

Abstract
In light of the central role of inflammation in normal wound repair and regeneration, we hypothesize that the preponderance of human‐specific genes expressed in human inflammatory cells is commensurate with the genetic versatility of inflammatory response and the emergence of injuries associated with uniquely hominid behaviors, like a bipedal posture and the use of tools, weapons and fire. The hypothesis underscores the need to study human‐specific signaling pathways in experimental models of injury and infers that a selection of human‐specific genes, driven in part by the response to injury, may have facilitated the emergence of multifunctional genes expressed in other tissues.
The fact that new genes emerge during speciation is not unexpected, although their significance to the injury response remains largely enigmatic. Yet, for over 60 years, a tenet of modern molecular biology has held that essential genes, like those needed for tissue repair, have significant protein and/or DNA sequence homology across multiple species.1 As a consequence, the contributions of human‐specific genes to the onset and progression of human disease have been largely understudied.2 In contrast, the genetic mechanisms to explain the emergence of these taxonomically restricted genes are well described. They include complete and/or partial gene duplication, gene rearrangement, silencing, mutagenesis and, the endogenization of foreign DNA.3
If an increased incidence of injury can be associated with or be the result of newly acquired behaviors, then it is reasonable to presume that plasticity in the inflammatory response can contribute to the emergence of new genes. This raises the possibility that selection pressures conferred by injury, and specifically the inflammatory responses that are tied to injury, might drive the adaptation of newly emerged genes to new functions in inflammatory cells. To this end, it is noteworthy that the ancestors of homo sapiens acquired three new injury‐prone behaviors that are hallmarks of the species. First, when early hominid moved between trees and the savannah, they became erect and bipedal. Second, they learned to use tools and weapons so as to modify their environment for hunting and compete for habitation. Finally, when hominid harnessed fire and turned to cooking food, their gut microbiome was fundamentally altered. Presumably, the acquisition of these behaviors came with significant risk for trauma and burn injury: bipedal motion and tool/weapon‐making skills led to the appearance of new kinds of pressure and penetrating injuries while the use of fire, another uniquely human activity, was likely associated with accidental burn of skin and hair and smoke inhalation. Like no other species, some early human societies are even known for using injury response (scar formation) to confer reproductive advantage.
In modern humans, there are approximately 300 human‐specific genes that distinguish the human genome from that of great apes.4 There are perhaps another 2,000 that differentiate primates from other species.3 For the most part, however, the reasons for the retention of these genes in the human genome remain to be investigated. Neuroscientists posit that human‐specific genes were selected to regulate traits that are uniquely human: size of the human brain, cognition, memory, and consciousness. To others, their very existence has been enigmatic, their functions unknown and the reasons for their over‐representation in complex human disease,5 an interesting, albeit cursory problem in translational research. We hypothesize here that the opposite may be true, and that the very existence of human‐specific genes may help explain intrinsic problems in the representation of human inflammatory disease in animal models.
In an in silico survey of candidate taxonomically restricted genes, we were struck by the number of human‐specific genes that are expressed in blood‐derived cells, most often leukocytes and lymphocytes (Table 1). We also noted that the expression of these genes is often associated with the biology of injury, namely infection, inflammation, and tissue repair and regeneration. These genes include well‐known anti‐infection and human‐specific defensin genes (Table 1), some of which specifically localize to human neutrophils. They also include more recently uncovered genes, like CHRFAM7A, ARHGAP11B and TBC1D3 that are either linked injury responses like the cholinergic regulation of inflammation by the α7‐nicotinic acetyl choline receptor (α7−nAChR), to growth factor‐mediated signal transduction through rho‐mediated signal transduction or to the regulation of microvescle exocytosis and endocytosis, respectively (Table 1).
Table 1.
Human‐specific genes and inflammatory cells
| Gene name and/or gene family* | Gene ID (Ensembl) | Known, putative and/or inferred function(s)* | Immune and injury cell expression | Reference* |
|---|---|---|---|---|
| CHRFAM7A | ENSG00000166664 | Receptor antagonist | BM,WB, L, LN, T | J Leukoc Biol. 2015; 97:247 |
| TBC1D3 | ENSG00000274611 | Endocytosis/pinocytosis | BM,WB, L, LN, T | Genomics. 2006; 88:731 |
| ARHGAP11B | ENSG00000187951 | Rho GTPase activating | WB, L,LN | Science. 2015; 347:1465 |
| CCL18 | ENSG00000275385 | Immunoregulation | BM, WB, LN, T | Genomics. 1999; 55:353 |
| CCL23 (MIP‐3) | ENSG00000274736 | Leukocyte chemotaxis | BM, WB, LN, T | J Exp Med. 1997;185:1163 |
| NLGN4X (Neuroligin4X) | ENSG00000146938 | Tissue remodeling | BM, WB, LN, T | Nat Genet. 2003; 34:27 |
| Interleukin 26 | ENSG00000111536 | T cell responsiveness | BM, WB, LN, T | J Virol. 2000;74: 3881 |
| IFNL1 | ENSG00000182393 | Antiviral host defense | WB, LN | J Biol Chem. 2004;279:32269 |
| ANGPTL5 (Angiopoietin‐like) | ENSG00000187151 | Tissue regeneration | WB LN | J Hum Genet. 2003;48:159 |
| Alpha Defensin (N = 5; DEFA1B,A3, A4, A5, A6) | ENSG00000240247 ENSG00000239839 ENSG00000164821 ENSG00000164816 ENSG00000164822 | Antimicrobial peptides | BM,WB, L, LN, T | Physiol. Gen 2004; 20: 1 |
| Beta Defensin (N = 5; DEF4B, B104A, B104B, B114, B132) | ENSG00000177257 ENSG00000176782 ENSG00000177023 ENSG00000177684 ENSG00000186458 | Antimicrobial peptides | BM, WB, LN, T | Genome Biol. 2003;4:R31 |
| NPIP genes (N = 2; NPIP‐A1 and B11) | ENSG00000183426 ENSG00000254206 | Nuclear pore proteins | BM, WB, LN, T | Nature. 2001; 413:514 |
| CSAG (N = 2; CSAG1, CSAG2) | ENSG00000198930 ENSG00000268902 | Cell growth and tumor antigen | WB, LN | Gene. 1999;229:75 |
| c20orf203 (Alugen) | ENSG00000198547 | Neurodegeneration | WB | PLoS Comput Biol. 2010;6:e1000734 |
| SPANX genes (N = 9; SPANXB1, D, A2, C, A1, N3, N2, N5, N1) | ENSG00000227234 ENSG00000196406 ENSG00000203926 ENSG00000198573 ENSG00000198021 ENSG00000189252 ENSG00000268988ENSG00000204363 ENSG00000203923 | Nuclear proteins, spermatogenesis and cancer antigens | BM, WB, LN, T | Proc Natl Acad Sci USA.2004;101:3077 |
| VCX genes (N = 3; VCX2, VCX3A, VCX3B) | ENSG00000182583 ENSG00000177504 ENSG00000169059 ENSG00000205642 | mRNA stability reproduction | WB | Cancer Res. 2014; 74:4694 |
| VCY | ENSG00000129864 | Nuclear protein | BM, WB, LN, T | Mol Reprod Dev. 2008; 75:219 |
| OPN genes OPN1MW | ENSG00000268221 ENTREZ: 100534624 | Locus control for opsin trichromatic vision | BM, WB, LN, T | Trends Ecol Evol 2003; 18:198 |
| VN1R genes (VN1R3) | ENSG00000180663 (mouse, no rat) | Olfactory chemosensory | BM, WB, LN, T | Genome Res. 2010;20:10 |
| STRA6 | ENSG00000137868 | light adaptation | BM, WB, LN, T | PLoS One. 2014;9:e108388 |
| NBPF | ENSG00000162825 | Development | BM, WB, LN, T | Mol Biol Evol. 2005; 22:2265 |
| CT45 genes (N = 4; CT45A1, A2, A3, A4) | ENSG00000268940 ENSG00000271449 ENSG00000269096 ENSG00000271449 | B cell lymphoma | BL | Proc Natl Acad Sci USA. 2005;102:7940 |
*See www.gencards.org for details on genes, gene ontogeny, orthologs, paralogs, inferred and known functions and relative tissue expression.
BM, bone marrow; BL, B cell lymphoma; WB, whole blood; LN, lymph node; T, thymus; LL, leukocytes and lymphocytes as determined by microarray.
Numerous investigators have noted the significant variability that exists in the resilience of different species to different pathogens, but species differences in injury and scarring is largely underinvestigated or often minimized. Yet, trauma and burn surgeons attest to the intrinsic variability of the inflammatory response after burn and traumatic injury in humans that are otherwise indistinguishable.6 Interestingly, just as we noted the expression of human‐specific genes in human immune cells (Table 1), Long and colleagues noted the wide and variable representation of primate‐ and human‐specific protein‐coding sequences in brain development.3 We, therefore, considered the possibility that genes, originally selected to modulate human resilience to injury and infection, could have multifunctional biological effects, for example, when expressed in other tissues (Figure 1). In one such example, CHRFAM7A7 is a proinflammatory human‐specific gene that is a dominant negative inhibitor of the anti‐inflammatory α7‐nicotinic acetylcholine receptor (α7AChR) in peripheral tissues like macrophages. When expressed in the central nervous system, it presumably alters α7AChR activities on human cognition and memory. In other examples, the human antimicrobial defensins are highly multifunctional in different tissues8 and the human‐specific TBC1D3 gene regulates human macropinocytosis and the formation of human microvesicles.9
Figure 1.
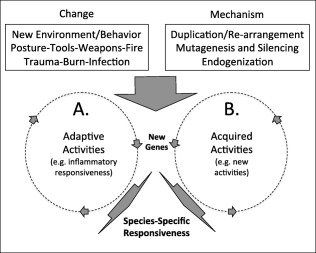
Injury, inflammation, and adaptation. New hominid behaviors, like the emergence of bipedal movement (trauma), harnessing of fire (burn injury) and the use of tools and weapons (injury/infection) is a positive selection for new genes. Positive adaptive selection enables new inflammatory activities (A) in response to new types of injuries. In this model, genes like CHRFAM7A, TBC1D3, and ARHGAP11B are initially selected in leukocytes and the in the human inflammatory response for their capacity to regulate ligand binding, internalization, and signaling, respectively. When these same genes are expressed in other tissues however (B), they can elicit new activities that also contribute to positive selection for example regulating neurotransmitter action (CHRFAM7A), cell–cell communication (TBC1D3) and progenitor cell growth (ARHGAP11B).
So what are the consequences of an anthropogenic injury hypothesis to better understanding the mechanistic basis to human disease, and particularly wound repair and regeneration? First, like most genes, the expression of human‐specific genes in different tissues is highly contextual. Their multifunctional activities are dependent on where and when they are expressed (Figure 1). In this proposed paradigm, a human‐specific gene in monocytes may have arisen to alter responsiveness after injury but may be retained in the human genome for unrelated activities, for example, in the central nervous system. Second, current experimental models of human disease do not test the contribution(s) of human‐specific genes to disease. To this end, the mouse still remains the most robust and experimentally tractable vertebrate animal model of human disease and it is possible to ask whether imparting mice with a human‐specific gene alters their injury response. Plasmids encoding human‐specific genes‐like ARHGAP11B have been injected into mice to generate a human phenotype.10 Alternatively, human genes (e.g., CHRFAM7A) that encode antagonists (e.g., for CHRNA7) can be studied in mice to determine if over expression mimics the proinflammatory and behavioral phenotype of target gene knockout (e.g., CHRNA7). Finally, endogenous human‐specific genes in circulating and resident human immune cells can be studied in mice after the transplantation and engraftment of human hemato‐lymphoid immune systems.11 With heterologous or autologous human skin grafts, it is even possible to create precision models of human diseases, including for wound repair and regeneration.
On a final note, the possibility that injury, inflammation, and infection responses contribute to the selection of species‐specific genes and regulatory pathways implies that they contribute factors to injury and wound healing responses in modern humans. If so, their study should be integrated into animal models of injury, wound healing and inflammation research. This is particularly relevant to “characteristically human” diseases like keloids and certain inflammatory bowel diseases that are difficult to model in animals and whose origins might trace to human behaviors like scarring and changes in the gut microbiome. Along this line, it is interesting to note that genomic expression data,12 albeit controversial,13, 14, 15 already points to the existence of characteristically human gene expression patterns in response to infection, trauma, and burn injury. We propose that while a better understanding of the contributions of species‐specific genes to the injury and inflammation response adds another level of complexity to biomedical research, it could lead to the identification of completely new drugs, new drug targets and the development of new therapeutics.
Acknowledgments
Conflict of Interest: The authors declare that they have no conflicts of interest.
Source of Funding: The biomedical hypothesis presented in this manuscript initially arose during Supplementary Research funding from NIGMS (NIGM78421) to AB and BPE through the American Recovery and Reinvestment Act (ARRA) and was further developed with support of the National Cancer Institute (CA170140) to BPE, the Hammond Fund (RC), the Division of Trauma Reinvestment Fund (AB, TC, RC and BPE), the American Surgical Association Foundation Research Fellowship (TC), and a Department of Defense grant (W81XWH‐10‐1‐0527) to RC that is administered by the American Burn Association.
References
- 1. Acharya D, Mukherjee D, Podder S, Ghosh TC. Investigating different duplication pattern of essential genes in mouse and human. PloS One 2015; 10: e0120784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. O'Bleness M, Searles VB, Varki A, Gagneux P, Sikela JM. Evolution of genetic and genomic features unique to the human lineage. Nat Rev Genet 2012; 13: 853–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang YE, Landback P, Vibranovski M, Long M. New genes expressed in human brains: implications for annotating evolving genomes. Bioessays 2012; 34: 982–91. [DOI] [PubMed] [Google Scholar]
- 4. Long M, VanKuren NW, Chen S, Vibranovski MD. New gene evolution: little did we know. Ann Rev Genet 2013; 47: 307–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cooper DN, Kehrer‐Sawatzki H. Exploring the potential relevance of human‐specific genes to complex disease. Hum Genomics 2011; 5: 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Harrison C. Sepsis: calming the cytokine storm. Nat Rev Drug Discov 2010; 9: 360–1. [DOI] [PubMed] [Google Scholar]
- 7. Costantini TW, Dang X, Coimbra R, Eliceiri BP, Baird A. CHRFAM7A, a human‐specific and partially duplicated alpha7‐nicotinic acetylcholine receptor gene with the potential to specify a human‐specific inflammatory response to injury. J Leukoc biol 2015; 97: 247–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheng DQ, Li Y, Huang JF. Molecular evolution of the primate alpha‐/theta‐defensin multigene family. PloS One 2014; 9: e97425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Frittoli E, Palamidessi A, Pizzigoni A, Lanzetti L, Garre M, Troglio F, et al. The primate‐specific protein TBC1D3 is required for optimal macropinocytosis in a novel ARF6‐dependent pathway. Mol Biol Cell 2008; 19: 1304–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Florio M, Albert M, Taverna E, Namba T, Brandl H, Lewitus E, et al. Human‐specific gene ARHGAP11B promotes basal progenitor amplification and neocortex expansion. Science 2015; 347: 1465–70. [DOI] [PubMed] [Google Scholar]
- 11. Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, et al. Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol 2014; 32: 364–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA 2013; 110: 3507–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Takao K, Miyakawa T. Genomic responses in mouse models greatly mimic human inflammatory diseases. Proc Natl Acad Sci USA 2015; 112: 1167–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Warren HS, Tompkins RG, Moldawer LL, Seok J, Xu W, Mindrinos MN, et al. Mice are not men. Proc Natl Acad Sci USA 2015; 112: E345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Osuchowski MF, Remick DG, Lederer JA, Lang CH, Aasen AO, Aibiki M, et al. Abandon the mouse research ship? Not just yet! Shock 2014; 41: 463–75. [DOI] [PMC free article] [PubMed] [Google Scholar]