

Abstract
The evolution of Type 1 diabetes (T1D) therapy has been marked by consecutive shifts, from insulin replacement to immunosuppressive drugs and targeted biologics (following the understanding that T1D is an autoimmune disease), and to more disease‐specific or patient‐oriented approaches such as antigen‐specific and cell‐based therapies, with a goal to provide efficacy, safety, and long‐term protection. At the same time, another important paradigm shift from treatment of new onset T1D patients to prevention in high‐risk individuals has taken place, based on the hypothesis that therapeutic approaches deemed sufficiently safe may show better efficacy if applied early enough to maintain endogenous β cell function, a concept supported by many preclinical studies. This new strategy has been made possible by capitalizing on a variety of biomarkers that can more reliably estimate the risk and rate of progression of the disease. More advanced (“omic”‐based) biomarkers that also shed light on the underlying contributors of disease for each individual will be helpful to guide the choice of the most appropriate therapies, or combinations thereof. In this review, we present current efforts to stratify patients according to biomarkers and current alternatives to conventional drug‐based therapies for T1D, with a special emphasis on cell‐based therapies, their status in the clinic and potential for treatment and/or prevention. Stem Cells 2016;34:809–819
Keywords: T cells, Cell therapy, Autoimmunity, Prevention, Type 1 diabetes, Immunoregulation, Antigen‐specific
Significance Statement.
This article summarizes the significance of the paradigm shift in the thinking about the treatment of Type 1 diabetes. Current treatment strategies are now directed toward prevention of disease progression to maintain endogenous beta cell function. The use of immunomodulating strategies including antigen‐ and cell‐based therapies as well as the need to identify new biomarkers that allow a measure of disease stage and time to onset of hyperglycemia for selection of appropriate patients to enroll in prevention trials are discussed.
Introduction
Type 1 diabetes (T1D), like its more common Type 2 counterpart, has been rising in prevalence and incidence primarily in Western countries 1, 2 (Fig. 1). Insulin replacement therapy has been the primary treatment of all forms of diabetes for almost 100 years, but inadequate control of its delivery has allowed a number of complications to markedly diminish the quality of life of affected individuals, and contributed to an increasingly intolerable financial burden. The realization that a subset of patients presents with an autoimmune form of insulin‐dependent diabetes was made in the 1970s 3. The initial model suggesting the potential pathogenesis of this disorder as a chronic autoimmune disease directed against β cells was proposed by George Eisenbarth in the mid‐1980s 4. This understanding led to subsequent attempts to develop more specific treatments for this autoimmune form of diabetes, initially with immunosuppressive therapies that had proven effective in other chronic autoimmune diseases, including cyclosporine A (CsA) or anti‐thymocyte globulin and prednisone 5, 6, 7. Despite initial suggestion of efficacy of CsA, no subsequent study has been able to confirm these initial results. In addition, the lack of lasting effects once CsA was withdrawn and the serious renal toxicity of the drug severely limited enthusiasm for this approach.
Figure 1.
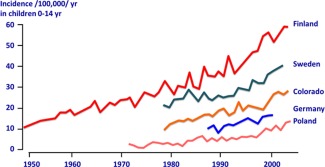
T1D incidence has doubled every 20 years. Data for Finland are from the Finnish National Public Health Institute; data for Sweden are from the Swedish Childhood Diabetes Registry; data for Colorado are from the Colorado IDDM Registry, the Barbara Davis Center for Childhood Diabetes, and SEARCH for Diabetes in Youth; data for Germany are a compilation of two reports; and data from Poland are from Diabetologia 2010;54:508‐515. Reprinted with permission from the Ann NY Acad Sci 2008;1150:1‐13, with additional modifications and permission from Marian Rewers and Jay Skyler.
Subsequent natural history studies have made the approach to treatment more complex as these studies have demonstrated that underlying autoimmune responses are present for varying periods of time, usually years, in genetically predisposed individuals before the appearance of overt hyperglycemia. Particular major histocompatibility class II haplotypes (HLA‐DR3/4, HLA‐DQ8) confer the greatest risk as genetic factors 8. In addition to serving as a diagnosis tool for T1D in new onset diabetic patients, circulating autoantibodies against β cell proteins (specificity and quantity) in at‐risk individuals, as well as abnormalities in the oral glucose tolerance test, can generally help predict the risk of, and time remaining, before the onset of hyperglycemia 9. These predictive data have raised the possibility of attempting therapeutic intervention before the onset of hyperglycemia in high‐risk individuals identified based on biomarkers like those mentioned above. Prevention of T1D may represent a viable alternative to an actual cure by permanently blocking the autoimmune response while there are sufficient β cells remaining, and may offer a more cost‐effective approach in the short‐term to deal with the alarming rise in the incidence of disease (Fig. 1). To make the matter even more complex, many patients may present a spontaneous but temporary remission after onset, known as the honeymoon period, possibly reflecting reduced stress on residual β cells after initial insulin treatment. This honeymoon period may perhaps represent a sweet window of opportunity (pun intended) to exploit for the use of intervention therapy.
In this review, we will discuss how our therapeutic arsenal to fend off this autoimmune disease has greatly diversified beyond traditional drugs and biologicals to include various forms of cell therapies, as well as other less conventional approaches. The field is witnessing a paradigm shift from immunosuppressive therapies applied after the onset of hyperglycemia (a time at which β cell function has generally been irreversibly lost) to prevention strategies attempting to shut off the autoimmune response and preserve β cell function in high‐risk individuals. This of course entails the use of reliable biomarkers to identify the most appropriate at‐risk subjects for such intervention trials, and perhaps also guide the type of therapy that should be employed, paving the way to more personalized therapies. Current studies using new techniques of transcriptomics and proteomics 10, 11, 12, 13 are attempting to more precisely stratify those at risk by identifying novel biomarkers that may be superior to those currently used to define the stage or rate of progression of disease, and thus help select appropriate subjects to enter into prevention trials. Although the strategies described in this review have all shown remarkable efficacy in preclinical models, it should be noted that little or no clinical efficacy data is available for most of them, whether they are evaluated in the treatment of recent onset patients after safety has been demonstrated or in prevention studies following safe but ineffective use in recent onset patients.
A Shift from Treatment to Prevention Is Driven by Biomarkers Guiding When and How to Intervene
The progress in identifying the patients who are at “high‐risk” and should be entered into prevention trials has been supported by an improved understanding of T1D disease processes that allows screening for at‐risk individuals and stratification of the individual's risk and time of progression to the development of hyperglycemia. The first level of screening is comprised of family history (number of relatives with T1D and degree of relationship) and HLA haplotype (HLA‐DR3/4 heterozygosy combined with HLA‐DQ8 conferring the highest known risk) 9. Although these risk factors are fixed from birth, new relatives may become diagnosed later and the relative risk re‐evaluated. These parameters have served to enroll young subjects into studies on how environmental factors influence disease progression (e.g., primary prevention studies examining diet alterations in genetically at‐risk babies with no evidence of autoimmunity, Table 1). These individuals can be closely and regularly monitored and undergo a second level of screening consisting of well‐established biomarkers such as circulating autoantibodies to β cell antigens insulin, GAD65, IA‐2, ZnT8, and IGRP 9, 14, which have served as good predictive tools 15, 16, 17, 18 and enrollment criteria for prevention studies. In vitro immunoassays performed on peripheral blood cells including T cell responses to β cell antigens or identification of diabetogenic T cells by tetramer staining complete this assessment of the breadth (how many autoantigens targeted) and amplitude (antibody titers or frequency of tetramer‐positive T cells) of the autoimmune response 14. More recently, biomarkers based on epigenetic changes have been discovered, such as circulating demethylated insulin DNA 19, 20 and differences in methylation level at specific CpG sites in immune cells 21. Increased levels of demethylated insulin DNA in the blood correlates with the extent of β cell damage 19, while the extent of insulitis may now be evaluated by refined imaging techniques 22.
Table 1.
Main clinical trials focused on the prevention of T1D
| Prevention trials | Drug | Type of study |
|---|---|---|
| Diet/supplement‐based prevention | ||
| NCT01055080 (FINDIA) | Baby diet alteration | Phase 1, primary prevention |
| NCT00570102 (MIP) | Baby diet alteration | Phase 2, primary prevention |
| NCT01115621 (BABYDIET) | Baby diet, delayed gluten | Phase 1, primary prevention |
| NCT00179777 (TRIGR) | Controlled diet in infants | Phase 2, primary prevention |
| NCT00333554 (NIDDK) | Omega‐3‐fatty acids | Phase 2, primary prevention |
| NCT00141986 (CDA) | Vitamin D | Phase 1, primary prevention |
| β cell antigen‐based prevention | ||
| NCT00004984 (DPT‐1) | Parenteral or oral insulin | Phase 2, secondary prevention |
| NCT00419562 (NIDDK) | Oral insulin | Phase 3, secondary prevention |
| ISRCTN76104595 (Pre‐POINT) | Oral insulin | Phase 1, primary prevention |
| NCT00654121 (BDR Trial) | Subcut. insulin (Actrapid HM) | Phase 2, secondary prevention |
| NCT00223613 (DIPP) | Intranasal insulin | Phase 3, secondary prevention |
| NCT00336674 (INIT‐II) | Intranasal insulin | Phase 2, secondary prevention |
| NCT01122446 (DIAPREV‐IT) | Diamyd (GAD‐Alum) | Phase 2, secondary prevention |
| Combinations of the above | ||
| NCT02387164 (DIAPREV‐IT2) | Diamyd (GAD‐Alum + Vit. D) | Phase 2, secondary prevention |
| Prevention using biological‐based immunotherapy | ||
| NCT01773707 (NIDDK, TN18) | Abatacept (CTLA4‐Ig) | Phase 2, secondary prevention |
| NCT01030861 (NIDDK, TN10) | Teplizumab (anti‐CD3) | Phase 2, secondary prevention |
| Cell‐based prevention | ||
| CoRD study (Sydney) | Umbilical cord blood | Phase 1, secondary prevention |
Note: Clinical trials are color‐shaded based on whether they are completed, ongoing, or planned.
These biomarkers of prediction are important to evaluate disease risk and rate of progression (indicating with a high degree of confidence if and approximately when a patient will progress to overt hyperglycemia unless the course of progression is altered by treatment), and, therefore, to determine when to treat. Individuals with a comparable risk level may experience a different rate of progression, according to their genetic makeup (other genes besides HLA) and their environment, which differentially affect mechanisms of immune tolerance and pathogenesis. As environmental factors change, the rate of progression may increase or decrease, placing the subject at greater risk or slowing down the development of the disease (Fig. 2). As a result of multiple etiological factors acting in concert, the disease can progress from very fast in the case of fulminant T1D 23 to very slow in the case of latent autoimmune diabetes in adults (LADA) 24. However, the most aggressive forms of disease, like fulminant T1D, might not benefit from prevention unless the trigger becomes well understood.
Figure 2.
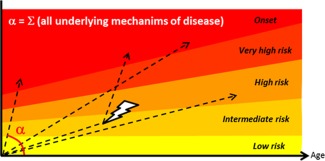
Rates of T1D disease progression. In T1D, earlier onset reflects a faster rate of progression through risk levels (represented by narrower height). The risk level can be evaluated according to family history and HLA haplotype, which are fixed at birth, as well as circulating anti‐β cell autoantibodies and certain metabolic measurements, which are dynamic 9. The rate of progression (α) takes into account all causes and factors that may contribute to pathogenesis, including (but not limited to) defective deletional tolerance, defective immune regulation, defective/delayed clearance of damaged β cells, viral infection (high tropism for β cells and/or molecular mimicry), and β cell intrinsic factors such as susceptibility to infection, apoptosis or dedifferentiation (Table 2). Progression through these stages may not be linear for all individuals, as precipitating events during life (depicted by a thunderbolt) may accelerate the rate, while other environmental changes may curb it. Furthermore, accumulation of genetic (prefixed) factors may cause an individual to start at an intermediate risk, but genetic analysis other than HLA haplotype is not yet routinely performed. Successful prevention of disease will require more advanced tools to evaluate the risk level, the rate of progression, and preferably, the nature of the deficiencies that contribute to the rate of progression (Table 2).
Because of the heterogeneity of etiological factors that may control the rate of progression, it is unlikely that patients stratified as having a similar risk will be equally responsive to a particular treatment (Table 2). Treating a high‐risk patient with the wrong drug would cost precious time during which β cells will continue to be destroyed. In prevention studies, as opposed to new onset cases, more time will be needed before it can be determined whether the treatment is effective. Conversely, treating a low‐risk patient, who may never advance to onset, even with the right prevention therapy, would involve unnecessary costs and risks. Thus, a third level of screening that is more sophisticated (using novel biomarkers featured in larger datasets) will be required to help determine how best to treat each patient by providing clues as to the underlying defects that need to be acted upon. A combination of genetic, transcriptomic, and proteomic tests performed on blood samples will likely be part of such screening in the future, and extensive research is being conducted to this end. Furthermore, advances in viromics have enabled the development of sensitive blood tests that can detect prior exposure to particular viruses, some of which have long been suspected to play a role as a trigger for the disease in some individuals 44. If these tests help confirm a link between these pathogens (which are not uncommon, and therefore would not be sufficient to induce T1D), then the prospect of vaccinating genetically at‐risk individuals becomes possible.
Table 2.
Factors contributing to the rate of disease progression
| Possible mechanisms (not mutually exclusive) | Possible causes or predisposing genes | Possible biomarkers | Possible therapeutic approach to prevent (and reverse) disease | References |
|---|---|---|---|---|
| Defective deletional tolerance: higher frequency of β cell‐reactive T cells (and B cells) | HLA‐DR, INS, PTPN22 polymorphism | HLA‐DR, selected SNP analysis, MHC tetramer analysis/ELISPOT | Antigen‐specific therapies | 8, 25, 26, 27, 28, 29 |
| Defective immune regulation: reduced number, responsiveness and/or function of regulatory T cells | IL2RA, IL2, CTLA4, IL10, PTPN22 polymorphism Vitamin D and/or fiber deficiency Foxp3 promoter methylation | Treg suppression, TSDR assay, STAT5 responsiveness | Expanding Tregs and/or boosting their function (in vivo or ex vivo); antigen‐specific therapies | 8, 25, 30, 31, 32, 33, 34, 35, 36 |
| Antigen‐presenting cells: hyperactivity under inflammatory conditions or defective tolerogenic properties | Genetic predispositions? Environmental factors? | Functional characterization of certain blood cells? | Blockade of specific costimulatory pathways or cytokines | 37, 38 |
| Response of β cells: apoptosis, stress ( ± generation of neo‐antigens), de‐differentiation or trans‐differentiation | IFIH1 polymorphism, inflammation, some unknown genetic determinants, inability to cope with excessive stress | Selected SNPs, circulating demethylated insulin DNA (also reflects β cell immune destruction); impaired glucose tolerance | Drugs increasing β cell replication, reducing β cell stress (imatinib?), or stabilizing β cell phenotype Anti‐inflammatory drugs? | 20, 39, 40, 41 |
| Defective/delayed clearance of damaged β cells / β cell antigens (disease initiation) | Genetic predispositions? | None (too early to detect)? | None at the moment | 42, 43 |
| Interaction with microbes: molecular mimicry (cross‐reactivity with β cell antigens), immune deviation or dysregulation | HLA‐DR, IFIH1, TLR7/8 polymorphism; Infection (e.g., enterovirus); Dysbiosis (imbalanced microbiome) | Infection history (difficult) Microbiome profiling | Probiotics? Vaccines? Anti‐inflammatory drugs? | 8, 35, 44, 45, 46, 47, 48 |
Combinations of genetic and environmental factors contribute to the initiation and progression of T1D disease, leading to various deficiencies at the level of both immune cells and β cells. Gene‐wide association studies have identified some 40 gene polymorphisms each contributing a small risk to the disease, some of which are listed here. The more deficiencies that exist in a particular individual, the faster the progression is expected to be. However, different patients are characterized by different combinations of such deficiencies, leading to substantial heterogeneity in how they progress and how rapidly. Biomarkers that assess not only the risk but also the underlying deficiencies will help inform the choice of prevention therapies to be applied to more homogeneous cohorts of patients for better efficacy. References are only provided as examples to illustrate the concepts.
In recent years, it has become clear that combination therapies, selected to address multiple underlying defects, will become more prominent in our effort to tackle T1D heterogeneity in both prevention and treatment. Such combination therapies are expected to be effective in larger cohorts of patients with overlapping defects. Although many of the immunosuppressive strategies have not been effective when administered post‐hyperglycemia, they might be appropriate to use at an earlier stage of disease where they may prove more efficacious. Besides efficacy and safety, the cost will need to be leveraged against the long‐term benefits. Expensive therapies that induce durable tolerance and protection may, however, save money in the long run. It is unusual for new drugs to be approved for prevention of disease without prior testing in new‐onset patients: the immunomodulatory drugs and cell‐based therapies described below are no exception to this rule.
Overview of the Current Landscape of Non‐Cell Therapies
Cell‐ or Pathway‐Neutralizing Biologics
Regulatory authorities have historically prioritized treatment of new onset patients because these cases are more pressing, the risk tolerance greater, and the studies shorter, smaller, and less expensive. In an attempt to replace the use of globally immunosuppressive drugs, a number of promising biologics have been evaluated in new onset patients, such as anti‐CD3 mAb 49, 50, 51, anti‐CD20 mAb 52, CTLA4‐Ig 53. These drugs showed significant but limited (transient) efficacy in new‐onset patients. These drugs are now being tested in high‐risk normoglycemic patients where they might have a more pronounced and durable effect in sustaining euglycemia (Table 1). These drugs were deemed safe enough by the US Food and Drug Administration to be used prophylactically at a dose unlikely to cause serious adverse events. Current studies using such biologics to prevent disease in high‐risk patients include two major TrialNet studies: TN10 (ClinicalTrial.gov NCT01030861) using Teplizumab (anti‐CD3 mAb) and TN18 (ClinicalTrial.gov NCT01773707) using Abatacept (CTLA4‐Ig). Many other biologics used to treat other autoimmune diseases are also being evaluated for T1D. However, these drugs remain relatively nonspecific and may still carry accrued risks of infections or malignancies in susceptible subjects.
Low Dose IL‐2: A Safer and More Selective Approach?
Because of different sensitivities, it was found that regulatory T cells (Tregs), a subset of T cells that protects from autoimmunity, are selectively stimulated by low doses of IL‐2, a T cell growth factor 54. This new approach is particularly noteworthy because of its safety profile, based on several published studies on the use of low dose IL‐2 to treat inflammatory diseases including chronic graft‐versus‐host disease (GvHD) 55, 56, 57 and hepatitis C virus‐induced vasculitis 58, 59, and preliminary studies on its use as a potential treatment of T1D 60. In addition, preclinical studies in non‐obese diabetic (NOD) mice showed that low dose IL‐2 administered after onset of hyperglycemia restored euglycemia in a majority of the treated mice 61. In each instance, low dose IL‐2 therapy was associated with a dose‐dependent increase in the number of circulating Tregs and a marked diminution of inflammatory cytokine expression in the serum of the treated mice or patients in the initial short‐term safety trial 60. An additional dose finding study to determine the optimal dose of IL‐2 required to increase the number and response of Tregs has been completed in T1D patients (ClinicalTrial.gov NCT01827735). Subsequently, an efficacy trial has begun in patients with recent onset T1D (ClinicalTrial.gov NCT01862120). Once these studies (in Table 3) are completed and the safety profile confirmed, a move to recruit high‐risk patients in low dose IL‐2 studies will be expected.
Table 3.
Main clinical trials using low‐dose IL‐2 or cell‐based therapies in recent onset T1D patients
| New onset trials | Drug | Type of study |
|---|---|---|
| Low‐dose IL‐2 | ||
| NCT01827735 (DILT1D) | Proleukin (IL‐2) | Phase 1/2, onset < 24 months |
| NCT02265809 (DILfrequency) | Aldesleukin (IL‐2) | Phase 1/2, onset < 60 months |
| NCT01353833 (DF‐IL2) | Aldesleukin (IL‐2) | Phase 1/2, onset < 24 months |
| NCT01862120 (DFIL2‐Child) | IL‐2 | Phase 2, recent onset |
| NCT02411253 (DIABIL‐2) | rhIL‐2 | Phase 2, recent onset |
| Cell‐based therapies | ||
| ISRCTN06128462 (Gdansk) | Polyclonal Tregs | Phase 1, onset < 2 months |
| NCT01210664 (UCSF) | Polyclonal Tregs | Phase 1, onset 3‐24 months |
| NCT00445913 (Pittsburgh) | Autologous DCs | Phase 1, long‐term T1D (5y+) |
| NCT02354911 (Pittsburgh) | Autologous DCs | Phase 2, new onset < 100d |
| NCT01068951 (Uppsala) | MSCs | Phase 1, new onset |
| NCT00690066 (Mesoblast) | Prochymal (MSCs) | Phase 2, onset 2‐20 wks |
| NCT02057211 (Uppsala) | MSCs | Phase 2, new onset < 3 weeks |
| NCT01322789 (Sao Paulo) | MSCs | Phase 1/2, new onset < 6 weeks |
| NCT00305344 (Florida) | Umbilical cord blood (UCB) | Phase 1/2, post‐onset |
| NCT00989547 (Munich) | Umbilical cord blood (UCB) | Phase 1, post‐onset |
| NCT01350219 (Tianhe) | UCB‐derived stem cells | Phase 2, post‐onset |
| NCT01996228 (Tianhe) | UCB‐derived stem cells | Phase 1/2, post‐onset |
| NCT00315133 (Sao Paulo) | Autologous HSCs | Phase 1/2, onset < 12 weeks |
| NCT01285934 (Northwestern) | Autologous HSCs | Phase 1/2, onset < 5 months |
Note: Clinical trials are color‐shaded based on whether they are completed, ongoing, or planned. Stem cells used for the generation of new β cells are not covered here. DCs: dendritic cells; HSCs: hematopoietic stem cells; MSCs: mesenchymal stem/stromal cells; Tregs: regulatory T cells.
However, the potential success of low dose IL‐2 therapy in T1D patients rests on two assumptions: (i) Tregs are functionally defective and (ii) IL‐2 production is impaired. Studies on whether CD4+ CD25+ Tregs are defective in T1D have yielded conflicting results (decreased frequency 62, decreased function 63, or normal frequency and function 64), which may reflect inadequate identification of Tregs by available markers; recruitment of patients different in age and disease progression and differences in experimental conditions. Follow‐up studies using more specific markers (FOXP3+ CD127low and demethylation of regulatory elements of the FOXP3 gene) showed that both the frequency and function of Tregs are normal in the blood of T1D patients, even though a transient decrease of suppressor activity may occur early after diagnosis 65, and in a subset of T1D patients 30. Studies from the Battaglia lab showed that reduced suppressive function of Tregs may be restricted to the pancreatic lymph nodes in patients with long lasting T1D 31. A defect in IL‐2 production by total peripheral blood mononuclear cells of patients with new onset T1D was reported several years ago 66 but never confirmed as a key immunological feature of T1D patients. A recent study showed that the T1D‐susceptibility IL2RA haplotype identified by rs12722495 is associated with decreased signaling via the IL‐2 pathway in both memory T cells and Tregs and that this is linked to diminished Treg function 32. However, this phenotype is limited to carriers of this single nucleotide polymorphism (SNP) and not to all individuals. Thus, it is likely that this treatment may benefit some patients more than others, again based on their underlying defects that contribute to disease.
A Wide Array of Approaches to Reestablish Antigen‐Specific Tolerance
The overall objective of this strategy is to deliver β cell antigens in particular ways such that their presentation in vivo results in elimination or inactivation of antigen‐specific diabetogenic T cells, or induction of antigen‐specific immunoregulatory populations, to confer durable protection from autoimmunity without compromising the general immunosurveillance for infectious agents and malignant cells. The traditional method has been to administer protein antigens via tolerogenic routes (mainly oral or intranasal insulin and GAD65/Alum), but this approach has not produced significant clinical benefit in recent onset patients 67. Because of lack of adverse side effects, these therapies are now being tested in secondary prevention trials (i.e., in patients with ongoing autoimmunity evidenced by circulating autoantibodies) (Table 1). It is worth pointing out that oral insulin has also been tested in a primary prevention trial (in young subject with no evidence of autoimmunity, Pre‐POINT trial, Table 1) and data suggest that insulin‐specific Tregs were induced at the highest dose 68. Antigens coupled with apoptotic cells have been known for several decades to be very tolerogenic and showed efficacy in preclinical models of T1D 69. This strategy has now been tested in patients with multiple sclerosis and was well tolerated 70. Massive apoptosis resulting from depletion of B cells and CD8+ T cells (using a short course of biologics) is accompanied by release of TGF‐β, which combined with exogenous antigens such as GAD65 peptides, supports the generation of protective Tregs, because CD4+ T cells are left untouched and available for conversion 71. This promising approach validated in mouse models of T1D and multiple sclerosis remains to be tested for safety in humans.
A less conventional alternative to protein antigen delivery lets the body produce specific antigens in cells or sites amenable for tolerance induction following gene transfer 72. Plasmid DNA encoding autoantigens such as insulin or its InsB9‐23 immunodominant peptide prevented disease in NOD mice 73, 74, 75 and was given to recent‐onset T1D patients in a phase 1 trial 76. Data from this trial demonstrated both safety and diminution of insulin‐reactive CD8+ T cells, thus tolerogenic DNA vaccines merit consideration for prevention trials. Delivery of autoantigens by viral vectors used for gene therapy has also been explored 77, 78. One legitimate concern when using viral components is the inadvertent activation of antigen‐specific effector T cells that could exacerbate β cell autoimmunity, especially if expression with ubiquitous promoters is allowed in professional antigen‐presenting cells (APCs) than can mature and become immunogenic 79. The insertion of a microRNA‐142 target sequence to abrogate transgene expression in professional APCs and other hematopoietic lineage cells, together with the use of a liver‐specific promoter resulted in a lentiviral vector, which specifically targets the antigens to hepatocytes 79, 80. Treatment with such a vector encoding the InsB9‐23 peptide‐induced some antigen‐specific effector T cells but also antigen‐specific CD4+ FoxP3+ Tregs, which halted islet immune cell infiltration, and protected mice from T1D 79. When combined with a single suboptimal dose of anti‐CD3 mAb, it was effective in reversing hyperglycemia after onset in a Treg‐dependent manner 79. The use of nonintegrating forms of lentiviral vectors will offer an additional level of safety when implementing such an approach clinically 81.
So far, antigen‐specific therapies for T1D have been proved efficient in mouse models and to be among the safest in patients, but evidence for clinical efficacy is lacking. One possible reason may have to do with the choice of β cell antigens used, which is limited in two aspects: (1) only a single antigen (mostly insulin or GAD65) is used despite the evidence of epitope spreading reflected by different types of autoantibodies, and (2) only native antigens are used while it has become increasingly clear that many diabetogenic T cells respond to post‐translationally modified or processed neo antigens. Accomplishing long‐term antigen‐specific tolerance, whether it is with Tregs or other regulatory cells will require these issues to be addressed.
Cell‐Based Therapies: Current and Future Applications
Cell‐based therapies are individualized approaches that currently involve the transfer of autologous cells that have immunoregulatory properties and can provide a counterbalance for effector T cells that mediate β cell destruction. While certain drugs aim at expanding and potentiating Tregs in vivo (see low dose IL‐2 above), cell‐based therapies generally involve Tregs or cells that have the ability to induce or potentiate such immunoregulatory populations in vivo. We will also discuss the use of different types of stem cells as part of cell‐based therapies to block autoimmune responses, but we will not cover the generation of new β cells from stem cells for transplantation, which is reviewed elsewhere 82.
Regulatory T Cells (Tregs)
Several preclinical animal studies have established that the adoptive transfer of Tregs can prevent various autoimmune diseases, T1D included. However, only a few studies showed that Treg cell transfer is efficacious in reverting active disease and, when it occurred, the transferred cells needed to be antigen‐specific 83, 84, 85. Based on this evidence, Tregs are now being used in phase 1/2 studies in patients with autoimmune diseases 86. Increasing doses of autologous ex vivo–expanded polyclonal CD4+CD25+ Tregs have been used safely in newly diagnosed T1D patients 87. These studies were instrumental in demonstrating the safety and feasibility of such a complex approach of personalized medicine, but efficacy has still to be demonstrated in larger trials.
Based on the data in animal models, to be of any therapeutic use, Tregs have to be transferred prior to overt hyperglycemia or have to be antigen‐specific. It has been, up to now, difficult to obtain sufficient antigen‐specific CD4+ FoxP3+ Tregs, but this might be more feasible by inducing Tregs de novo in vitro. Such an approach has been used in the context of allogeneic hematopoietic stem cell transplantation to prevent GvHD where host‐specific Tr1 cells were generated in vitro from donor peripheral blood and transferred to transplanted hosts 88. The induction of self‐specific Tr1 cells in NOD mice in vivo is feasible and they protect from diabetes development 89. However, the generation of human diabetes‐related antigen‐specific Tregs in vitro has yet to be achieved. Studies on adoptive T cell therapy have demonstrated the possibility of engineering T cells using lentivirus, either by expressing a relevant (β cell antigen‐specific) T cell receptor into polyclonal Tregs 90 or by overexpressing FoxP3 in T cells 91, potentially in antigen‐specific T cells that have been enriched and ex vivo expanded.
Regulatory B Cells (Bregs)
Recently, the IL‐10–producing regulatory Bregs have attracted attention as being altered in autoimmune diseases and thus represent another potential tool for cell therapy 92. As with Tregs, their numbers and function might be compromised in T1D patients 93, indicating the possibility of using Breg therapy, alone or in concert with Tregs. However, the importance of antigen‐specificity in this case is not clear, and considering that we are still at the beginning of cell therapy with Tregs, using Bregs is even more futuristic.
Dendritic Cells
As the most specialized of APCs, dendritic cells (DCs) have long been a candidate of choice for their ability to engage T cells through presentation of β cell antigens, and under a tolerogenic phenotype, to achieve deletion or inactivation of diabetogenic T cells, converting them into Tregs or restimulating preexisting Tregs. DC infusions have shown remarkable efficacy in numerous preclinical studies, even in the absence of exogenously provided antigens 94. The first clinical trial using autologous DCs in recent onset T1D patients demonstrated both safety and the potential to induce Bregs 95. In this phase 1 study, the first of its kind for the treatment of autoimmune diseases, the monocyte‐derived DCs were locked in a nonimmunogenic state by silencing important costimulatory molecules (CD40, CD80, and CD86), but were not provided exogenous antigen. Autoantigen expression and maturation stage are two crucial considerations, because immunogenic DCs expressing β cell antigens could boost autoimmune T cell responses against β cells. The antigen‐specific therapies previously described rely on the acquisition and presentation of relevant antigens by tolerogenic APCs that are not that well characterized but possibly comprising different subsets of DCs and other types of APCs. In the case of the recent DC trial, the mechanism of action is not completely understood as no exogenous antigen was provided, and whether these DCs could pick up and present relevant autoantigens in vivo remains unclear. Furthermore, there is evidence that DCs expressing costimulatory molecules but not inflammatory cytokines (termed semimature DCs) may induce tolerance as well 96.
Another phase 1 trial employing tolerogenic DCs pulsed with citrullinated peptides for the treatment of rheumatoid arthritis has demonstrated safety as well as immunological responses reflective of regulation 97, suggesting that provision of autoantigens may not lead to exacerbated responses as long as the DCs are maintained tolerogenic, which in this case was achieved by pretreatment with an NF‐κB inhibitor. An alternative or complementary approach to silencing the expression of costimulatory genes is the overexpression of tolerogenic products for which the list is long and includes immunoregulatory cytokines, inhibitory ligands, and metabolism‐altering enzymes 98. A safe and clinically viable way to overexpress genes and achieve a significant therapeutic outcome is by mRNA electroporation, which has been used widely for autologous DC therapy in cell therapy of cancer 99, 100. This method of modification allows for coexpression of multiple products of interest in the same cell and at the same time, including relevant antigens for added specificity if desired 101. Although transient, expression of genes in DCs by mRNA is sufficient to induce long‐lasting responses resulting in prevention of T1D in mice 102. A follow‐up trial of the first safety study of autologous DCs in T1D patients is set to begin in the near future.
Mesenchymal Stem/Stromal Cells
Mesenchymal stem/stromal cells (MSCs) are endowed with regenerative and immunosuppressive properties that have fueled their popularity in cell therapy, yet controversies remain regarding their name and definition 103. Although they can generally suppress immune responses on their own in a nonspecific manner, they have also been shown to induce or expand Tregs, including in preclinical T1D studies 104, 105, 106. Because MSCs are nonprofessional APCs, it is unclear if and how they specifically interact with Tregs and diabetogenic T cells, and their effect may be indirect through inflammation relief 107. Unlike DCs, MSCs are nonimmunogenic, and can provide protection even in an allogeneic host, which makes them attractive for the clinic 108. They have been proven to be well‐tolerated in T1D patients whether they were isolated by bone marrow aspiration 109 or from adipose tissue 110, and associated with improvement of disease parameters such as C‐peptide preservation. Careful characterization of the phenotype and properties of MSCs used in cell therapy is crucial to demonstrate consistency between studies and draw meaningful conclusions, regardless of the source of the cells, isolation, and culture conditions (Table 4). Two follow‐up clinical trials are currently recruiting in Sweden and Brazil to demonstrate long‐term efficacy (Table 3). All trials so far are being conducted in recently diagnosed T1D patients using MSCs that are either autologous or from first‐degree relatives. Although no serious side effects have been reported so far, there remains a concern that slow growing tumors may appear in the long term in some patients receiving autologous cells, according to some preclinical studies 108.
Table 4.
Phenotype of MSCs used in T1D studies
| Source | CD11c | CD14 | CD29 | CD31 | CD34 | CD44 | CD45 | CD73 | CD90 | CD105 | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mouse bone marrow | − | − | + | − | + | − | + | + | 104 | ||
| Mouse bone marrow | + | + | − | + | − | + | 108 | ||||
| Mouse adipose tissue | − | − | + | − | + | + | + | 105 | |||
| Human bone marrow | − | − | − | − | + | + | + | 109 | |||
| Human bone marrow | − | + | − | + | − | + | + | + | [*] | ||
| Human adipose tissue | − | + | + | 110 |
Note: Additional characterization may include ability to terminally differentiate (e.g., adipocytes) or to suppress T cell responses. [*] Prochymal MSCs (NCT00690066).
Umbilical Cord Blood
Although this approach is limited to a few individuals who have banked samples, umbilical cord blood (UCB) is a great source of abundant MSCs and Tregs, which might work in synergy when infused into patients. As many important self‐reactive Tregs appear to be released early in life 111, these Tregs may also include more antigen‐specific Tregs of relevance. Autologous UCB infusion was found to be safe but did not have any significant therapeutic effect despite increased numbers of Tregs 112, 113 and even with oral docosahexaenoic acid and vitamin D supplementation 114. Another phase 1 trial, also in new onset pediatric subjects, is well under way in Germany (Table 3). As previously mentioned, it is possible that therapies involving Tregs would be more effective when applied prior to disease onset. In that model, the Cord Reinfusion in Diabetes (CoRD) study, enrolling high‐risk children with banked UCB, was recently initiated in Australia and represents the first cell‐based therapy used for secondary prevention of diabetes 115. A distinct process is being tested in China, whereby lymphocytes are obtained from the blood by leukapheresis, “reeducated” ex vivo in contact with UCB‐derived stem cells, and then reinfused into the patient. These studies suggest that this treatment improved preservation of β cell function without notable adverse effects, but caution must be exercised in the interpretation of these studies, which were improperly controlled 116.
Hematopoietic Stem Cells
Perhaps the most drastic of all cell therapies consists of a major immunological reset with the transient ablation of circulating T cells and their replacement with hematopoietic stem cells (HSCs). Multiple completed studies have involved autologous HSCs mobilized from peripheral blood and administered after a nonmyeloablative regimen consisting of cyclophosphamide and anti‐thymocyte globulin 117, 118, 119. This treatment performed in new‐onset T1D patients demonstrated a remarkable ability to normalize glycemia in a majority of the subjects. Independence from insulin lasted between several months and several years, up to 3‐4 years (as reported in the last meeting of the Immunology of Diabetes Society), and a larger trial is now enrolling patients (ClinicalTrial.gov NCT01285934). However, this therapy is fraught with considerable side effects associated with the nonmyeloablative regimen 117, 118, 119, which make this approach unattractive in its current form to many prospective patients and precludes its use for prevention. Furthermore, the contribution of HSCs in maintaining normoglycemia is unclear as the immunosuppressive effect of the regimen may account for part if not all of the therapeutic benefit. Finally, new T cells (and other immune cells) generated from autologous HSCs would still carry any inherent genetic defects that may play a role in disease etiology. Transplantation of bone marrow‐derived allogeneic HSCs with induction of mixed chimerism has also been tested in a minority of patients with autoimmunity, including T1D 120. The use of HSCs in combination with islet transplant to induce chimerism and immunological tolerance has been tested in a recent trial at the University of Miami based on campath‐1H and infusion of donor CD34+ HSCs (ClinicalTrials.gov NCT00315614), but did not show any significant benefit. Although allogeneic HSCs from a compatible and healthy donor may help correct some genetic abnormalities, this must be preceded by high doses of chemotherapy and radiation to ablate the patient's bone marrow and is followed by prolonged immunosuppression to prevent GvHD. The consequent transplant‐related morbidity and mortality limit this approach to patients with concomitant hematological malignancies 121. It should be noted that preclinical data with purified allogeneic CD34+ CD90+ HSCs showed complete reversion of T1D in the absence of GvHD 122. In addition, novel biologicals are under investigation for use as safer and less toxic drugs to myeloablate the patient's bone marrow 123. Thus, as safer and more effective HSC transplantation protocols become available, allogeneic HSCs might also be indicated in T1D. A more detailed review of the different applications of stem cells (including MSCs and UCB) to treat T1D can be found elsewhere 124.
In parallel to efforts in generating insulin‐producing cells from stem cells (embryonic stem cells or induced pluripotent stem cells) 82, there is an expanded interest in growing tissues specialized in tolerance induction, such as thymic tissue 125, 126. Although much can be learned from these studies, the clinical implementation of such advances is elusive, including where and how to implant the new tissue.
Conclusion
A variety of original therapeutic strategies for treating or preventing T1D have emerged in the past decade, with the latest approaches clearly dominated by cell‐based therapies. As the least expensive and most conventional therapies have failed to deliver efficient and durable protection from diabetogenic immune responses, testing of more expensive, and individualized therapies has become justified as long as preclinical studies indicate a strong prospect of durable efficacy achieved with a minimal number of treatments. Strategies that are more antigen‐specific and less immunosuppressive tend to have the best safety profile, and their poor efficacy in new onset patients should not discourage evaluation in prevention trials in high‐risk patients in which they might perform surprisingly well. The enrollment of subjects for prevention studies should be guided by more refined biomarkers, which may help the diabetes community to better understand the underlying defects behind the autoimmune response in each patient, and better tailor the treatment type, dose, and timing. When appropriate, two or more of these therapies may be combined in order to address multiple defects and benefit a larger number of patients. A clear advantage of cell‐based therapies is that they can perform multiple tasks. For example, one can envision tolerogenic APCs engineered to express selected β cell antigens (to specifically engage diabetogenic T cells), additional immunoregulatory ligands or cytokines (to potentiate T cell deletion or Treg induction), homing molecules (for targeting to inflamed islets or their draining lymph nodes), anti‐inflammatory cytokines (to quench inflammation), and even growth factors to promote β cell replication.
Author Contributions
All authors wrote and edited the manuscript
Acknowledgments
RJC is currently funded by the National Institutes of Health (NIH), the Diabetes & Endocrinology Research Center (Columbia University), and the Irving Institute for Clinical and Translational Research; MB by the Italian Ministry of Health, the European Community, the Cariplo Foundation, and the Juvenile Diabetes Research Foundation (JDRF); MGR by the Italian Telethon Foundation and JDRF; and CGF by the NIH and JDRF.
References
- 1. Diamond Project Group . Incidence and trends of childhood Type 1 diabetes worldwide 1990‐1999. Diabet Med 2006;23:857–866. [DOI] [PubMed] [Google Scholar]
- 2. Patterson CC, Dahlquist GG, Gyurus E et al. Incidence trends for childhood type 1 diabetes in Europe during 1989‐2003 and predicted new cases 2005‐20: A multicentre prospective registration study. Lancet 2009;373:2027–2033. [DOI] [PubMed] [Google Scholar]
- 3. Bottazzo GF, Florin‐Christensen A, Doniach D. Islet‐cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet 1974;2:1279–1283. [DOI] [PubMed] [Google Scholar]
- 4. Eisenbarth GS. Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med 1986;314:1360–1368. [DOI] [PubMed] [Google Scholar]
- 5. Feutren G, Papoz L, Assan R et al. Cyclosporin increases the rate and length of remissions in insulin‐dependent diabetes of recent onset. Results of a multicentre double‐blind trial. Lancet. 1986;2:119–124. [DOI] [PubMed] [Google Scholar]
- 6. Cyclosporin‐induced remission of IDDM after early intervention . Association of 1 yr of cyclosporin treatment with enhanced insulin secretion. The Canadian‐European Randomized Control Trial Group. Diabetes 1988;37:1574–1582. [PubMed] [Google Scholar]
- 7. Eisenbarth GS, Srikanta S, Jackson R et al. Anti‐thymocyte globulin and prednisone immunotherapy of recent onset type 1 diabetes mellitus. Diabetes Res 1985;2:271–276. [PubMed] [Google Scholar]
- 8. Todd JA. Etiology of type 1 diabetes. Immunity 2010;32:457–467. [DOI] [PubMed] [Google Scholar]
- 9. Ziegler AG, Nepom GT. Prediction and pathogenesis in type 1 diabetes. Immunity 2010;32:468–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kaizer EC, Glaser CL, Chaussabel D et al. Gene expression in peripheral blood mononuclear cells from children with diabetes. J Clin Endocrinol Metab 2007;92:3705–3711. [DOI] [PubMed] [Google Scholar]
- 11. Jin Y, She JX. Novel biomarkers in type 1 diabetes. Rev Diabet Stud 2012;9:224–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jin Y, Sharma A, Bai S et al. Risk of type 1 diabetes progression in islet autoantibody‐positive children can be further stratified using expression patterns of multiple genes implicated in peripheral blood lymphocyte activation and function. Diabetes 2014;63:2506–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kallionpaa H, Elo LL, Laajala E et al. Innate immune activity is detected prior to seroconversion in children with HLA‐conferred type 1 diabetes susceptibility. Diabetes 2014;63:2402–2414. [DOI] [PubMed] [Google Scholar]
- 14. Tooley JE, Herold KC. Biomarkers in type 1 diabetes: Application to the clinical trial setting. Curr Opin Endocrinol Diabetes Obes 2014;21:287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kulmala P, Savola K, Petersen JS et al. Prediction of insulin‐dependent diabetes mellitus in siblings of children with diabetes. A population‐based study. The Childhood Diabetes in Finland Study Group. J Clin Invest 1998;101:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. LaGasse JM, Brantley MS, Leech NJ et al. Successful prospective prediction of type 1 diabetes in schoolchildren through multiple defined autoantibodies: An 8‐year follow‐up of the Washington State Diabetes Prediction Study. Diabetes Care 2002;25:505–511. [DOI] [PubMed] [Google Scholar]
- 17. Bingley PJ, Gale EA, European Nicotinamide Diabetes Intervention Trial G . Progression to type 1 diabetes in islet cell antibody‐positive relatives in the European Nicotinamide Diabetes Intervention Trial: The role of additional immune, genetic and metabolic markers of risk. Diabetologia 2006;49:881–890. [DOI] [PubMed] [Google Scholar]
- 18. Sosenko JM, Krischer JP, Palmer JP et al. A risk score for type 1 diabetes derived from autoantibody‐positive participants in the diabetes prevention trial‐type 1. Diabetes Care 2008;31:528–533. [DOI] [PubMed] [Google Scholar]
- 19. Akirav EM, Lebastchi J, Galvan EM et al. Detection of beta cell death in diabetes using differentially methylated circulating DNA. Proc Natl Acad Sci USA 2011;108:19018–19023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Herold KC, Usmani‐Brown S, Ghazi T et al. Beta cell death and dysfunction during type 1 diabetes development in at‐risk individuals. J Clin Invest 2015;125:1163–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rakyan VK, Beyan H, Down TA et al. Identification of type 1 diabetes‐associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet 2011;7:e1002300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gaglia JL, Harisinghani M, Aganj I et al. Noninvasive mapping of pancreatic inflammation in recent‐onset type‐1 diabetes patients. Proc Natl Acad Sci USA 2015;112:2139–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shibasaki S, Imagawa A, Hanafusa T. Fulminant type 1 diabetes mellitus: A new class of type 1 diabetes. Adv Exp Med Biol 2012;771:20–23. [DOI] [PubMed] [Google Scholar]
- 24. Naik RG, Brooks‐Worrell BM, Palmer JP. Latent autoimmune diabetes in adults. J Clin Endocrinol Metab 2009;94:4635–4644. [DOI] [PubMed] [Google Scholar]
- 25. Cerosaletti K, Buckner JH. Protein tyrosine phosphatases and type 1 diabetes: Genetic and functional implications of PTPN2 and PTPN22. Rev Diabet Stud 2012;9:188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Herold KC, Brooks‐Worrell B, Palmer J et al. Validity and reproducibility of measurement of islet autoreactivity by T‐cell assays in subjects with early type 1 diabetes. Diabetes 2009;58:2588–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rich SS, Concannon P. Role of type 1 diabetes‐associated SNPs on autoantibody positivity in the type 1 diabetes genetics consortium: Overview. Diabetes Care 2015;38(Suppl 2):S1–S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pugliese A, Zeller M, Fernandez A, Jr. et al. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR‐IDDM2 susceptibility locus for type 1 diabetes. Nat Genet 1997;15:293–297. [DOI] [PubMed] [Google Scholar]
- 29. Vafiadis P, Bennett ST, Todd JA et al. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat Genet 1997;15:289–292. [DOI] [PubMed] [Google Scholar]
- 30. Lawson JM, Tremble J, Dayan C et al. Increased resistance to CD4+CD25hi regulatory T cell‐mediated suppression in patients with type 1 diabetes. Clin Exp Immunol 2008;154:353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ferraro A, Socci C, Stabilini A et al. Expansion of Th17 cells and functional defects in T regulatory cells are key features of the pancreatic lymph nodes in patients with type 1 diabetes. Diabetes 2011;60:2903–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Garg G, Tyler JR, Yang JH et al. Type 1 diabetes‐associated IL2RA variation lowers IL‐2 signaling and contributes to diminished CD4+CD25 + regulatory T cell function. J Immunol 2012;188:4644–4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schneider A, Rieck M, Sanda S et al. The effector T cells of diabetic subjects are resistant to regulation via CD4 + FOXP3 + regulatory T cells. J Immunol 2008;181:7350–7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. de St Groth BF. Regulatory T‐cell abnormalities and the global epidemic of immuno‐inflammatory disease. Immunol Cell Biol 2012;90:256–259. [DOI] [PubMed] [Google Scholar]
- 35. Adamczak DM, Nowak JK, Frydrychowicz M et al. The role of Toll‐like receptors and vitamin D in diabetes mellitus type 1‐‐A review. Scand J Immunol 2014;80:75–84. [DOI] [PubMed] [Google Scholar]
- 36. McClymont SA, Putnam AL, Lee MR et al. Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J Immunol 2011;186:3918–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mollah ZU, Pai S, Moore C et al. Abnormal NF‐kappa B function characterizes human type 1 diabetes dendritic cells and monocytes. J Immunol 2008;180:3166–3175. [DOI] [PubMed] [Google Scholar]
- 38. Badami E, Sorini C, Coccia M et al. Defective differentiation of regulatory FoxP3 + T cells by small‐intestinal dendritic cells in patients with type 1 diabetes. Diabetes 2011;60:2120–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fisher MM, Watkins RA, Blum J et al. Elevations in circulating methylated and unmethylated preproinsulin DNA in new‐onset type 1 diabetes. Diabetes 2015;64:3867–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McGinty JW, Marre ML, Bajzik V et al. T cell epitopes and post‐translationally modified epitopes in type 1 diabetes. Curr Diabetes Rep 2015;15:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Talchai C, Xuan S, Lin HV et al. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012;150:1223–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Diana J, Simoni Y, Furio L et al. Crosstalk between neutrophils, B‐1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat Med 2013;19:65–73. [DOI] [PubMed] [Google Scholar]
- 43. Mathis D, Vence L, Benoist C. Beta‐cell death during progression to diabetes. Nature 2001;414:792–798. [DOI] [PubMed] [Google Scholar]
- 44. Xu GJ, Kula T, Xu Q et al. Viral immunology. Comprehensive serological profiling of human populations using a synthetic human virome. Science 2015;348:aaa0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ferreira RC, Guo H, Coulson RM et al. A type I interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes 2014;63:2538–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Krogvold L, Edwin B, Buanes T et al. Detection of a low‐grade enteroviral infection in the islets of langerhans of living patients newly diagnosed with type 1 diabetes. Diabetes 2015;64:1682–1687. [DOI] [PubMed] [Google Scholar]
- 47. Pane JA, Coulson BS. Lessons from the mouse: Potential contribution of bystander lymphocyte activation by viruses to human type 1 diabetes. Diabetologia 2015;58:1149–1159. [DOI] [PubMed] [Google Scholar]
- 48. Kostic AD, Gevers D, Siljander H et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 2015;17:260–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Herold KC, Hagopian W, Auger JA et al. Anti‐CD3 monoclonal antibody in new‐onset type 1 diabetes mellitus. N Engl J Med 2002;346:1692–1698. [DOI] [PubMed] [Google Scholar]
- 50. Herold KC, Gitelman SE, Masharani U et al. A single course of anti‐CD3 monoclonal antibody hOKT3gamma1(Ala‐Ala) results in improvement in C‐peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes 2005;54:1763–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Keymeulen B, Vandemeulebroucke E, Ziegler AG et al. Insulin needs after CD3‐antibody therapy in new‐onset type 1 diabetes. N Engl J Med 2005;352:2598–2608. [DOI] [PubMed] [Google Scholar]
- 52. Pescovitz MD, Greenbaum CJ, Krause‐Steinrauf H et al. Rituximab, B‐lymphocyte depletion, and preservation of beta‐cell function. New Engl J Med 2009;361:2143–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Orban T, Bundy B, Becker DJ et al. Co‐stimulation modulation with abatacept in patients with recent‐onset type 1 diabetes: A randomised, double‐blind, placebo‐controlled trial. Lancet 2011;378:412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yu A, Snowhite I, Vendrame F et al. Selective IL‐2 responsiveness of regulatory T cells through multiple intrinsic mechanisms supports the use of low‐dose IL‐2 therapy in type 1 diabetes. Diabetes 2015;64:2172–2183. [DOI] [PubMed] [Google Scholar]
- 55. Koreth J, Matsuoka K, Kim HT et al. Interleukin‐2 and regulatory T cells in graft‐versus‐host disease. N Engl J Med 2011;365:2055–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Matsuoka K, Koreth J, Kim HT et al. Low‐dose interleukin‐2 therapy restores regulatory T cell homeostasis in patients with chronic graft‐versus‐host disease. Sci Transl Med 2013;5:179ra143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kennedy‐Nasser AA, Ku S, Castillo‐Caro P et al. Ultra low‐dose IL‐2 for GVHD prophylaxis after allogeneic hematopoietic stem cell transplantation mediates expansion of regulatory T cells without diminishing antiviral and antileukemic activity. Clin Cancer Res 2014;20:2215–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Saadoun D, Rosenzwajg M, Joly F et al. Regulatory T‐cell responses to low‐dose interleukin‐2 in HCV‐induced vasculitis. N Engl J Med 2011;365:2067–2077. [DOI] [PubMed] [Google Scholar]
- 59. Oo YH, Mutimer D, Adams DH. Low‐dose interleukin‐2 and HCV‐induced vasculitis. N Engl J Med 2012;366:1353–1354; author reply 1354. [DOI] [PubMed] [Google Scholar]
- 60. Hartemann A, Bensimon G, Payan CA et al. Low‐dose interleukin 2 in patients with type 1 diabetes: A phase 1/2 randomised, double‐blind, placebo‐controlled trial. Lancet Diabetes Endocrinol 2013;1:295–305. [DOI] [PubMed] [Google Scholar]
- 61. Grinberg‐Bleyer Y, Baeyens A, You S et al. IL‐2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med 2010;207:1871–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kukreja A, Cost G, Marker J et al. Multiple immuno‐regulatory defects in type‐1 diabetes. J Clin Invest 2002;109:131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lindley S, Dayan C, Bishop A et al. Defective suppressor function in CD4(+)CD25(+) T‐cells from patients with type 1 diabetes. Diabetes 2005;54:92–99. [DOI] [PubMed] [Google Scholar]
- 64. Putnam AL, Vendrame F, Dotta F et al. CD4+CD25 high regulatory T cells in human autoimmune diabetes. J Autoimmun 2005;24:55–62. [DOI] [PubMed] [Google Scholar]
- 65. Hughson A, Bromberg I, Johnson B et al. Uncoupling of proliferation and cytokines from suppression within the CD4+CD25+Foxp3 + T‐cell compartment in the 1st year of human type 1 diabetes. Diabetes 2011;60:2125–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kaye WA, Adri MN, Soeldner JS et al. Acquired defect in interleukin‐2 production in patients with type I diabetes mellitus. N Engl J Med 1986;315:920–924. [DOI] [PubMed] [Google Scholar]
- 67. Coppieters KT, Harrison LC, von Herrath MG. Trials in type 1 diabetes: Antigen‐specific therapies. Clin Immunol 2013;149:345–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bonifacio E, Ziegler AG, Klingensmith G et al. Effects of high‐dose oral insulin on immune responses in children at high risk for type 1 diabetes: The pre‐POINT randomized clinical trial. JAMA 2015;313:1541–1549. [DOI] [PubMed] [Google Scholar]
- 69. Prasad S, Xu D, Miller SD. Tolerance strategies employing antigen‐coupled apoptotic cells and carboxylated PLG nanoparticles for the treatment of type 1 diabetes. Rev Diabet Stud 2012;9:319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lutterotti A, Yousef S, Sputtek A et al. Antigen‐specific tolerance by autologous myelin peptide‐coupled cells: A phase 1 trial in multiple sclerosis. Sci Transl Med 2013;5:188ra175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kasagi S, Zhang P, Che L et al. In vivo‐generated antigen‐specific regulatory T cells treat autoimmunity without compromising antibacterial immune response. Sci Transl Med 2014;6:241ra278. [DOI] [PubMed] [Google Scholar]
- 72. Wasserfall CH, Herzog RW. Gene therapy approaches to induce tolerance in autoimmunity by reshaping the immune system. Curr Opin Investig Drugs 2009;10:1143–1150. [PubMed] [Google Scholar]
- 73. Bot A, Smith D, Bot S et al. Plasmid vaccination with insulin B chain prevents autoimmune diabetes in nonobese diabetic mice. J Immunol 2001;167:2950–2955. [DOI] [PubMed] [Google Scholar]
- 74. Chang Y, Yap S, Ge X et al. DNA vaccination with an insulin construct and a chimeric protein binding to both CTLA4 and CD40 ameliorates type 1 diabetes in NOD mice. Gene Ther 2005;12:1679–1685. [DOI] [PubMed] [Google Scholar]
- 75. Solvason N, Lou YP, Peters W et al. Improved efficacy of a tolerizing DNA vaccine for reversal of hyperglycemia through enhancement of gene expression and localization to intracellular sites. J Immunol 2008;181:8298–8307. [DOI] [PubMed] [Google Scholar]
- 76. Roep BO, Solvason N, Gottlieb PA et al. Plasmid‐encoded proinsulin preserves C‐peptide while specifically reducing proinsulin‐specific CD8(+) T cells in type 1 diabetes. Sci Transl Med 2013;5:191ra182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Han G, Wang R, Chen G et al. Gene delivery GAD 500 autoantigen by AAV serotype 1 prevented diabetes in NOD mice: Transduction efficiency do not play important roles. Immunol Lett 2008;115:110–116. [DOI] [PubMed] [Google Scholar]
- 78. Luth S, Huber S, Schramm C et al. Ectopic expression of neural autoantigen in mouse liver suppresses experimental autoimmune neuroinflammation by inducing antigen‐specific Tregs. J Clin Invest 2008;118:3403–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Akbarpour M, Goudy KS, Cantore A et al. Insulin B chain 9‐23 gene transfer to hepatocytes protects from type 1 diabetes by inducing Ag‐specific FoxP3 + Tregs. Sci Transl Med 2015;7:289ra281. [DOI] [PubMed] [Google Scholar]
- 80. Brown BD, Venneri MA, Zingale A et al. Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat Med 2006;12:585–591. [DOI] [PubMed] [Google Scholar]
- 81. Annoni A, Goudy K, Akbarpour M et al. Immune responses in liver‐directed lentiviral gene therapy. Transl Res 2013;161:230–240. [DOI] [PubMed] [Google Scholar]
- 82. Johannesson B, Sui L, Freytes DO et al. Toward beta cell replacement for diabetes. EMBO J 2015;34:841–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Roncarolo MG, Battaglia M. Regulatory T‐cell immunotherapy for tolerance to self antigens and alloantigens in humans. Nat Rev Immunol 2007;7:585–598. [DOI] [PubMed] [Google Scholar]
- 84. Tang Q, Henriksen KJ, Bi M et al. In vitro‐expanded antigen‐specific regulatory T cells suppress autoimmune diabetes. J Exp Med 2004;199:1455–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Tarbell KV, Yamazaki S, Olson K et al. CD25 + CD4 + T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J Exp Med 2004;199:1467–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Trzonkowski P, Bacchetta R, Battaglia M et al. Hurdles in therapy with regulatory T cells. Sci Transl Med 2015;7:304ps318. [DOI] [PubMed] [Google Scholar]
- 87. Marek‐Trzonkowska N, Mysliwiec M, Dobyszuk A et al. Therapy of type 1 diabetes with CD4(+)CD25(high)CD127‐regulatory T cells prolongs survival of pancreatic islets ‐ Results of one year follow‐up. Clin Immunol 2014;153:23–30. [DOI] [PubMed] [Google Scholar]
- 88. Bacchetta R, Lucarelli B, Sartirana C et al. Immunological outcome in haploidentical‐HSC transplanted patients treated with IL‐10‐anergized donor T cells. Front Immunol 2014;5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Battaglia M, Stabilini A, Draghici E et al. Induction of tolerance in type 1 diabetes via both CD4+CD25 + T regulatory cells and T regulatory type 1 cells. Diabetes 2006;55:1571–1580. [DOI] [PubMed] [Google Scholar]
- 90. Brusko TM, Koya RC, Zhu S et al. Human antigen‐specific regulatory T cells generated by T cell receptor gene transfer. PloS One 2010;5:e11726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Allan SE, Alstad AN, Merindol N et al. Generation of potent and stable human CD4 + T regulatory cells by activation‐independent expression of FOXP3. Mol Ther 2008;16:194–202. [DOI] [PubMed] [Google Scholar]
- 92. Tedder TF. B10 cells: A functionally defined regulatory B cell subset. J Immunol 2015;194:1395–1401. [DOI] [PubMed] [Google Scholar]
- 93. Kleffel S, Vergani A, Tezza S et al. Interleukin‐10 + regulatory B cells arise within antigen‐experienced CD40 + B cells to maintain tolerance to islet autoantigens. Diabetes 2015;64:158–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Creusot RJ, Giannoukakis N, Trucco M et al. It's time to bring dendritic cell therapy to type 1 diabetes. Diabetes 2014;63:20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Giannoukakis N, Phillips B, Finegold D et al. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care 2011;34:2026–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Gordon JR, Ma Y, Churchman L et al. Regulatory dendritic cells for immunotherapy in immunologic diseases. Front Immunol 2014;5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Benham H, Nel HJ, Law SC et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype‐positive rheumatoid arthritis patients. Sci Transl Med 2015;7:290ra287. [DOI] [PubMed] [Google Scholar]
- 98. Schinnerling K, Soto L, Garcia‐Gonzalez P et al. Skewing dendritic cell differentiation towards a tolerogenic state for recovery of tolerance in rheumatoid arthritis. Autoimmun Rev 2015;14:517–527. [DOI] [PubMed] [Google Scholar]
- 99. Kyte JA, Gaudernack G. Immuno‐gene therapy of cancer with tumour‐mRNA transfected dendritic cells. Cancer Immunol Immunother 2006;55:1432–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Shurin MR, Gregory M, Morris JC et al. Genetically modified dendritic cells in cancer immunotherapy: A better tomorrow? Expert Opin Biol Ther 2010;10:1539–1553. [DOI] [PubMed] [Google Scholar]
- 101. Bonehill A, Tuyaerts S, Van Nuffel A et al. Enhancing the T‐cell stimulatory capacity of human dendritic cells by co‐electroporation with CD40L, CD70 and constitutively active TLR4 encoding mRNA. Mol Ther 2008;16:1170–1180. [DOI] [PubMed] [Google Scholar]
- 102. Creusot RJ, Chang P, Healey DG et al. A short pulse of IL‐4 delivered by DCs electroporated with modified mRNA can both prevent and treat autoimmune diabetes in NOD mice. Mol Ther 2010;18:2112–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Bianco P. “Mesenchymal” stem cells. Annu Rev Cell Dev Biol 2014;30:677–704. [DOI] [PubMed] [Google Scholar]
- 104. Madec AM, Mallone R, Afonso G et al. Mesenchymal stem cells protect NOD mice from diabetes by inducing regulatory T cells. Diabetologia 2009;52:1391–1399. [DOI] [PubMed] [Google Scholar]
- 105. Bassi EJ, Moraes‐Vieira PM, Moreira‐Sa CS et al. Immune regulatory properties of allogeneic adipose‐derived mesenchymal stem cells in the treatment of experimental autoimmune diabetes. Diabetes 2012;61:2534–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kota DJ, Wiggins LL, Yoon N et al. TSG‐6 produced by hMSCs delays the onset of autoimmune diabetes by suppressing Th1 development and enhancing tolerogenicity. Diabetes 2013;62:2048–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Burr SP, Dazzi F, Garden OA. Mesenchymal stromal cells and regulatory T cells: The Yin and Yang of peripheral tolerance? Immunol Cell Biol 2013;91:12–18. [DOI] [PubMed] [Google Scholar]
- 108. Fiorina P, Jurewicz M, Augello A et al. Immunomodulatory function of bone marrow‐derived mesenchymal stem cells in experimental autoimmune type 1 diabetes. J Immunol 2009;183:993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Carlsson PO, Schwarcz E, Korsgren O et al. Preserved beta‐cell function in type 1 diabetes by mesenchymal stromal cells. Diabetes 2015;64:587–592. [DOI] [PubMed] [Google Scholar]
- 110. Thakkar UG, Trivedi HL, Vanikar AV et al. Insulin‐secreting adipose‐derived mesenchymal stromal cells with bone marrow‐derived hematopoietic stem cells from autologous and allogenic sources for type 1 diabetes mellitus. Cytotherapy 2015;17:940–947. [DOI] [PubMed] [Google Scholar]
- 111. Yang S, Fujikado N, Kolodin D et al. Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self‐tolerance. Science 2015;348:589–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Haller MJ, Wasserfall CH, Hulme MA et al. Autologous umbilical cord blood transfusion in young children with type 1 diabetes fails to preserve C‐peptide. Diabetes Care 2011;34:2567–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Giannopoulou EZ, Puff R, Beyerlein A et al. Effect of a single autologous cord blood infusion on beta‐cell and immune function in children with new onset type 1 diabetes: A non‐randomized, controlled trial. Pediatr Diabetes 2014;15:100–109. [DOI] [PubMed] [Google Scholar]
- 114. Haller MJ, Wasserfall CH, Hulme MA et al. Autologous umbilical cord blood infusion followed by oral docosahexaenoic acid and vitamin D supplementation for C‐peptide preservation in children with Type 1 diabetes. Biol Blood Marrow Transpl 2013;19:1126–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Han MX, Craig ME. Research using autologous cord blood ‐ Time for a policy change. Med J Aust 2013;199:288–299. [DOI] [PubMed] [Google Scholar]
- 116. Zhao Y, Jiang Z, Zhao T et al. Reversal of type 1 diabetes via islet beta cell regeneration following immune modulation by cord blood‐derived multipotent stem cells. BMC Med 2012;10:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Voltarelli JC, Couri CE, Stracieri AB et al. Autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus. JAMA 2007;297:1568–1576. [DOI] [PubMed] [Google Scholar]
- 118. Couri CE, Oliveira MC, Stracieri AB et al. C‐peptide levels and insulin independence following autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus. JAMA 2009;301:1573–1579. [DOI] [PubMed] [Google Scholar]
- 119. D'Addio F, Valderrama Vasquez A, Ben Nasr M et al. Autologous nonmyeloablative hematopoietic stem cell transplantation in new‐onset type 1 diabetes: A multicenter analysis. Diabetes 2014;63:3041–3046. [DOI] [PubMed] [Google Scholar]
- 120. Pasquini MC, Voltarelli J, Atkins HL et al. Transplantation for autoimmune diseases in North and South America: A report of the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transpl 2012;18:1471–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Battaglia M. Experiments by nature: Lessons on type 1 diabetes. Tissue Antigens 2014;83:1–9. [DOI] [PubMed] [Google Scholar]
- 122. Beilhack GF, Landa RR, Masek MA et al. Prevention of type 1 diabetes with major histocompatibility complex‐compatible and nonmarrow ablative hematopoietic stem cell transplants. Diabetes 2005;54:1770–1779. [DOI] [PubMed] [Google Scholar]
- 123. Czechowicz A, Kraft D, Weissman IL et al. Efficient transplantation via antibody‐based clearance of hematopoietic stem cell niches. Science 2007;318:1296–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Fiorina P, Voltarelli J, Zavazava N. Immunological applications of stem cells in type 1 diabetes. Endocr Rev 2011;32:725–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Sun X, Xu J, Lu H et al. Directed differentiation of human embryonic stem cells into thymic epithelial progenitor‐like cells reconstitutes the thymic microenvironment in vivo. Cell Stem Cell 2013;13:230–236. [DOI] [PubMed] [Google Scholar]
- 126. Parent AV, Russ HA, Khan IS et al. Generation of functional thymic epithelium from human embryonic stem cells that supports host T cell development. Cell Stem Cell 2013;13:219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]