment with puromycin. Cells expressing the miR-155 construct had a significant increase in miR-155 expression as measured by qRT-PCR relative to NTC cells (Figure 1A).
Figure 1. miR-155 over-expression leads to an increase in mutation frequency in vitro and suppression of DNA repair factors.
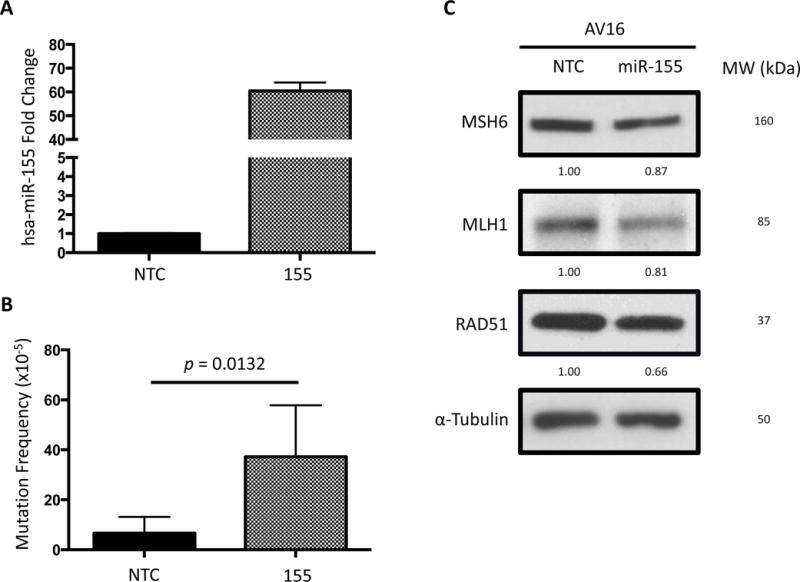
(A) miR-155 over-expression in AV16 mouse epithelial cells by lentiviral infection is confirmed by qRT-PCR. (B) Mutation frequency is measured using a cII mutation reporter gene in the presence or absence of miR-155 over-expression. Error bars are calculated based on standard error of the mean and p-value by unpaired t-test (n=5). (C) RAD51, MLH1, and MSH6 Western blots measure translational repression by miR-155 over-expression. Quantifications are normalized to the NTC bands.
AV16 NTC and 155 cells were collected, DNA was isolated, and mutation frequency was analyzed to determine mutation frequency in vitro. Because the reporter genes are carried in a recoverable lambda phage-based shuttle vector, incubation of DNA with bacterial packaging extracts allowed for a temperature-dependent mutation screen in indicator bacteria. We determined that over-expression of pre-miR-155 in AV16 cells resulted in a two-fold increase in mutation frequency relative to the AV16 NTC cells (Figure 1B). This demonstrates that miR-155 over-expression drives an increase in mutation frequency in this reporter system.
Since several DNA repair factors have been identified as bona fide miR-155 targets including HR facilitator RAD51 and mismatch repair factor MLH1, we set out to confirm their repression upon over-expression of miR-155 in the AV16 cells. Both of these targets were suppressed at the protein level by Western blot in this system (Figure 1C), which could contribute to the observed increase in mutation frequency, in keeping with previous reports [15, 16].
miR-155 over-expression increases mutation frequency in vivo
Once miR-155 driven mutagenesis was confirmed in our in vitro system, we examined the role of miR-155 in maintaining genomic integrity in vivo. We utilized a conditional miR-155 knock-in mouse model. Briefly, a “tet-off” tTA-miR-155 sequence was cloned into the ROSA26 locus behind a loxP-floxed stop cassette, and mice were generated in a B6 background. These mice, miR-155LSLtTA, were crossed with a Nestin-driven Cre mouse model to generate a tetracycline-regulated Nestin-Cre loxP miR-155 knock-in mouse model (NesCre8; miR-155LSLtTA) [9]. Nestin-Cre allows for spatially controlled expression of miR-155 in tissues where Nestin is constitutively expressed, which includes lymph and nervous system tissues. We crossed these animals with a transgenic mutation reporter mouse model (AV) carrying the cII and λsupFG1 reporter genes [37]. Resulting animals (155KI) were genotyped for NesCre8, miR-155LSLtTA, and λsupFG1 and were raised in the absence of doxycycline, enabling the over-expression of miR-155 in Nestin-expressing tissues. At 8 weeks of age, animals were sacrificed and tissues were harvested. This time point was chosen for humane reasons as these animals typically develop severely debilitating lymphoma beginning around 10 weeks of age [9].
To confirm miR-155 over-expression in specific organs, we isolated RNA from spleen and brain tissue of control (WT) or 155KI animals and analyzed miR-155 expression by qRT-PCR. As expected, both spleen and brain displayed significant over-expression of miR-155 (Figure 2A, B), correlating with a Nestin-driven expression pattern. We chose to analyze the spleen due to the infiltration of lymphocytes, as previously reported, resulting in enlarged spleen size (Figure 2C) [9]. The brain was chosen to confirm findings in another tissue in the absence of pathological response to miR-155 over-expression.
Figure 2. miR-155 over-expression in vivo leads to an increase in mutation frequency.
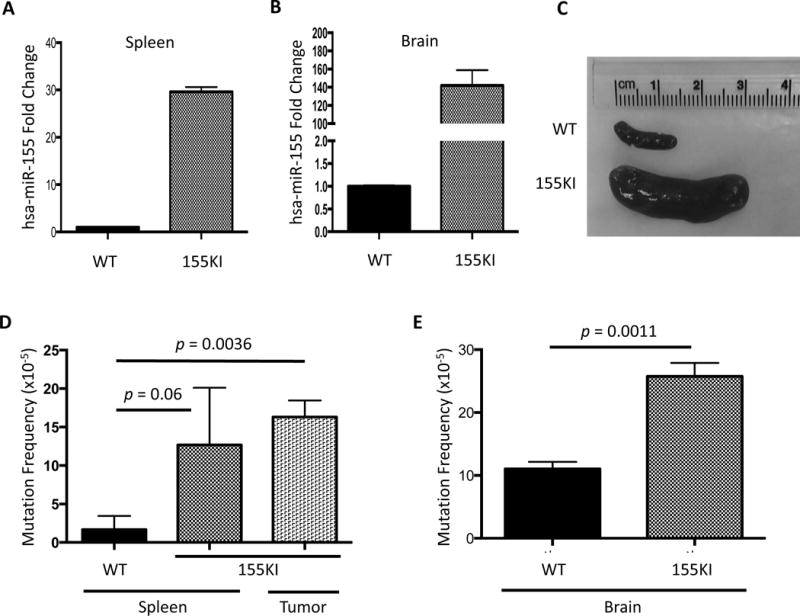
miR-155 over-expression was confirmed by qRT-PCR in the (A) spleen and (B) brain. (C) Representative images of spleens from WT and 155KI animals prior to DNA isolation. (D) Mutation frequency is measured by supFG1 mutation reporter gene in the presence or absence of miR-155 over-expression in the spleen or in lymphadenopathy tissue (lymphoma), or (E) brain at 8 weeks of age. In both D and E, error bars are calculated based on standard error of the mean and p-value is calculated by unpaired t-test (WT spleen: n=3; 155KI spleen: n=4; 155KI tumor: n=2; WT brain: n=4; 155KI brain: n=3).
To determine whether there was an in vivo effect on mutation frequency, we utilized a λsupFG1 mutation reporter assay. DNA was isolated from spleen tissue as well as lymphadenopathy (tumor) tissue and analyzed for the frequency of mutation in the presence or absence of miR-155 over-expression. We utilized a blue/white bacterial plaque screen to quantify the mutation frequency in the λsupFG1 gene. We found that 155KI mice had at least a six-fold increase in mutation frequency in the spleen and representative tumors at the 8-week time point (Figure 2D).
Importantly, we analyzed the mutation frequency in brain tissue as well, where Nestin is highly expressed, to test whether the increase in mutation frequency could be directly attributed to the over-expression of miR-155 and rather than a secondary mutagenic effect that might accompany the induced lymphomagenesis. Indeed, in brain tissue, where no significant pathology is observed [9], we detected a two-fold increase in mutation frequency in 155KI mice when compared to controls (Figure 2E). It is important to note that the baseline mutation frequency in the 8-week old brain is roughly five-fold higher than in the spleen. Additional work is necessary to conclusively determine the source of this difference; however, it is possible that due to the highly metabolic nature of the nervous tissue, a higher level of oxidative stress is observed, accounting for the difference in baseline mutation. In fact, it has been demonstrated that higher levels of oxidative base damage are observed at baseline in the rodent brain compared to the spleen [38–40]. Nonetheless, these data suggest that the over-expression of miR-155 leads to increased mutagenesis in vivo.
We next evaluated whether the miR-155-induced mutations showed a pattern consistent with deficient mismatch repair. We analyzed the sequences of the mutations observed within the λsupFG1 reporter gene. A hallmark of mismatch repair deficiency is an increase in single-nucleotide insertion/deletion mutations [22]. In an analysis of λsupFG1 gene mutations from spleen samples, we did not see a significant shift in mutation pattern between the control and 155KI spleen mutations (Figure S1), suggesting that suppression of another mechanism of DNA repair other than mismatch repair is likely playing a greater role in generating mutation than mismatch repair deficiency. This result is further supported by analysis of the miR-155 induced mutations in the HPRT gene in vitro observed by Croce and colleagues [16]. It is important to note that the λsupFG1 mutation assay is highly sensitive to mismatch repair deficiency and readily reports small insertion and deletion mutations (due to runs of G:C base pairs in the supFG1 gene), but it will inherently underestimate the level of large deletion mutagenesis induced by DSBs because large deletions inactivate the λ shuttle vector and so cannot be counted. Therefore, we analyzed DSBs by Comet assay as well, described later, to explore the hypothesis that miR-155 over-expression might be altering DSB repair and contributing to the observed increase in mutation.
miR-155 over-expression drives increased NHEJ activity and decreased HR activity
DSB repair can be divided into two main pathways, NHEJ and HR, both of which help to maintain genomic integrity. Pathway choice between HR and NHEJ is a complicated and not entirely understood process. NHEJ is the simpler of the two pathways and can take place in the absence of a homologous template, but it is also the more error-prone, occasionally resulting in deleterious mutations. Since miR-155 has been shown to target factors associated with DSB repair, we analyzed the level of baseline double strand breaks by neutral comet assay in AV16 155 cells compared to NTC cells. We found that over-expression of miR-155 resulted in a small, but statistically significant decrease in DSBs (Figure 3A).
Figure 3. Double-strand break repair activity correlates with miR-155 expression.
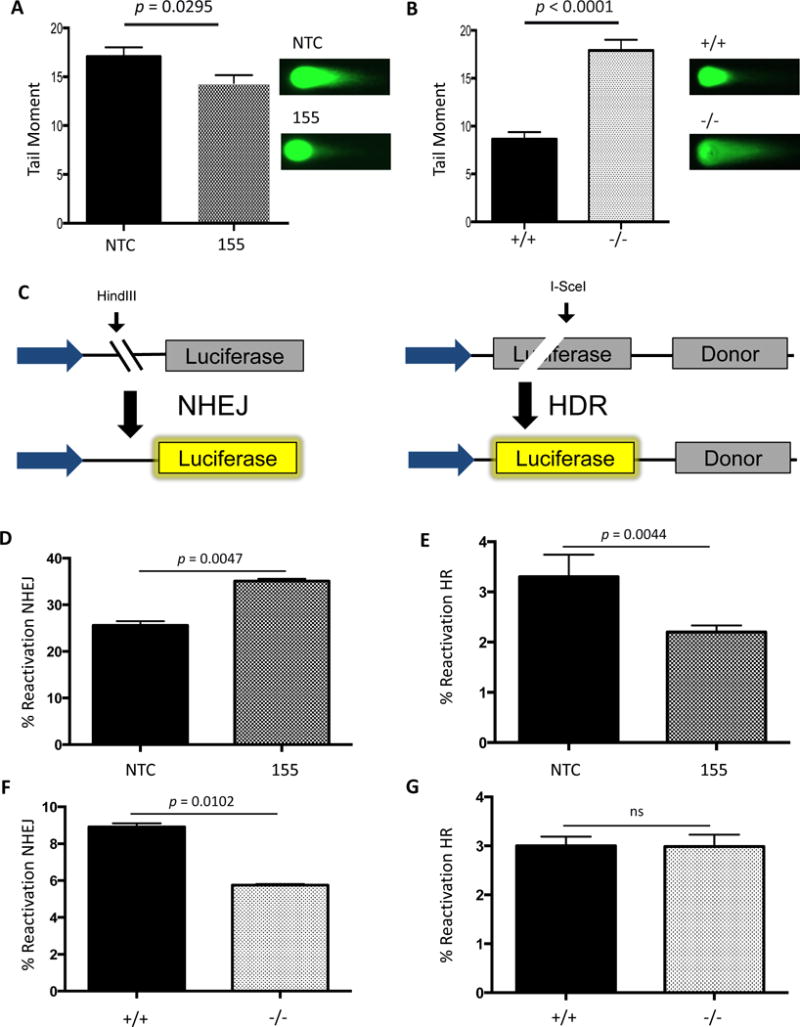
(A) AV16 cells constitutively expressing NTC (black) or 155 (grey) and (B) MEFs either WT (black) or KO (grey) for the BIC locus containing the miR-155 gene were analyzed for double-strand breaks using the neutral Comet assay. Error bars were calculated as standard error of the mean. P-value was calculated by unpaired t-test (n=3). (C) Schematic representation of the luciferase reactivation assay measuring NHEJ or HR activity. NHEJ or HR luciferase reactivation was compared to a positive control to determine percent reactivation. A Renilla luciferase plasmid was used to control for transfection efficiency. (D) AV16 NTC and 155 cells were analyzed for NHEJ and (E) HR activity using luciferase reactivation assays. MEFs WT or KO for miR-155 were analyzed for (F) NHEJ and (G) HR activity using the luciferase reactivation assays.
Conversely, to measure DSBs in the absence of miR-155, we examined the level of DSBs in a B-cell Induced Cluster (BIC) knockout (BICKO) mouse embryonic fibroblast (MEF) cell line. BIC, also known as MIR155 host gene, is a gene that lacks an open reading frame and, as the name suggests, houses the primary miR-155 genomic sequence. BIC is transcribed by RNA polymerase II as a long non-coding RNA molecule that is ultimately processed into mature miR-155. In the absence of miR-155 expression, we found a significant increase in the level of DSBs at baseline (Figure 3B). Importantly, we did not observe a change in the mutation frequency in the spleen when BICKO animals were crossed to the transgenic mutation reporter animals (data not shown) suggesting that although miR-155 loss seems to delay DNA repair or promote DSBs, this does not appear to lead to increased point mutagenesis. Additionally, it suggests that the over-expression of miR-155 and the loss of miR-155 function act differently in modulating DSB repair. Taken together, these results suggest that tight regulation of miR-155 expression is necessary to maintain genomic integrity.
To determine whether these changes in DSB levels were due to a change in the efficiency of DSB repair, we utilized a panel of luciferase-based reporter assays engineered to score repair of DSBs through HR or NHEJ (Figure 3C). In one assay, HR is measured based on restoration of luciferase expression following an I-SceI cut within a luciferase gene. A template copy of luciferase without a promoter for HR-based repair is located on the same plasmid. As a result, reactivation of luciferase activity provides a measure of HR activity. In the second assay, NHEJ activity is measured in a similar fashion except the HindIII site used to create a DSB is located between the promoter and the luciferase gene and there is no homologous template within the plasmid. When the enzyme cuts, luciferase cannot be expressed, but if the DSB is repaired by NHEJ, expression is restored. Control matched pair cell lines reported as expected to validate these assays. Specifically, BRCA2 modulation did not show any effect on NHEJ reporter activity in the DLD1 or PEO1 matched pair cell lines (Figure S2A). In contrast, BRCA2 knockout in DLD1 cells resulted in impaired HR reporter activity (Figure S2B) whereas BRCA2 complementation in the PEO1 cells yielded a marked increase in HR activity (Figure S2B). Similarly, Ku80 complementation of the xrs6 cell line did not induce changes in HR reporter activity (Figure S2B), but showed dramatic decreases in NHEJ reporter activity (Figure S2A). Taken together, these results demonstrate that these luciferase-based NHEJ and HR assays are reliable tools to analyze pathway-specific DNA repair across cell lines.
We found that AV16 155 cells show a decrease in HR when compared to the AV16 NTC cells as assayed by the luciferase HR reporter (Figure 3D). We also measured HR activity using the U2OS-DR-GFP system. Validation of this system showed that BRCA2 and RAD51 knockdown (Figure S2C) demonstrated HR impairment (Figure S2D), while XRCC4 knockdown (Figure S2C) did not show any impairment in HR, as expected (Figure S2D). Similar to the luciferase HR assay, we observed a decrease in HR efficiency upon miR-155 over-expression by pre-miR-155 transfection, confirmed by FOXO3a repression (Figure S2C), in the U2OS DR-GFP cells (Figure S2D). In contrast, an increase in NHEJ was observed in AV16 155 cells when compared to the repair activity in AV16 NTC cells (Figures 3E). The increase in NHEJ activity in the setting of miR-155 over-expression correlates with an observed decrease in unresolved DSBs but, because of the error prone nature of NHEJ, might also contribute to the observed increase in mutation frequency. To test this directly, we utilized the U2OS-DR-EJ cells [20] that contain an RFP reporter to measure total mutagenic end-joining. Upon miR-155 over-expression, we observed an increase in mutagenic end-joining as a function of miR-155 expression (Figure S2E). To further probe the mutagenic mechanism of miR-155, we examined the MMEJ pathway by host cell reactivation of the EJ2 plasmid [34]. To validate this assay, we analyzed the xrs6 cells compared with the xrs6 + Ku80 cells and observed a decrease in MMEJ activity (Figure S2F), which is consistent with previous reports of Ku80 suppressing MMEJ [34, 41–43] Using the EJ2 plasmid, we compared 155KI MEFs to their wild-type controls. However, we did not identify a change in MMEJ activity in this system with miR-155 over-expression (Figure S2G). Taken together these results indicate miR-155 over-expression induces a functional decrease in HR efficiency with a concurrent increase in NHEJ activity.
In contrast, when we examined the level of NHEJ in BICKO cells lacking any miR-155 expression, we found the opposite effect. In the absence of miR-155, NHEJ activity is reduced compared to wild-type function (Figure 3F), while MMEJ activity is up-regulated (Figure S2H) as likely a compensatory mechanism. With respect to HR, however, we did not observe a change in activity in the absence of miR-155 (Figure 3G), suggesting that HR factors may already be expressed at a sufficient level in the setting of normal miR-155 expression and that the absence of miR-155 therefore does not yield any further functional increase in HR.
miR-155 over-expression results in a decrease in DNA polymerase delta expression
To further elucidate the role of miR-155 in NHEJ activity, we analyzed changes in mRNA expression upon miR-155 over-expression. We hypothesized that over-expression of miR-155 might lead to altered expression of an NHEJ factor to mediate the measured increase in activity. Using an Illumina mouse whole genome beadchip microarray, we compared mRNA levels in AV16 155 cells to those in AV16 NTC. Surprisingly, we did not see a difference in mRNA expression of any NHEJ repair factors on the array (Table 1); however, we did find nearly 1,200 genes with at least a 20% change in expression in cells over-expressing miR-155 compared to control cells (Figure 4A). Most notably, we observed a decrease in all four subunits of polymerase delta mRNA in miR-155 over-expressing cells (Table 2) and validated these findings using qRT-PCR (Figure S3). Polymerase delta is a high-fidelity polymerase that functions in replication and DNA repair. None of the polymerase delta subunits are predicted targets of miR-155; however, each subunit has at least one putative binding site within its promoter for the transcription factor FOXO3a, an established target of miR-155. Importantly, we identified several established transcriptional targets of FOXO3a that were also down-regulated in our microarray, consistent with a functional effect of FOXO3a repression by miR-155 (Table 3). To explore the possibility of FOXO3a regulating polymerase delta expression, we used Western blot analysis to examine levels of FOXO3a and POLD1, the catalytic subunit of polymerase delta, in AV16 155 cells compared to AV16 NTC cells. We found that both FOXO3a and POLD1 expression are reduced in cells over-expressing miR-155 (Figure 4B). Additionally, we knocked down FOXO3a expression using siRNAs in the AV16 parent cells and found that POLD1 expression is subsequently reduced (Figure 4C), but did not observe any effect on the NHEJ pathway (Figure S4). Taken together, these results suggest that miR-155 suppresses the expression of the transcription factor FOXO3a, subsequently reducing levels of polymerase delta (Figure 4D).
Table 1.
Relatively Unchanged Levels of NHEJ Factors (p>0.05)
| GENE | Fold Change (155 vs. NTC) |
Fold Change (KO vs. Control) |
|---|---|---|
| Dclre1c | −1.03 | −1.02 |
| Dntt | −1.00 | 1.03 |
| Lig4 | 1.03 | 1.07 |
| Nhej1 | 1.02 | 1.03 |
| Prkdc | 1.01 | 1.00 |
| Rad50 | −1.01 | 1.02 |
| Xrcc4 | 1.02 | 1.01 |
| Xrcc5 | −1.02 | 1.00 |
| Xrcc6 | −1.08 | −1.05 |
Figure 4. Suppression of polymerase delta expression upon miR-155 over-expression is likely mediated by the FOXO3a transcription factor.
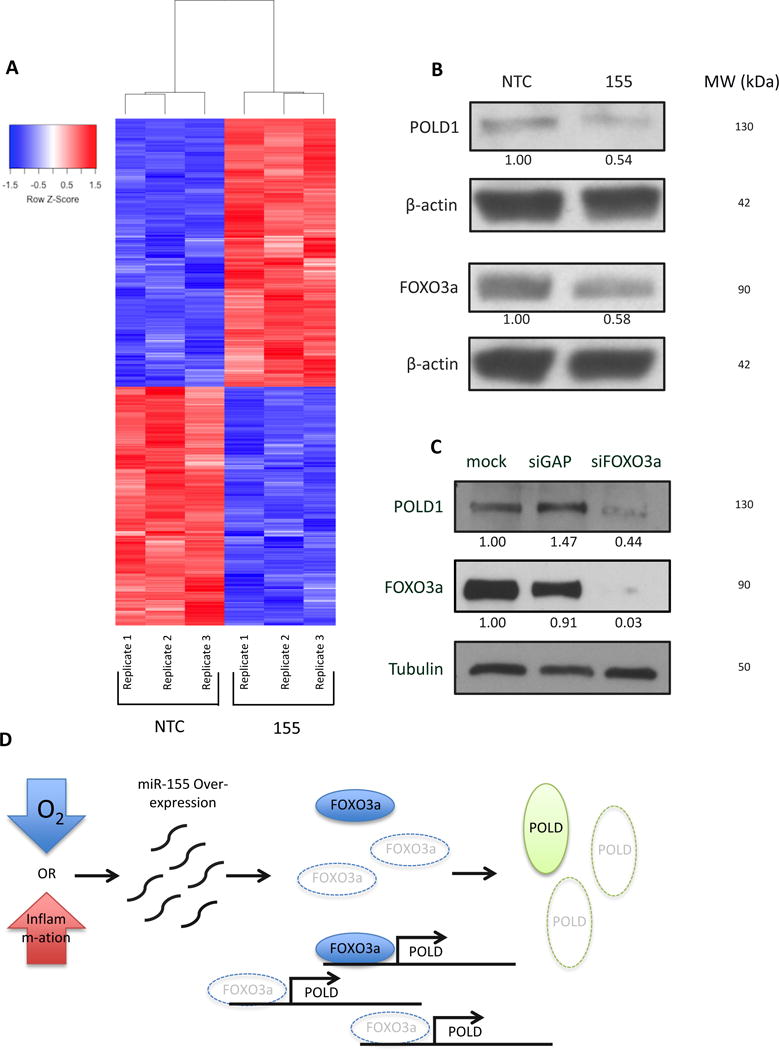
(A) Microarray analysis reveals differential gene expression changes upon miR-155 over-expression. (B) Western blot to analyze expression of FOXO3a and POLD1 in presence of miR-155 over-expression in AV16 cells. (C) siRNA knockdown of FOXO3a in AV16 cells and analysis of POLD1 expression by Western blot. (D) Schematic representation of miR-155 over-expression repressing FOXO3a expression and preventing FOXO3a from activating POLD expression, ultimately leading to an increase mutation frequency.
Table 2.
Down-regulated Double Strand Break Repair Genes (p<0.05)
| GENE | Fold Change (155 vs. NTC) |
p-value (155 vs. NTC) |
|---|---|---|
| Pold4 | −1.93 | 1.99653E-06 |
| Pold3 | −1.35 | 0.014425434 |
| Pold1 | −1.35 | 0.004880594 |
| Fen1 | −1.22 | 0.00420546 |
| Mre11a | −1.17 | 0.004091406 |
| Rpa2 | −1.17 | 0.017108579 |
| Rpa3 | −1.16 | 0.019565474 |
| Hus1 | −1.16 | 0.000687722 |
| Pold2 | −1.14 | 0.057739236 |
| Rad1 | −1.14 | 0.000274311 |
| Rad51 | −1.13 | 0.004452602 |
| Rad51C | −1.13 | 0.014211263 |
| Poll | −1.11 | 0.022761465 |
| Check2 | −1.11 | 0.012247363 |
Table 3.
Known FOXO3a targets down-regulated upon miR-155 over-expression (p<0.05)
| SYMBOL | Fold Change (155 vs. NTC) |
p-value (155 vs. NTC) |
|---|---|---|
| Bcl2l11 | −1.31 | 0.001974636 |
| Foxm1 | −1.36 | 0.004424737 |
| Gadd45a | −1.54 | 0.007134949 |
Discussion
We have demonstrated that the over-expression of miR-155 is sufficient to drive an increase in mutation frequency both in vitro and in vivo. Additionally, we confirmed prior reports that miR-155 over-expression mediates the repression of key DNA repair factors, MLH1 and RAD51, and we discovered that miR-155 suppresses the expression of all four subunits of the DNA polymerase, polymerase delta. Our data correlates with previously published work indicating that miR-155 is capable of targeting select mismatch repair factors and that its over-expression can increase the mutation rate in vitro in colon cancer cells [15, 16]. However, our analysis of mutation patterns suggests that our observed increase in mutation frequency driven by miR-155 may not simply reflect a mismatch repair deficiency but rather that other mechanisms likely contribute to this observed mutagenesis.
We also explored the role of miR-155 in regulating HR activity. As mentioned, miR-155 targets RAD51, an important factor in HR. We found that as a result of miR-155 over-expression, HR activity is reduced. At the same time, we found that NHEJ displays an increase in activity. A relative increase in NHEJ in preference to HR is another possible explanation for the observed increase in mutation frequency as NHEJ is a more error-prone DSB repair pathway than is HR.
Upon further investigation, we discovered that over-expression of miR-155 also results in a decrease in polymerase delta mRNA and protein expression. This decrease correlates with suppression of FOXO3a, a transcription factor and known miR-155 target [1, 10, 44]. Importantly, there are putative binding sites for FOXO3a within the promoter of all four subunits of polymerase delta, suggesting the possibility that FOXO3a may be involved in the regulation of polymerase delta expression. To confirm this hypothesis, we found that experimental suppression of FOXO3a by siRNA transfection results in a decrease in polymerase delta expression, demonstrating that the FOXO3a transcription factor promotes expression of polymerase delta. Although known FOXO3a targets are down-regulated in our miR-155 over-expression system, we find that FOXM1, an oncogenic transcription factor that is typically up-regulated upon FOXO3a repression [45], is down-regulated. Despite seeming contradictory to the FOXO3a-FOXM1 axis in DNA repair and cancer [46], this suppression of FOXM1 might also contribute to genomic instability as FOXM1 has been shown to be an important mediator of genomic integrity. In fact, it has been suggested that repression of FOXM1 delays the G2 phase of the cell cycle and can cause chromosomal instability [47]. It is possible that a delay in the G2 phase in the presence of functionally stunted HR and over-active NHEJ might further support the increased mutagenesis observed with miR-155 over-expression, though further studies are warranted to test this hypothesis.
Additional evidence supporting the relationship between miR-155, FOXO3a, and polymerase delta comes from phenotypic observations of transgenic mice. In fact, although POLD1 knock-out mice are embryonic lethal [48], FOXO3a knock-out mice are viable and are prone to develop malignancies [49–51]. Interestingly, the 155KI animals are also prone to malignancy, namely a lymphoma-like disorder, which has been previously reported [9]. These observations support the hypothesis of a functional relationship between miR-155, FOXO3a, and polymerase delta.
Although direct experimental down-regulation of polymerase delta has not been evaluated, several studies have shown that aberrant polymerase delta expression or function can cause increased levels of mutagenesis [48, 52–54]. Therefore, the miR-155-mediated decrease in polymerase delta expression that we have observed is likely a key contributor to the increased genomic instability that is produced upon over-expression of miR-155. We hypothesize that compensation for a decrease in high-fidelity polymerase delta might involve the increased activity of other, more error-prone translesion polymerases, contributing to the observed increase in mutation frequency with miR-155 over-expression. It is tempting to speculate that previously reported hypoxic induction of miR-155 [1] might promote genomic instability in hypoxia by the suppression of high-fidelity polymerase delta coupled with concurrent HIF-1α-dependent over-expression of polymerase iota [55], a translesion polymerase. Another possibility is that reduced polymerase delta expression is compensated for by an increase in activity of polymerase theta, which mediates MMEJ [56, 57] and might contribute to an increase in mutation frequency. Both of these hypotheses will require further study.
microRNAs are quickly being identified as key regulators of genomic stability and understanding their function mechanistically as it relates to tumorigenesis will become a crucial part of personalized treatment. In addition to our evidence for the role of miR-155 in maintaining genomic integrity, a recent study demonstrated that three other microRNAs, miR-148b*, miR-193b*, and miR-1255b, play important roles in regulating the timing of HR to prevent loss of heterozygosity during the G1 phase of the cell cycle [58]. It is likely that several other microRNAs exist to regulate DNA repair pathways and ensure fidelity of the DNA damage response.
Elucidating the role of miR-155 in promoting tumorigenesis is an important step in developing adjuvant therapies to sensitize miR-155 over-expressing tumors to chemotherapeutic agents. As an oncomiR, miR-155 is an attractive target for personalized medicine. Importantly, miR-155 is not the first miRNA to be explored as a therapeutic target or agent. In fact, a phase IIa clinical trial using an anti-miR-122 agent has been positively evaluated as a treatment for Hepatitis C, and a phase I trial is ongoing using a miR-34 mimic to treat unresectable liver cancer [59, 60]. Although further study is necessary to determine the effects of an antagomiR on preventing genomic instability, miR-155 is a promising target candidate for improving efficacy of current therapy, and our group is currently pursuing strategies to selectively inhibit miR-155 in tumors [61].
Supplementary Material
Implications.
Taken together, miR-155 over-expression drives an increase in mutation frequency via multifaceted impact on DNA damage response and DNA repair pathways.
Acknowledgments
We thank Denise Hegan for her technical assistance and Frank Slack for providing reagents.
This work was supported by NIH R01 ES005775 to PMG.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to disclose.
Contributor Information
Jennifer R. Czochor, Email: jennifer.czochor@yale.edu.
Parker Sulkowski, Email: parker.sulkowski@yale.edu.
References
- 1.Babar IA, et al. Inhibition of hypoxia-induced miR-155 radiosensitizes hypoxic lung cancer cells. Cancer Biol Ther. 2011;12(10):908–14. doi: 10.4161/cbt.12.10.17681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bruning U, et al. MicroRNA-155 promotes resolution of hypoxia-inducible factor 1alpha activity during prolonged hypoxia. Mol Cell Biol. 31(19):4087–96. doi: 10.1128/MCB.01276-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Papaconstantinou IG, et al. Expression of microRNAs in patients with pancreatic cancer and its prognostic significance. Pancreas. 2013;42(1):67–71. doi: 10.1097/MPA.0b013e3182592ba7. [DOI] [PubMed] [Google Scholar]
- 4.Shibuya H, et al. Clinicopathological and prognostic value of microRNA-21 and microRNA-155 in colorectal cancer. Oncology. 2010;79(3–4):313–20. doi: 10.1159/000323283. [DOI] [PubMed] [Google Scholar]
- 5.Yanaihara N, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9(3):189–98. doi: 10.1016/j.ccr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 6.Iorio MV, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65(16):7065–70. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 7.Lee EJ, et al. Expression profiling identifies microRNA signature in pancreatic cancer. Int J Cancer. 2007;120(5):1046–54. doi: 10.1002/ijc.22394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eis PS, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A. 2005;102(10):3627–32. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Babar IA, et al. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc Natl Acad Sci U S A. 2012;109(26):E1695–704. doi: 10.1073/pnas.1201516109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kong W, et al. MicroRNA-155 regulates cell survival, growth, and chemosensitivity by targeting FOXO3a in breast cancer. J Biol Chem. 2010;285(23):17869–79. doi: 10.1074/jbc.M110.101055. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.O’Connell RM, et al. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci U S A. 2009;106(17):7113–8. doi: 10.1073/pnas.0902636106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, et al. MicroRNA Regulation of Ionizing Radiation-Induced Premature Senescence. Int J Radiat Oncol Biol Phys. 2011;81(3):839–48. doi: 10.1016/j.ijrobp.2010.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang S, et al. MicroRNA-155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res. 70(8):3119–27. doi: 10.1158/0008-5472.CAN-09-4250. [DOI] [PubMed] [Google Scholar]
- 14.Gasparini P, et al. Protective role of miR-155 in breast cancer through RAD51 targeting impairs homologous recombination after irradiation. Proc Natl Acad Sci U S A. 2014;111(12):4536–41. doi: 10.1073/pnas.1402604111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valeri N, et al. Modulation of mismatch repair and genomic stability by miR-155. Proc Natl Acad Sci U S A. 2010;107(15):6982–7. doi: 10.1073/pnas.1002472107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tili E, et al. Mutator activity induced by microRNA-155 (miR-155) links inflammation and cancer. Proc Natl Acad Sci U S A. 2011;108(12):4908–13. doi: 10.1073/pnas.1101795108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rogers FA, et al. Peptide conjugates for chromosomal gene targeting by triplex-forming oligonucleotides. Nucleic Acids Res. 2004;32(22):6595–604. doi: 10.1093/nar/gkh998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singleton BK, et al. Molecular and biochemical characterization of xrs mutants defective in Ku80., in. Mol Cell Biol. 1997:1264–1273. doi: 10.1128/mcb.17.3.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakai W, et al. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009;69(16):6381–6. doi: 10.1158/0008-5472.CAN-09-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bindra RS, et al. Development of an assay to measure mutagenic non-homologous end-joining repair activity in mammalian cells. Nucleic Acids Res. 2013;41(11):e115. doi: 10.1093/nar/gkt255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakanishi K, et al. Homologous recombination assay for interstrand cross-link repair. Methods Mol Biol. 2011;745:283–91. doi: 10.1007/978-1-61779-129-1_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hegan DC, et al. Differing patterns of genetic instability in mice deficient in the mismatch repair genes Pms2, Mlh1, Msh2, Msh3 and Msh6. Carcinogenesis. 2006;27(12):2402–8. doi: 10.1093/carcin/bgl079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Narayanan L, et al. Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2. Proc Natl Acad Sci U S A. 1997;94(7):3122–7. doi: 10.1073/pnas.94.7.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leach EG, et al. Frequent spontaneous deletions at a shuttle vector locus in transgenic mice. Mutagenesis. 1996;11(1):49–56. doi: 10.1093/mutage/11.1.49. [DOI] [PubMed] [Google Scholar]
- 25.Thai TH, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316(5824):604–8. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 26.Crosby ME, et al. MicroRNA regulation of DNA repair gene expression in hypoxic stress. Cancer Res. 2009;69(3):1221–9. doi: 10.1158/0008-5472.CAN-08-2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagel ZD, et al. Multiplexed DNA repair assays for multiple lesions and multiple doses via transcription inhibition and transcriptional mutagenesis. Proc Natl Acad Sci U S A. 2014;111(18):E1823–32. doi: 10.1073/pnas.1401182111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bau DT, et al. Breast cancer risk and the DNA double-strand break end-joining capacity of nonhomologous end-joining genes are affected by BRCA1. Cancer Res. 2004;64(14):5013–9. doi: 10.1158/0008-5472.CAN-04-0403. [DOI] [PubMed] [Google Scholar]
- 29.Li YH, et al. Inhibition of non-homologous end joining repair impairs pancreatic cancer growth and enhances radiation response. PLoS One. 2012;7(6):e39588. doi: 10.1371/journal.pone.0039588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sears CR, Turchi JJ. Complex cisplatin-double strand break (DSB) lesions directly impair cellular non-homologous end-joining (NHEJ) independent of downstream damage response (DDR) pathways. J Biol Chem. 2012;287(29):24263–72. doi: 10.1074/jbc.M112.344911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang HC, et al. Ataxia telangiectasia mutated and checkpoint kinase 2 regulate BRCA1 to promote the fidelity of DNA end-joining. Cancer Res. 2006;66(3):1391–400. doi: 10.1158/0008-5472.CAN-05-3270. [DOI] [PubMed] [Google Scholar]
- 32.Zhong Q, et al. BRCA1 facilitates microhomology-mediated end joining of DNA double strand breaks. J Biol Chem. 2002;277(32):28641–7. doi: 10.1074/jbc.M200748200. [DOI] [PubMed] [Google Scholar]
- 33.Soong CP, et al. Development of a novel method to create double-strand break repair fingerprints using next-generation sequencing. DNA Repair (Amst) 2015;26:44–53. doi: 10.1016/j.dnarep.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 34.Bennardo N, et al. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4(6):e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fattah FJ, et al. A role for XLF in DNA repair and recombination in human somatic cells. DNA Repair (Amst) 2014;15:39–53. doi: 10.1016/j.dnarep.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaushik Tiwari M, Rogers FA. XPD-dependent activation of apoptosis in response to triplex-induced DNA damage. Nucleic Acids Res. 2013;41(19):8979–94. doi: 10.1093/nar/gkt670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rogers FA, et al. Targeted gene modification of hematopoietic progenitor cells in mice following systemic administration of a PNA-peptide conjugate. Mol Ther. 2012;20(1):109–18. doi: 10.1038/mt.2011.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirano T, et al. 8-hydroxyguanine levels in nuclear DNA and its repair activity in rat organs associated with age. J Gerontol A Biol Sci Med Sci. 1996;51(5):B303–7. doi: 10.1093/gerona/51a.5.b303. [DOI] [PubMed] [Google Scholar]
- 39.Russo MT, et al. Different DNA repair strategies to combat the threat from 8-oxoguanine. Mutat Res. 2007;614(1–2):69–76. doi: 10.1016/j.mrfmmm.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 40.Russo MT, et al. Accumulation of the oxidative base lesion 8-hydroxyguanine in DNA of tumor-prone mice defective in both the Myh and Ogg1 DNA glycosylases. Cancer Res. 2004;64(13):4411–4. doi: 10.1158/0008-5472.CAN-04-0355. [DOI] [PubMed] [Google Scholar]
- 41.McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 2008;24(11):529–38. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nussenzweig A, Nussenzweig MC. A backup DNA repair pathway moves to the forefront. Cell. 2007;131(2):223–5. doi: 10.1016/j.cell.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 43.Truong LN, et al. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad Sci U S A. 2013;110(19):7720–5. doi: 10.1073/pnas.1213431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ling N, et al. microRNA-155 regulates cell proliferation and invasion by targeting FOXO3a in glioma. Oncol Rep. 2013;30(5):2111–8. doi: 10.3892/or.2013.2685. [DOI] [PubMed] [Google Scholar]
- 45.McGovern UB, et al. Gefitinib (Iressa) represses FOXM1 expression via FOXO3a in breast cancer. Mol Cancer Ther. 2009;8(3):582–91. doi: 10.1158/1535-7163.MCT-08-0805. [DOI] [PubMed] [Google Scholar]
- 46.Gomes AR, Zhao F, Lam EW. Role and regulation of the forkhead transcription factors FOXO3a and FOXM1 in carcinogenesis and drug resistance. Chin J Cancer. 2013;32(7):365–70. doi: 10.5732/cjc.012.10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laoukili J, et al. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol. 2005;7(2):126–36. doi: 10.1038/ncb1217. [DOI] [PubMed] [Google Scholar]
- 48.Uchimura A, et al. DNA polymerase delta is required for early mammalian embryogenesis. PLoS One. 2009;4(1):e4184. doi: 10.1371/journal.pone.0004184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Castrillon DH, et al. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science. 2003;301(5630):215–8. doi: 10.1126/science.1086336. [DOI] [PubMed] [Google Scholar]
- 50.Paik JH, et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128(2):309–23. doi: 10.1016/j.cell.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tothova Z, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128(2):325–39. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 52.Albertson TM, et al. DNA polymerase epsilon and delta proofreading suppress discrete mutator and cancer phenotypes in mice. Proc Natl Acad Sci U S A. 2009;106(40):17101–4. doi: 10.1073/pnas.0907147106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goldsby RE, et al. High incidence of epithelial cancers in mice deficient for DNA polymerase delta proofreading. Proc Natl Acad Sci U S A. 2002;99(24):15560–5. doi: 10.1073/pnas.232340999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Venkatesan RN, et al. Mutator phenotypes caused by substitution at a conserved motif A residue in eukaryotic DNA polymerase delta. J Biol Chem. 2006;281(7):4486–94. doi: 10.1074/jbc.M510245200. [DOI] [PubMed] [Google Scholar]
- 55.Ito A, et al. Hypoxia-inducible factor-1 mediates the expression of DNA polymerase iota in human tumor cells. Biochem Biophys Res Commun. 2006;351(1):306–11. doi: 10.1016/j.bbrc.2006.10.048. [DOI] [PubMed] [Google Scholar]
- 56.Ceccaldi R, et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature. 2015;518(7538):258–62. doi: 10.1038/nature14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kent T, et al. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase theta. Nat Struct Mol Biol. 2015 doi: 10.1038/nsmb.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi YE, et al. MicroRNAs down-regulate homologous recombination in the G1 phase of cycling cells to maintain genomic stability. Elife. 2014;3:e02445. doi: 10.7554/eLife.02445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Janssen HL, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368(18):1685–94. doi: 10.1056/NEJMoa1209026. [DOI] [PubMed] [Google Scholar]
- 60.Shibata C, Otsuka Motoyuki, Kishikawa Takahiro, Yoshikawa Takeshi, Ohno Motoko, Takata Akemi, Koike Kazuhiko. Current status of miRNA-targeting therapeutics and preclinical studies against gastroenterological carcinoma. Molecular and Cellular Therapies. 2013;1(5) doi: 10.1186/2052-8426-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheng CJ, et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature. 2015;518(7537):107–10. doi: 10.1038/nature13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.