

Abstract
The regulation of transcription is a fundamental process underlying the determination of cell identity and its maintenance during development. In the last decades, most of the transcription factors, which have to be expressed at the right place and at the right time for the proper development of the fly embryo, have been identified. However, mostly because of the lack of methods to visualize transcription as the embryo develops, their coordinated spatiotemporal dynamics remains largely unexplored. Efforts have been made to decipher the transcription process with single molecule resolution at the single cell level. Recently, the fluorescent labeling of nascent RNA in developing fly embryos allowed the direct visualization of ongoing transcription at single loci within each nucleus. Together with powerful imaging and quantitative data analysis, these new methods provide unprecedented insights into the temporal dynamics of the transcription process and its intrinsic noise. Focusing on the Drosophila embryo, we discuss how the detection of single RNA molecules enhanced our comprehension of the transcription process and we outline the potential next steps made possible by these new imaging tools. In combination with genetics and theoretical analysis, these new imaging methods will aid the search for the mechanisms responsible for the robustness of development. WIREs Dev Biol 2016, 5:296–310. doi: 10.1002/wdev.221
For further resources related to this article, please visit the WIREs website.
INTRODUCTION
The Drosophila embryo has been used for decades as an excellent model to understand how cell identity is determined and maintained during development. Powerful genetics demonstrated that most of the patterning in this system relies on the activity of key transcription factor networks, which have to be expressed at the right place and at the right time for proper development. These networks include: (1) the initial morphogenic transcription factors Bicoid and Dorsal and their downstream networks, respectively, essential for Antero‐Posterior (AP)1 and Dorso‐Ventral (DV)2, 3 patterning, (2) the transcription cascade responsible for the myogenic program4, 5 or (3) the temporal cascades patterning neural fates.6, 7 To understand the coordinated spatiotemporal expression of genes during development, it is now essential to obtain quantitative measurements of the interplay between the molecular components involved.
Until recently, like in most developmental systems, the process of gene expression in the Drosophila embryo has been mainly monitored by in situ hybridization detecting mRNA on fixed samples, using radioactive8 or fluorescent9, 10 complementary RNA probes. Combined with genetics and efficient imaging, this approach provided a rather exhaustive insight into the precision and noise involved in the spatial regulation of gene expression and its role in patterning.11, 12, 13, 14, 15 However, given the extremely rapid development of the embryo (Figure 1(a)), it became clear that approaches extracting only static clues could not provide a precise understanding of the temporal dynamics of transcription15, 16 and even more of its intrinsic molecular noise.17, 18, 19 In a few cases, it has been possible to reconstitute temporal progression by identifying the precise developmental stage of the samples with the help of additional quantitative markers, such as density and the size of the nuclei in the case of early development15, 20 or cell membrane invagination as a time scale marker during nuclear cycle (nc) 14.16, 21 However, each case was limited by marker‐dependent discrete time resolution and required the analysis of many samples for significant statistics. Given these limitations, there was a need to develop new methods, in which time would be a directly accessible dimension, to follow the transcription process in living organisms. The first steps toward this goal have recently been achieved in the very early embryo to describe the temporal dynamics of the response to the Bicoid morphogen.22, 23 Besides providing new insights into the Bicoid system, these recent studies provided unprecedented insight into the dynamics of the transcription process in the developing fly embryo. The goal of this review is to describe these new methods, the insights so far and the prospects. We focus on the Drosophila embryo because it was the first multicellular organism in which these approaches were implemented. However, there is no technical limitation that should restrict these methods to this particular model system as highlighted by their recent use in zebrafish embryos.24 Many of the points raised here are not specifically limited to the fly embryo and can be of general interest to study gene expression during development.
Figure 1.
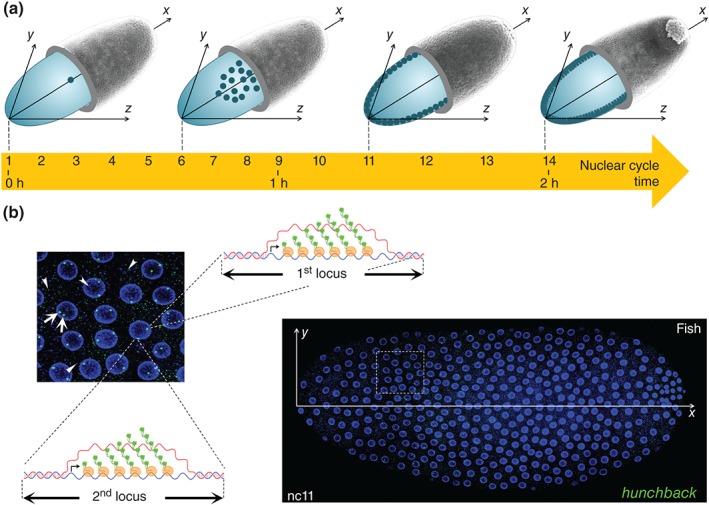
Detecting ongoing transcription in young Drosophila embryos. (a) The young fruit fly embryo is a unique cell of ellipsoidal shape. At egg laying, the nucleus of the fertilized egg undergoes 13 rapid divisions over a two hour period. These divisions first occur in the center of the embryo. At nuclear cycle 6 (nc 6), nuclei start their migration from the center to the periphery of the cell and spread in a single layer at the surface of the cell to give rise after about one hour of development (nc 8–nc 9) to the syncytial blastoderm. Once at the periphery, nuclei continue to divide for 5 more rapid divisions before reaching the long nc14 during which the cellularization process occurs. The first hints of zygotic transcription are detected at nc8. (b) On the right, nuclei (blue, nuclear envelope labeled with WGA‐AlexaFluor‐63315) are visualized at the surface of the whole embryo at nc11 and on the left, a close up of expressing nuclei (taken from the dashed square on the right). Expression of a given gene of interest (here hunchback) can be detected by RNA FISH with fluorescently labeled anti‐sense RNA probes. Expression is revealed by two type of staining: speckle‐like dots (arrow heads) corresponding to single mRNA and bright intense foci (arrow) corresponding to the accumulation of several nascent pre‐mRNAs at their site of synthesis, as schematically diagrammed for the two hunchback loci.
INSIGHTS INTO GENE EXPRESSION FROM RNA FISH
Despite its intrinsic limitations, the RNA fluorescent in situ hybridization (RNA FISH) approach has been used successfully to tackle the dynamics of gene activation in syncytial nuclei or embryonic cells. Two types of fluorescent signals are generally observed in RNA FISH: (1) very small, speckle‐like fluorescent signals scattered in both the nucleus and the cytoplasm (Figure 1(b), arrow heads); (2) bright intense foci specifically detected in the nucleus (Figure 1(b), arrows). The former are not observed with intronic probes and correspond to the detection of single mature mRNA.25, 26, 27, 28 The latter are generally observed with both intronic and non‐intronic probes and thus correspond to premature mRNA. In embryos at an early stage (before nc 14), the maximum number of bright foci in expressing nuclei correlates with the number of loci encoding the RNA that is being detected.15 As fluorescent signals from a single mRNA are much weaker, these bright foci likely correspond to the accumulation of several nascent pre‐mRNAs at their site of transcription in the nucleus.15, 16, 26 Direct support for this assumption comes from experiments of double DNA‐RNA FISH performed in cultured cells25 or early mouse embryos26 showing that bright foci observed with RNA‐FISH in nuclei colocalize with their DNA locus. The double RNA‐DNA FISH cannot been achieved on whole mount Drosophila embryos (which are only fixed on slides at the end of the procedure) but the converging indirect evidence mentioned above, argues that the nuclear bright dots detected by RNA‐FISH directly correspond to the ongoing transcription process at each locus. Given the absence of homologue pairing in the early embryo,29 the two sites of transcription from homologous loci in diploid embryos are generally largely distant from each other, giving rise to two significantly distinguishable bright dots (Figure 1(b)). In rare cases, four distinct bright dots corresponding to ongoing transcription at the four sister chromatids are observed in nuclei, in which the loci of interest have already been replicated.26 Conveniently, as chromosomes adopt a Rabl configuration in the early embryo with apical centromeres and basal telomeres, the two homologous loci are generally localized in the same confocal Z‐stack, which considerably facilitates image acquisition.30 Importantly, the lack of pairing of homologous chromosomes in the early embryo is an exception and homologous chromosomes are generally paired in most Drosophila tissues.29 Therefore, ongoing transcription in older Drosophila tissues is generally detected as a single bright nuclear dot by RNA‐FISH irrespective of the homologous loci copy number.
In summary, RNA‐FISH in Drosophila embryos allows the detection of single mature mRNA in the cytoplasm and in the nucleus. In addition, it also allows for the detection of the accumulation of nascent premature transcripts at their site of synthesis inside the nucleus and it is thus an excellent marker to detect ongoing transcriptional activity at a given locus.27 The observed data are consistent with a transcription process occurring with periods of promoter activity during which several polymerases are transcribing the gene. This period of activity can either be temporally continuous or discontinuous, i.e. eventually interrupted by periods of inactivity during which no polymerases initiate transcription (Figure 2). Such periods of fluctuating activity, called ‘bursts’ or ‘pulses’ of transcription, have been described in several prokaryotic and eukaryotic living systems.33, 34, 35, 36, 37 A recent study using the MS2 approach to label RNA (see below) and focusing on different stages during preaggregative development in Dictyostellium indicates that in this system developmentally regulated genes are generally expressed in such pulses, while housekeeping genes strongly reduce the magnitude of their pulses during development presumably to better tune transcription at the single‐cell level.38 In the fly embryo, bursting has recently been directly observed with a similar approach for even‐skipped expression at nc 14.39
Figure 2.
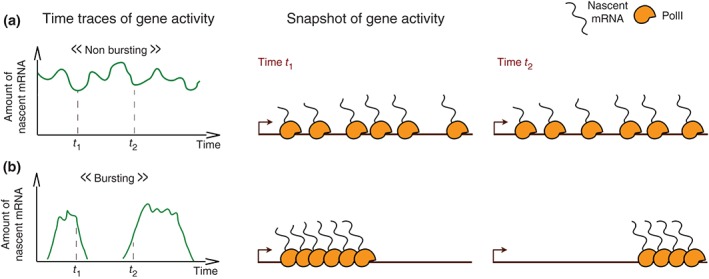
Bursting or nonbursting gene activity, the two types of transcription dynamics of a promoter. A continuum between the two extreme situations portrayed here can be found for different genes. (a) The amount of nascent RNA produced at the promoter fluctuates around a given positive value. The RNA PolII initiates transcription at an average constant rate. The promoter is active and nonbursting. (b) The activity of the promoter (measured as the amount of nascent RNA produced at a given time) alternates between periods of strong production (bursts) and periods of inactivity. The promoter is bursting. The characteristics of a bursting promoter include the frequency of the bursts, the intensity of the bursts, and their duration.31, 32
NOISE IN GENE EXPRESSION DURING DEVELOPMENT
Gene expression is intrinsically a noisy process, which contributes to cell‐to‐cell variability at the protein level within a homogenous population of cells.17, 40 Recent studies using quantitative RNA‐FISH revealed that genetically identical cells in identical environments can produce very different amounts of RNA (Figure 3(a)).28, 41 As these fluctuations are not correlated with any studied parameter, it is assumed that this expression is stochastic. This heterogeneity can be such that each cell in the population expresses a different, but constant amount of RNA over a long period of time (time‐invariant heterogeneity) or alternatively that the amounts of RNA produced by a given cell fluctuate over the same period (time‐fluctuating heterogeneity). Although these two types of scenarios have very different temporal dynamics, they show almost identical distributions of RNA per cell among the population at a given time (Figure 3(a), right) and thus the snap‐shot view provided by RNA‐FISH does not discriminate between these two scenarios. Mathematical modeling can be used to successfully interpret RNA‐FISH data and provide evidence for transcriptional bursting, such as in the case of the endogenous c‐Fos proto‐oncogene regulation by transcription factors.42
Figure 3.
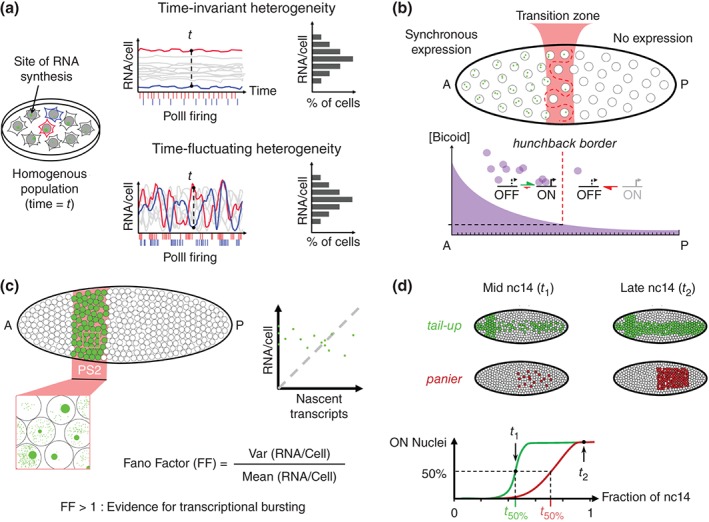
Noise in gene expression from single cells to Drosophila embryos, as detected in fixed tissues. (a) Hypothetical situation in which an isogenic population of cells within a homogenous environment exhibits cell‐to‐cell variability in the gene expression pattern at a given time. Time‐invariant heterogeneity and time‐fluctuating heterogeneity are two very distinct scenarios which could explain this variability. However, as at a given time the distributions of mRNA/cell are very similar for both scenarios, and thus the snapshot analysis on fixed tissues cannot provide a direct insight into the underlying transcriptional dynamics. (b) Transcriptional activation of the hunchback (hb) gene by the Bicoid transcription factor gradient. Transcription of hb is highly synchronous (a majority of bi‐allelic expression) in the anterior half of the early Drosophila embryo whereas transcription in not detected in the posterior half. While the transition zone is narrow, most of the active nuclei within this zone express only one allele and the border separating expressing from nonexpressing nuclei in the transition zone is not rectilinear.15 (c) The transcription from the Sex combs reduced (Scr) gene within the parasegment 2 (PS2) at nc14 embryo using a single mRNA resolution. A high variability in the distribution of nascent versus mature mRNA is observed among those cells. The absence of correlation between the amount of nascent transcript and the total amount of mRNA per cell argues for transcriptional bursting of Scr.27 The Fano factor is an expression of this cell‐to‐cell variability, and values greater than 1 indicate bursting. (c) Transcription dynamics of the two Decapentaplegic target genes, tail‐up and panier at nc14. The two genes are first heterogeneously expressed among nuclei and reach a more synchronous expression at the end of nc14. The time interval it takes for tail‐up to be expressed in 50% of the nuclei is shorter than for panier. The length of the transition period from heterogeneous to synchronous expression is anti‐correlated to the amount of RNA PolII pausing at the promoter.21
Stochasticity in the transcription process is even more important when considering the expression of developmental genes in Drosophila embryos, in which cells must integrate positional information and/or information about different environments varying in both space and time. Surprisingly, the few attempts addressing this question in Drosophila embryos indicate different mechanisms depending on the situation. In the case of the transcriptional response downstream of the Bicoid morphogen (Figure 3(b)), expression of the hunchback target gene monitored by RNA‐FISH indicates that the two alleles of the gene are active simultaneously (synchrony) in more than 60% of the anterior nuclei while they are completely silent in posterior nuclei. This synchrony is acquired extremely rapidly (in less than 30 min) and the transition zone between the two domains in which more variability is observed, spans only over 10% of the egg length.15 Interestingly, the high level of synchrony observed could suggest, that the transcription process at the hunchback loci at this stage of development, consists of a single pulse during each interphase, which does not fit with a bursty mode of transcription (Figure 2).
Evidence for transcriptional bursting was provided by analyzing expression of the Hox gene Sex combs reduced in the parasegment 2 at a slightly later stage of development27 (Figure 3(c)). Interestingly in this case, expression of the gene is very different among adjacent cells at a given time, with some cells exhibiting (1) low amounts of single mature mRNA but clearly detectable ongoing transcription at the locus, (2) high amounts of single mature mRNA but no trace of ongoing transcription, and (3) intermediate levels of single mature mRNA as well as ongoing transcription. A quantitative analysis indicated that there was no correlation between the amounts of mature mRNA and nascent mRNA per cell (Figure 3(c), right) arguing that the heterogeneity observed among cells was likely time‐fluctuating and that transcription in this case would occur in bursts. Importantly, the detection of bright foci at an active locus indicates that several polymerases are transcribing the gene one after the other (Figure 1(b), cartoons). This technical limitation could restrict the detection of ongoing transcription by RNA‐FISH to bursty genes when compared to nonbursty genes expressed at low levels for which it would be difficult to distinguish single mature mRNA from single nascent pre‐mRNA (Figure 2).
A third example concerned the onset of gene expression along the DV axis of the embryo during cellularization (nc14). The detection of nascent transcription of 14 genes using RNA‐FISH indicated that some of these genes are expressed uniformly (synchronously) in most cells of the expression domain, while other genes are expressed much more heterogeneously (stochastically).16 The analysis of a large number of embryos combined with morphological staging provided access to the dynamics of the transcription process throughout the cycle and indicated that while only a few genes were synchronously expressed at the onset of the cycle, most genes had acquired synchrony 30 min later (Figure 3(d)). The heterogeneity observed for most genes at the beginning of the cycle was interpreted as a transition period required for each gene to reach a coordinated expression in all cells of the expression domain. The length of this transition period is anti‐correlated to the abundance of paused polymerases at promoters16 and reciprocally, changing the amount of paused polymerase modifies the transition period required for synchronous expression.21 Polymerase pausing appears thus critical to reduce the time it takes to reach synchrony. Whether heterogeneous expression at early cycle 14 reflects time‐invariant (not bursting) or time‐fluctuating (bursting) expression remains to be clarified.
THE FLUORESCENT TAGGING OF RNA IN LIVING EMBRYOS
Although extremely powerful for single cell analysis, RNA FISH requires fixation of the sample and can thus only provide a snap‐shot view of the transcription process at a given time (Figure 4(a)). Benefiting from the pioneer work of R. Singer,44 several systems have been developed to fluorescently tag RNA in living cells. These new approaches take advantage of strong interactions between a specific Coat Protein (CP) and its RNA stem loop (SL) generally derived from viral systems. These include the MS2 RNA SL and the MCP from the MS2 bacteriophage,44 the PP7 RNA SL and the PCP from the PP7 bacteriophage45 or the Bbox SL and the λ phage N protein.46 Provided that the fluorescently tagged CP is expressed in the cell, it will bind strongly to RNA carrying its cognate target SL (Figure 4(b)). These RNA SL are artificially introduced in reporter genes and generally multimerized from 6 to 24 copies to allow the accumulation of fluorescence from bound CP above the background level. This approach has been widely used over the last decade to fluorescently label RNA in living systems.47 It has been very useful to study the movement of asymmetrically distributed RNA during Drosophila development including oogenesis48, 49 or the development of the tracheal system in the larvae.50 It has also been used in cultured cells to shed light on the mechanisms of RNA synthesis or processing, including the initiation,51, 52 elongation, pausing, and termination52 of transcription or splicing.53, 54 These studies provided estimates of key mechanistic parameters such as the duration of splicing events53, 54 or the elongation rate of the polymerase in various models.51, 52
Figure 4.
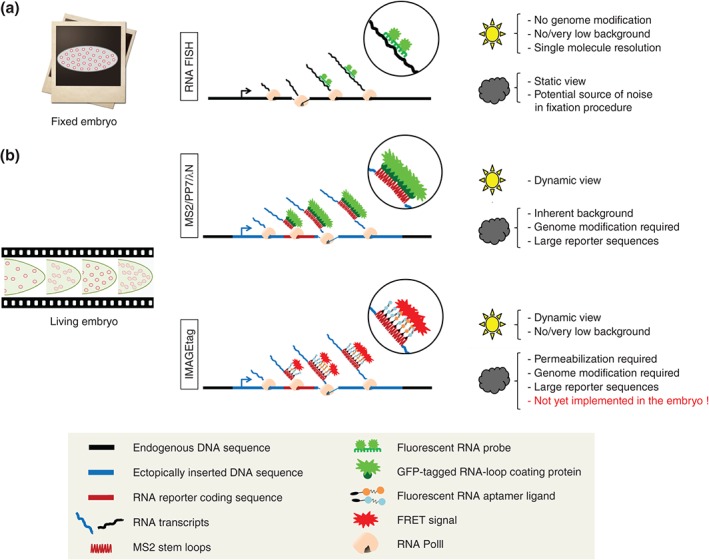
Available and potential methods to fluorescently‐tag nascent RNA in living embryos. (a) Detection of RNA in fixed embryos by RNA FISH only offers a snapshot view. However, it allows studying expression of endogenous loci without genome modification. In addition, it is mostly background free and can provide single molecule resolution. The sun icon indicates advantages of the method; the cloud indicates disadvantages. (b) Available live‐imaging methods provide access to the temporal dynamics of the transcription process. However, they require genome editing and the insertion of exogenous RNA reporter sequences, which can potentially bias the behavior of the promoter or the processing of the RNA. In addition, so far, these methods are limited by an inherent background of fluorescent signals for quantitative analysis. This limitation can potentially be overcome by new approaches such as the IMAGEtag, which combine the tagging of the RNA with FRET.43
Although these approaches provided an unprecedented insight into the dynamics of the gene expression process, they have limitations:
First, unlike RNA‐FISH, which enables one to directly detect RNA at endogenous loci, the fluorescent tagging of RNA requires genome modification and in most cases the handling of large reporter sequences. In the first attempts in mammalian cells, these reporter sequences were inserted in multiple copies (up to 200) at a single chromosomal location to gain sensitivity.52, 55 However, as the heterochromatin state of such repeats56 could affect the transcriptional behavior of the transgene,57 it is certainly wiser, when possible, to work with single‐copy insertions. Similarly, the extent to which the insertion of an exogenous RNA reporter sequence (Figure 4(b)) could affect the transcriptional behavior of the transgene or the processing of the transcript remains to be clarified. In particular the structure of the RNA loops could affect the polymerase elongation rate and the DNA sequence of the MS2 transgene could itself modify the initiation of transcription. Additionally, the MS2 loops could alter the mRNA lifetime. It is noteworthy that the CRISPR/Cas9 technology58, 59, 60 should now allow the detection of ongoing transcription at endogenous loci and limit in part side effects from reporters.
Second, a major limitation for quantitative analysis of RNA fluorescent tagging is the existence of an inherent fluorescent background, which is due to the presence of unbound or nonspecifically bound fluorescent CP. It is thus essential to express these fluorescent CP at low levels, the critical parameter that needs to be finely tuned being the signal‐to‐noise ratio (SNR). The signal is controlled by the number of fluorescent CP binding sites, whereas the noise or background is due to the fluorescent CP that are not bound to the RNA but freely diffusing through the cell. To diminish background without affecting too much the physical parameters of the interaction between the RNA and the fluorescent proteins so that they can remain negligible in comparison to the dynamics of the transcription process itself, an appropriate balance between fluorescent CP concentration and the amount of binding sites has to be found. This can be achieved by tight control of the fluorescent CP expression level (using the Gal4/UAS system or heat‐shock‐induced expression) or possibly by removing the nuclear localization signal from the fluorescent CP as shown to be surprisingly efficient to increase the SNR.22
Another possibility to reduce the SNR is to take advantage of bimolecular fluorescent complementation (BiFC), with a fluorescent protein split into two nonfluorescent fragments fused to two RNA binding proteins with distinct specificity (e.g., MCP/MS2 and PCP/PP7). When expressed in the same cell, the two free coat proteins are not fluorescent, which allows for considerable reduction of the background fluorescent signal. A complete mature fluorescent protein is only reconstituted, when the two coat proteins are brought close to each other by binding to their corresponding motifs that are adjacent in the RNA.61 The major limitation of this system is the time it takes to mature the reconstituted fluorophore (12–36 h) and it is therefore not appropriate to image real‐time transcription, which is a much more rapid process. Recently, live‐imaging of transcription with almost no fluorescent background has been described. This system is based on RNA aptamers called ImageTags, which can be expressed from a given promoter in a reporter.43 The RNAs containing a string of aptamers are fluorescently tagged when the cells are incubated with a mix of aptamer ligands conjugated to either one or the other component of a fluorescent energy transfer (FRET) pair, such as Cy3 or Cy5 (Figure 4(b)). The ImageTags have not yet been used to image transcription in multicellular organisms. In fly embryos, the difficulty to introduce the aptamer ligands without perturbing the development of the embryo will not be easy to solve. Nevertheless, FRET, which is much more rapid, is probably more suitable than BiFC for imaging rapid processes such as transcription. It is certainly worth exploring the possibility to fluorescently tag RNA in living embryos using RNA coat proteins fused to fluorescent molecules that would constitute a FRET pair, to generate background free tagging of RNA. Although, this approach has not yet been developed, it could certainly provide a powerful path to the increase of the SNR.62
THE POSITION OF THE RNA SLS INFLUENCES THE STRENGTH OF THE SIGNAL AND ITS PERSISTENCE
These approaches to fluorescently tag RNA in living cells or organisms provide an almost direct access to the dynamics of promoter activity. The possibility to detect nascent mRNA in vivo simplifies the estimation of the kinetics parameters, as opposed to more complex indirect inference from protein concentrations. However, the interpretability of the fluorescent signal depends on the details of the probe design, specifically the position of the RNA SL with respect to the gene sequence. In particular, in addition to the ratio of the number of bound and freely diffusing fluorescent CP molecules, the SNR also depends on the position of the RNA SLs with respect to the transcribed sequence.
The sequence coding for the RNA SL could be inserted anywhere in the transcribed sequence. However, to stay close to the natural context, it is certainly better to insert them into nontranslated sequences. This could be either in the 5′ untranslated region (UTR) of the transcribed sequence (Figure 5(a)), in introns (Figure 5(b)) or in the 3′ UTR of the transcribed region (Figure 5(c)). As shown in Figure 5, assuming the same activity period of the promoter, these various configurations produce different fluorescent time traces: (1) when the RNA SL is localized in the 5′UTR, a strong fluorescent signal is rapidly observed but its persistence is significantly longer than the activity period of the promoter (Figure 5(a)); (2) when the RNA SL is localized in the intron, the fluorescent signal will only account for premature mRNA and should in theory provide a better surrogate of the transcription initiation process. Unfortunately in this case, the signal will also depend on the efficiency of splicing events and dual‐color single‐molecule RNA imaging in cultured cells indicated recently that splicing at the same gene might occur stochastically both before and after transcription release.53 Given this stochasticity of splicing (which could depend on the gene context), it will be difficult to easily infer information concerning the promoter characteristics (bursting or nonbursting behavior, average size and frequency of bursts) from the fluorescent signal emerging from such constructs. The signal measured with intronic probes is a convolution of the signal coming from transcription initiation and stochastic splicing: deconvoluting the signal requires quantification of the splicing dynamics for each specific intron; (3) when the RNA SL is inserted in the 3′UTR, the fluorescent signal is much weaker and thus much more sensitive to the background. As multiple RNAPs are bound to one gene at a given time, the 5′ positioning of the MS2 cassette leads to a larger number of labeled transcripts that can cumulate along the gene length, leading to a higher intensity of the fluorescence signal. However, in the 3′ case, the signal persistence is closer to the activity period of the promoter.
Figure 5.
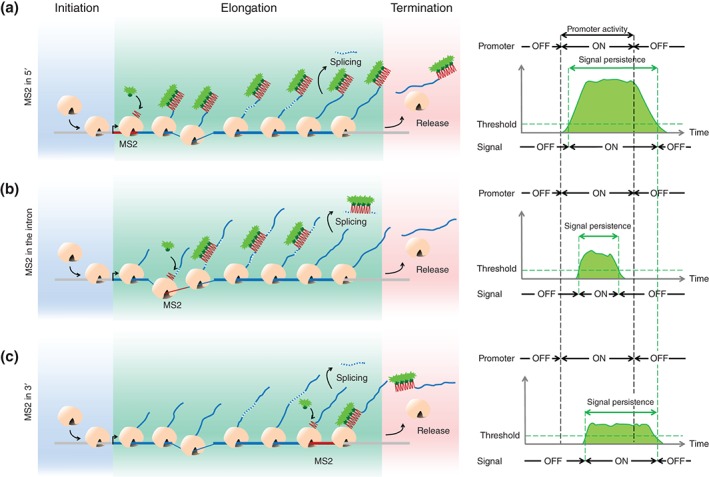
The position of the MS2 SLs within the transcribed sequence impact on the signal intensity and its persistence. On the left: snapshots of the gene at a given time during the ON state of the promoter (burst). On the right: fluorescent intensity at the locus as a function of time during the burst of transcription. (a) The insertion of the SL tagging sequence at a 5′ position in the gene allows accumulation of labeled transcripts along the whole length of the transcribed locus. This produces a strong fluorescent signal whose initiation time closely coincides with the initiation time of the burst of transcription. However, since this strong fluorescent signal will take longer to decay once the promoter is turned OFF, the overall persistence of the signal will be longer than the ON‐time period of the promoter (right panel). (b) The insertion of the SL tagging sequence inside an intron leads to a longer delay between the onset of the transcription period and signal detection, which depends on the position of the intron relative to the transcription initiation site. Moreover, in this case, the persistence of the signal will also depend on the splicing process. Since splicing takes place during the transcription process or after release,53 the signal persistence will not properly reflect the real activity period of the promoter (right panel). (c) When the SL tagging sequence is inserted at a 3′ position in the transcribed locus, the delay between the onset of the promoter activity period and the detection of the signal will be even longer than in previous cases. The intensity of the signal is also much lower and therefore more sensitive to the signal‐to‐noise ratio than in the previous cases. However, in this case, because the buffering time is shorter, the persistence of the signal is closer to the activity period of the promoter (right panel). See Movie S1, Supporting Information, for a movie of the transcription process.
USING MODELING TO DETERMINE THE OPTIMAL POSITION OF THE RNA SLS
Insertions of the RNA SL at different positions have already been engineered to study the hunchback promoter in the very early Drosophila embryo.22, 23 The different positioning of the MS2 cassette inspired us to explore how each of the constructs would impact the reliability of the output signal.
Our goal was to assess the advantages and disadvantages of each of these constructs with regards to the fidelity and precision in measuring the transcription process itself. To do this, we modeled a promoter cycle (input) and compared its theoretical expression state to the calculated fluorescent signals expected for each construct (readout). Recent single‐cell temporal measurements of a short‐lived luciferase reporter gene under the control of a promoter in mouse fibroblast cells have quantitatively confirmed that transcription initiation in eukaryotes is not well described in terms of just activation and inactivation of the gene.63 Thus, following previous work,64 we modeled a promoter cycle that accounts for irreversible transitions between a number of inactive states before the gene can be turned on again (Figure 6). We then built a theoretical model in which we considered two variants of the SL insertion: one with 24 SL within a 1.3 kb cassette at the 3′ end of the transcribed sequence and another with the same cassette placed at the 5′ end.
Figure 6.
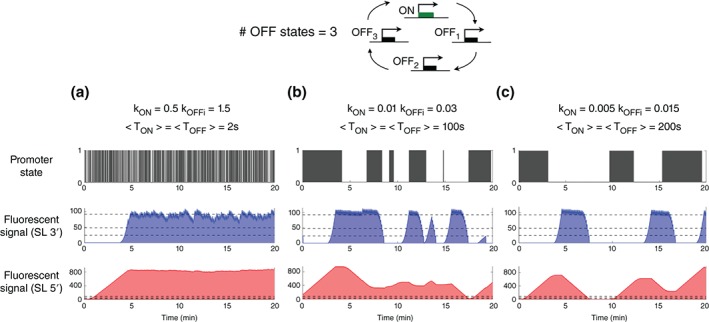
A lower sensitivity to noise background with 5’ insertions but a better correlation between promoter activity and signal readout with 3’ insertions. Simulations were performed assuming an irreversible promoter cycle (with three inactive states) model for transcription activation and deactivation at different frequencies of ON/OFF transitions (high (A), intermediate (B), and low (C)). For each case, we show: the change in the state of promoter activity with the distributions of ON (state 1) and OFF (state 0) waiting times as collected at the promoter (top panel), the simulation of the fluorescent signal detected when the SL tagging sequence is inserted in a 3’ (middle panel) or 5’ (bottom panel) position of the transcribed sequence, assuming a SL length of ~1.3 kb, a transcribed length of 5.4 kb, and an occupancy of ~0.15 kb for one PolII transcribing with a velocity of ~25 bp/s22. It is assumed that the concentration of PolIIs is not a limiting factor and that they can bind at every moment when the gene is in the ON state. Multiple PolII molecules can constitutively transcribe the gene during one ON event. The horizontal dashed lines in the middle and bottom panels indicate the background levels frequently encountered. The results do not qualitatively depend on the number of inactive states used, and the reversible two state telegraph model65 gives qualitatively the same results.
We calculated fluorescent time traces for the 3′ and 5′ constructs within this model with different values of switching rates between the two possible states of promoter activity: ON (in which mRNA is produced) and the OFF states of the promoter. These different values of switching ON/OFF rates of the promoter activity elicit very different bursting behaviors, with different burst frequencies and burst length (Figure 6(a)–(c)). The sensitivity of the readout on the transcription dynamics depends on the background (represented as the three dashed lines in Figure 6 corresponding to typical values of the background fluorescent signal due to unbound fluorescent CP). As in the 3′ case the whole gene is transcribed before the SL cassette, the time during which SLs accumulate GFP signal (‘buffering time,’ t buff= length of the transcribed genomic sequence accumulating fluorescently tagged RNA/polymerase velocity) is much smaller (t buff = 52 s) than with the 5′ construct (t buff = 268 s), resulting in a much stronger signal for the 5′ construct. On the one hand, the approximately fivefold increase of the 5′ signal makes it insensitive to the background level of unbound fluorescent CP, whereas the SNR for the 3′ construct will strongly depend on the unbound fluorescent CP levels. On the other hand, the strong accumulation of the signal in the 5′ case hinders the detection of transient OFF events, preventing the direct collection of ON and OFF duration statistics from the time traces (Figure 6). Qualitatively, if the transitions between the ON→OFF and OFF→ON states (T ON + T OFF) occur faster than the buffering time of a given construct, the fluorescent signal will not be able to follow these transitions. For example, if the kinetics of promoter switching is on the order of 100s for the gene length used in Figure 6, only the 3′ construct and not the 5′ one will show bursting events. In summary, the fluctuations of the fluorescent signal from the 3′ construct are very sensitive to background levels and have low SNR but are closer to the transcription dynamics of the promoter for a larger range of frequencies (k ON and k OFFi). The 5′ construct is much less sensitive to background, but its readout is only closely correlated to the promoter dynamics in a narrower frequency range.
AUTOCORRELATION ANALYSIS
The evaluation of ON and OFF switching rates directly from the durations of the ON and OFF fluorescent periods is often not reliable because it requires sampling the OFF times, which strongly depends on the background level. Another option would be to take the derivative of the signal to identify rising and decaying phases. However, this procedure further enhances the noise and precludes this approach.
Rather than evaluating the switching rates directly from the ON and OFF periods of the fluorescent signal, another approach based on the computation of autocorrelation functions (Figure 7) allows for an efficient readout of these two parameters with reduced sensitivity to noise. In situations where the autocorrelation function of the promoter dynamics follows closely the autocorrelation function of the fluorescent signal, we can infer the ON and OFF rates of the promoter dynamics from the fluorescent signal (readout). In these cases, the characteristic decay time of the fluorescent signal autocorrelation function will be a measure of the relaxation time of the promoter activity, which is explicitly related to the average duration of the ON and OFF states. The ratio between the ON and OFF switching rates can be evaluated from the steady‐state value of the gene expression signal, given that the fluorescent signal can be calibrated. If the measurements are performed in a non‐saturated regime, there is a linear relation between the fluorescence signal and the number of molecules. Calibration of the signal expected from a single RNA SL allows for the conversion of the fluorescence signal into the number of RNA SL and facilitates learning the ratio of the effective OFF and ON rates from the mean value of the fluorescence signal. Garcia et al. calibrated the fluorescence signal in terms of mRNA numbers by comparing the MS2‐MCP fluorescence spatial profile with the corresponding FISH profile22 exploiting the single‐molecule resolution of FISH.26
Figure 7.
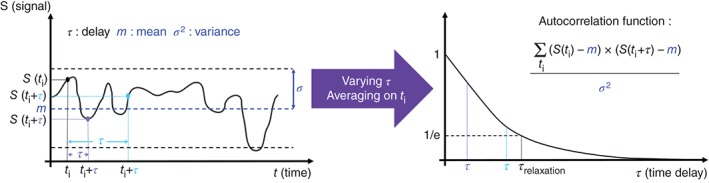
The autocorrelation function. The autocorrelation function determines the degree of correlation existing in any fluctuating signal (mean m and variance σ 2). The signal S(t) is multiplied for any time ti with its value at ti + τ, where τ is a delay time. The autocorrelation function at a given time delay is defined as the average of the overlap between the values of the signal taken at two time points at a fixed delay. This function is used to compare the fluctuating signal at two different times in order to quantify temporal correlations. The autocorrelation function describes the persistence of the signal (for example the promoter state). This persistence is characterized by the relaxation time of the autocorrelation function, which is the time at which the autocorrelation function of the signal decays by 1/e, and which represents the average time scale on which the signal has a similar value to its initial value.
As seen from Figure 8, the autocorrelation function of the fluorescent signal of the 3′ construct follows the correlation function of the underlying promoter dynamics more closely than of the 5′ construct (compare the differences between the black curves and the blue curves in Figure 8(a) with the differences between the black curves and the red curves in Figure 8(b)). In addition, the ability to discriminate between different switching timescales is inversely proportional to the length of the gene (compare Figure 8(b) with Figure 8(c)). For a long gene, the 5′ construct will accumulate more fluorescent loops, thus allowing the detection of switching rates only at low frequencies and hereby limiting the dynamic range of the measurement. In the 3′ case, the difference between the promoter activity and the fluorescent signal comes from the length of the SL cassette. Shorter cassettes in principle allow for better estimation of the kinetic parameters even in the case of high‐frequency switching promoter activity. However a shorter SL cassette also implies a lower fluorescence signal imposing a very low detection limit.
Figure 8.
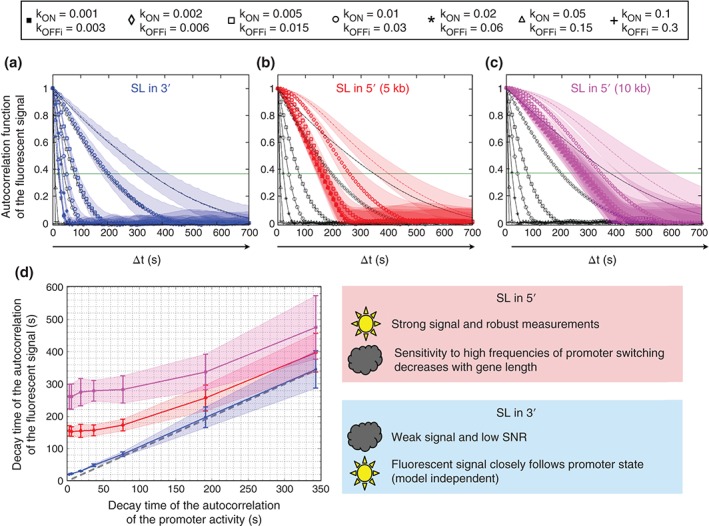
The fluctuations of the promoter cycle and of the fluorescent signal are better correlated when the SL tagging is inserted in 3′ of the transcribed sequence. The black curves in panels a, b, and c represent the autocorrelation function of the promoter activity simulated as a multi‐off state model with seven different conditions (cartoon) corresponding to different k ON rate and k OFF rates. The fluorescent signal was calculated for three different constructs and each of the seven conditions of the promoter cycle model. The autocorrelation functions corresponding to the fluorescent signal (readout, color) were compared to the autocorrelation of the promoter state (input, black). The SL sequence for fluorescent tagging of the RNA was inserted in the 3′ end of a 5 kb‐long transcribed sequence (a, blue), in the 5′ end of a 5 kb‐long transcribed sequence (b, red) or in the 5′ end of a 10 kb‐long transcribed sequence (c, pink). When the SL tagging sequence is inserted in the 3′ end of the transcribed sequence, the autocorrelation of the promoter cycle and the calculated fluorescent signal are almost superimposed. The similarity is weaker when the SL tagging sequence is inserted in the 5′ end and the difference increases with the length of the transcribed sequence. (d) Comparison of the decay time of the promoter activity autocorrelation and the decay time of the fluorescent readout autocorrelation, for the situations described in a (blue), b (red), and c (pink). The dashed gray line indicates perfect agreement indicative of an exact readout. The autocorrelation functions where calculated on 25 different computed traces 104 s long (2.8 hours). The shadows in all panels are standard deviations.
An inherent limitation to the autocorrelation approach is the length of the time window (from 5 to 15 min) of the interphases 10–13 in the early embryo. The computation of the autocorrelation functions requires statistics (the variability of the correlation function for a given time lag depends on the number of measurements available for this time lag) and the switching rates are extracted assuming a stationary process. The temporal resolution of the fluorescent signal can be increased by changing the imaging modality, for example using light sheet microscopy or by imaging a lower number of nuclei. However, if the timescale of the relaxation of the promoter dynamics is of the order of the interphase duration (5–20 min), the autocorrelation approach will not work. Yet, if the interphase duration is significantly longer than the autocorrelation time, using a microscope with greater temporal resolution could permit direct observation of the transcription process. Lastly, even if the autocorrelation approach is not sensitive to uncorrelated noise such as the temporally uncorrelated noise of the camera or instantaneous fluctuations of the CP concentration, the autocorrelation function remains sensitive to any temporally correlated noise, such as a drift of the focus in the experimental setup.
CONCLUSIONS AND PERSPECTIVES
New methods based on the fluorescent tagging of RNA in living cells allow for the visualization of ongoing transcription in living organisms. These new methods promise to provide insight into the dynamics of the interplay between the transcription factor networks underlying the determination and maintenance of cell identity during development. Insights from these new methods are expected at several levels. First, at the mechanistic level, these new methods make it possible to use the fly embryo as a test tube for studying the process of transcription and the details of its kinetics. Notably, bursting has been directly observed for the even‐skipped promoter,39 the polymerase velocity has been directly measured using two reporters with MS2 SL cassette insertions at different positions within the transcribed sequence22 and the Bicoid transcription factor has been shown to lengthen the activity periods of the hunchback promoter.23 Second, by providing temporal information about gene expression in each single cell in the context of the whole organism, these new methods will allow for the collective analysis of these developmental processes and will shed light on their robustness. Finally, besides determining the role of transcription factor networks in development, these approaches should also help to analyze how transcriptional memory and chromatin dynamics contribute throughout cell divisions to these processes.
Movies describing the temporal dynamics of the transcription state of a given locus in each nucleus of the living embryo generate a huge amount of data that requires analysis at different scales in space and in time. The core methods are now available but clearly using them to provide a quantitative understanding of development will require contributions from biologists to generate the material to be analyzed, biophysicists to provide input into the new imaging technologies and theoreticians for data analysis and modeling.
Supporting information
Movie S1. Dynamics of fluorescent RNA accumulation at a transcribed locus: simulated kinetics of the fluorescent signal produced at the site of transcription when the MS2 SL are located in the 5' UTR (top) or in the 3' UTR (bottom) of the transcribed region. On the horizontal axis: the 24 MS2 SL (1.3 Kb) are represented in red whereas the rest of the gene (5.4 Kb) is represented in blue. Individual black boxes represent individual PolII moving along the gene with a speed of 90bp/s [22]. A PolII molecules can bind to the promoter and start producing an RNA when the promoter is ON. The kinetics of the promoter is simulated by a two state telegraph model with an ON rate of 0.05 1/s and an OFF rate of 0.05 1/s. On the vertical axis: each green dot represents a fluorescence unit formed after the transcription of a MS2 SL. At each time‐point, the total amount of fluorescence accumulating at the locus being transcribed is the sum of the fluorescent signals coming from each individual fluorescent transcript at the locus
ACKNOWLEDGMENTS
We thank John Gerhart and the three anonymous reviewers for insightful suggestions on the manuscript. We apologize to those whose important work could not be cited due to space constraints. Research performed by the authors was supported by the 29311 ARC Subvention Fixe (ND), the ANR‐11‐LABX‐0044 DEEP Labex (ND), the ANR‐11‐BSV2‐0024 Axomorph (ND and AMW), a Marie Curie MCCIG grant No. 303561 (AMW) and the PSL ANR‐10‐IDEX‐0001‐02.
Conflict of interest: The authors have declared no conflicts of interest for this article.
REFERENCES
- 1. Chen H, Xu Z, Mei C, Yu D, Small S. A system of repressor gradients spatially organizes the boundaries of bicoid‐dependent target genes. Cell 2012, 149:618–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stathopoulos A, Levine M. Genomic regulatory networks and animal development. Dev Cell 2005, 9:449–462. [DOI] [PubMed] [Google Scholar]
- 3. Zeitlinger J, Zinzen RP, Stark A, Kellis M, Zhang H, Young RA, Levine M. Whole‐genome ChIP–chip analysis of dorsal, twist, and snail suggests integration of diverse patterning processes in the drosophila embryo. Genes Dev 2007, 21:385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Joussineau C, Bataillé L, Jagla T, Jagla K. Chapter 11 – Diversification of muscle types in drosophila: upstream and downstream of identity genes In: Serge P, François P, eds. Current Topics in Developmental Biology, vol. 98 Cambridge, MA: Academic Press Elsevier Inc.; 2012, 277–301. [DOI] [PubMed] [Google Scholar]
- 5. Junion G, Spivakov M, Girardot C, Braun M, Gustafson EH, Birney E, Furlong Eileen EM. A transcription factor collective defines cardiac cell fate and reflects lineage history. Cell 2012, 148:473–486. [DOI] [PubMed] [Google Scholar]
- 6. Bayraktar OA, Doe CQ. Combinatorial temporal patterning in progenitors expands neural diversity. Nature 2013, 498:449–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li X, Erclik T, Bertet C, Chen Z, Voutev R, Venkatesh S, Morante J, Celik A, Desplan C. Temporal patterning of Drosophila medulla neuroblasts controls neural fates. Nature 2013, 498:456–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Levine M, Hafen E, Garber RL, Gehring WJ. Spatial distribution of Antennapedia transcripts during Drosophila development. EMBO J 1983, 2:2037–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kosman D, Mizutani CM, Lemons D, Cox WG, McGinnis W, Bier E. Multiplex detection of RNA expression in drosophila embryos. Science 2004, 305:846. [DOI] [PubMed] [Google Scholar]
- 10. Tautz D, Pfeifle C. A non‐radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma 1989, 98:81–85. [DOI] [PubMed] [Google Scholar]
- 11. Gregor T, Tank DW, Wieschaus EF, Bialek W. Probing the limits to positional information. Cell 2007, 130:153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. He F, Wen Y, Deng J, Lin X, Lu LJ, Jiao R, Ma J. Probing Intrinsic properties of a robust morphogen gradient in drosophila. Dev Cell 2008, 15:558–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Perry MW, Boettiger AN, Levine M. Multiple enhancers ensure precision of gap gene‐expression patterns in the Drosophila embryo. Proc Natl Acad Sci 2011, 108:13570–13575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Perry MW, Bothma JP, Luu RD, Levine M. Precision of hunchback expression in the Drosophila embryo. Curr Biol 2012, 22:2247–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Porcher A, Abu‐Arish A, Huart S, Roelens B, Fradin C, Dostatni N. The time to measure positional information: maternal Hunchback is required for the synchrony of the Bicoid transcriptional response at the onset of zygotic transcription. Development 2010, 137:2795–2804. [DOI] [PubMed] [Google Scholar]
- 16. Boettiger AN, Levine M. Synchronous and stochastic patterns of gene activation in the Drosophila embryo. Science 2009, 325:471–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science 2002, 297:1183–1186. [DOI] [PubMed] [Google Scholar]
- 18. Munsky B, Neuert G, van Oudenaarden A. Using gene expression noise to understand gene regulation. Science 2012, 336:183–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ozbudak EM, Thattai M, Kurtser I, Grossman AD, van Oudenaarden A. Regulation of noise in the expression of a single gene. Nat Genet 2002, 31:69–73. [DOI] [PubMed] [Google Scholar]
- 20. Coppey M, Boettiger AN, Berezhkovskii AM, Shvartsman SY. Nuclear trapping shapes the terminal gradient in the Drosophila embryo. Curr Biol 2008, 18:915–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lagha M, Bothma JP, Esposito E, Ng S, Stefanik L, Tsui C, Johnston J, Chen K, Gilmour DS, Zeitlinger J, et al. Paused Pol II coordinates tissue morphogenesis in the Drosophila embryo. Cell 2013, 153:976–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Garcia HG, Tikhonov M, Lin A, Gregor T. Quantitative imaging of transcription in living Drosophila embryos links polymerase activity to patterning. Curr Biol 2013, 23:2140–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lucas T, Ferraro T, Roelens B, De Las Heras Chanes J, Walczak AM, Coppey M, Dostatni N. Live imaging of bicoid‐dependent transcription in Drosophila embryos. Curr Biol 2013, 23:2135–2139. [DOI] [PubMed] [Google Scholar]
- 24. Campbell PD, Chao JA, Singer RH, Marlow FL. Dynamic visualization of transcription and RNA subcellular localization in zebrafish. Development 2015, 142:1368–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boettiger AN, Levine M. Rapid transcription fosters coordinate snail expression in the Drosophila embryo. Cell Rep 2013, 3:8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Little SC, Tikhonov M, Gregor T. Precise developmental gene expression arises from globally stochastic transcriptional activity. Cell 2013, 154:789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Paré A, Lemons D, Kosman D, Beaver W, Freund Y, McGinnis W. Visualization of individual Scr mRNAs during Drosophila embryogenesis yields evidence for transcriptional bursting. Curr Biol 2009, 19:2037–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zenklusen D, Larson DR, Singer RH. Single‐RNA counting reveals alternative modes of gene expression in yeast. Nat Struct Mol Biol 2008, 15:1263–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hiraoka Y, Dernburg AF, Parmelee SJ, Rykowski MC, Agard DA, Sedat JW. The onset of homologous chromosome pairing during Drosophila melanogaster embryogenesis. J Cell Biol 1993, 120:591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wilkie GS, Shermoen AW, O'Farrell PH, Davis I. Transcribed genes are localized according to chromosomal position within polarized Drosophila embryonic nuclei. Curr Biol 1999, 9:1263–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cai L, Friedman N, Xie XS. Stochastic protein expression in individual cells at the single molecule level. Nature 2006, 440:358–362. [DOI] [PubMed] [Google Scholar]
- 32. Hornos JE, Schultz D, Innocentini GC, Wang J, Walczak AM, Onuchic JN, Wolynes PG. Self‐regulating gene: an exact solution. Phys Rev E Stat Nonlin Soft Matter Phys 2005, 72:051907. [DOI] [PubMed] [Google Scholar]
- 33. Chubb JR, Trcek T, Shenoy SM, Singer RH. Transcriptional pulsing of a developmental gene. Curr Biol 2006, 16:1018–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Elf J, Li G‐W, Xie XS. Probing transcription factor dynamics at the single‐molecule level in a living cell. Science 2007, 316:1191–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Golding I, Cox EC. Eukaryotic transcription: what does it mean for a gene to be ‘on’? Curr Biol 2006, 16:R371–R373. [DOI] [PubMed] [Google Scholar]
- 36. Golding I, Paulsson J, Zawilski SM, Cox EC. Real‐time kinetics of gene activity in individual bacteria. Cell 2005, 123:1025–1036. [DOI] [PubMed] [Google Scholar]
- 37. Walczak AM, Sasai M, Wolynes PG. Self‐consistent proteomic field theory of stochastic gene switches. Biophys J 2005, 88:828–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Muramoto T, Cannon D, Gierliński M, Corrigan A, Barton GJ, Chubb JR. Live imaging of nascent RNA dynamics reveals distinct types of transcriptional pulse regulation. Proc Natl Acad Sci USA 2012, 109:7350–7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bothma JP, Garcia HG, Esposito E, Schlissel G, Gregor T, Levine M. Dynamic regulation of eve stripe 2 expression reveals transcriptional bursts in living Drosophila embryos. Proc Natl Acad Sci USA 2014, 111:10598–10603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Blake WJ, Kaern M, Cantor CR, Collins JJ. Noise in eukaryotic gene expression. Nature 2003, 422:633–637. [DOI] [PubMed] [Google Scholar]
- 41. Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol 2006, 4:e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Senecal A, Munsky B, Proux F, Ly N, Braye FE, Zimmer C, Mueller F, Darzacq X. Transcription factors modulate c‐Fos transcriptional bursts. Cell Rep 2014, 8:75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shin I, Ray J, Gupta V, Ilgu M, Beasley J, Bendickson L, Mehanovic S, Kraus GA, Nilsen‐Hamilton M. Live‐cell imaging of Pol II promoter activity to monitor gene expression with RNA IMAGEtag reporters. Nucleic Acids Res 2014, 42:e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bertrand E, Chartrand P, Schaefer M, Shenoy SM, Singer RH, Long RM. Localization of ASH1 mRNA particles in living yeast. Mol Cell 1998, 2:437–445. [DOI] [PubMed] [Google Scholar]
- 45. Chao JA, Patskovsky Y, Almo SC, Singer RH. Structural basis for the coevolution of a viral RNA‐protein complex. Nat Struct Mol Biol 2008, 15:103–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Daigle N, Ellenberg J. [lambda]N‐GFP: an RNA reporter system for live‐cell imaging. Nat Methods 2007, 4:633–636. [DOI] [PubMed] [Google Scholar]
- 47. Gaspar I, Ephrussi A. Strength in numbers: quantitative single‐molecule RNA detection assays. WIREs Dev Biol 2015, 4:135–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Forrest KM, Gavis ER. Live imaging of endogenous RNA reveals a diffusion and entrapment mechanism for nanos mRNA localization in Drosophila. Curr Biol 2003, 13:1159–1168. [DOI] [PubMed] [Google Scholar]
- 49. Zimyanin VL, Belaya K, Pecreaux J, Gilchrist MJ, Clark A, Davis I, St Johnston D. In vivo imaging of oskar mRNA transport reveals the mechanism of posterior localization. Cell 2008, 134:843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. JayaNandanan N, Gavis ER, Riechmann V, Leptin M. A genetic in vivo system to detect asymmetrically distributed RNA. EMBO Rep 2011, 12:1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chubb JR, Liverpool TB. Bursts and pulses: insights from single cell studies into transcriptional mechanisms. Curr Opin Genet Dev 2010, 20:478–484. [DOI] [PubMed] [Google Scholar]
- 52. Darzacq X, Shav‐Tal Y, de Turris V, Brody Y, Shenoy SM, Phair RD, Singer RH. In vivo dynamics of RNA polymerase II transcription. Nat Struct Mol Biol 2007, 14:796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Coulon A, Ferguson ML, de Turris V, Palangat M, Chow CC, Larson DR. Kinetic competition during the transcription cycle results in stochastic RNA processing. Elife 2014, 3:e03939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Martin Robert M, Rino J, Carvalho C, Kirchhausen T, Carmo‐Fonseca M. Live‐cell visualization of pre‐mRNA splicing with single‐molecule sensitivity. Cell Rep 2013, 4:1144–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, Ried T, Shav‐Tal Y, Bertrand E, Singer RH, et al. From silencing to gene expression: real‐time analysis in single cells. Cell 2004, 116:683–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang F, Koyama N, Nishida H, Haraguchi T, Reith W, Tsukamoto T. The assembly and maintenance of heterochromatin initiated by transgene repeats are independent of the RNA interference pathway in mammalian cells. Mol Cell Biol 2006, 26:4028–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Maiuri P, Knezevich A, De Marco A, Mazza D, Kula A, McNally JG, Marcello A. Fast transcription rates of RNA polymerase II in human cells. EMBO Rep 2011, 12:1280–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bassett AR, Tibbit C, Ponting CP, Liu J‐L. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep 2013, 4:220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gratz SJ, Cummings AM, Nguyen JN, Hamm DC, Donohue LK, Harrison MM, Wildonger J, O'Connor‐Giles KM. Genome engineering of Drosophila with the CRISPR RNA‐guided Cas9 nuclease. Genetics 2013, 194:1029–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yu Z, Ren M, Wang Z, Zhang B, Rong YS, Jiao R, Gao G. Highly efficient genome modifications mediated by CRISPR/Cas9 in Drosophila. Genetics 2013, 195:289–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wu B, Chen J, Singer RH. Background free imaging of single mRNAs in live cells using split fluorescent proteins. Sci Rep 2014, 4:3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dictenberg J. Genetic encoding of fluorescent RNA ensures a bright future for visualizing nucleic acid dynamics. Trends Biotechnol 2012, 30:621–626. [DOI] [PubMed] [Google Scholar]
- 63. Suter DM, Molina N, Gatfield D, Schneider K, Schibler U, Naef F. Mammalian genes are transcribed with widely different bursting kinetics. Science 2011, 332:472–474. [DOI] [PubMed] [Google Scholar]
- 64. Zoller B. Modelling of transcription mechanisms in mammalian cells from kinetic measurements in single cells. PhD Thesis, EPFL, Lausanne, 2014.
- 65. Gardiner CW. Handbook of Stochastic Methods for Physics, Chemistry and the Natural Sciences. 3rd ed. Berlin and New York: Springer‐Verlag; 2004. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Dynamics of fluorescent RNA accumulation at a transcribed locus: simulated kinetics of the fluorescent signal produced at the site of transcription when the MS2 SL are located in the 5' UTR (top) or in the 3' UTR (bottom) of the transcribed region. On the horizontal axis: the 24 MS2 SL (1.3 Kb) are represented in red whereas the rest of the gene (5.4 Kb) is represented in blue. Individual black boxes represent individual PolII moving along the gene with a speed of 90bp/s [22]. A PolII molecules can bind to the promoter and start producing an RNA when the promoter is ON. The kinetics of the promoter is simulated by a two state telegraph model with an ON rate of 0.05 1/s and an OFF rate of 0.05 1/s. On the vertical axis: each green dot represents a fluorescence unit formed after the transcription of a MS2 SL. At each time‐point, the total amount of fluorescence accumulating at the locus being transcribed is the sum of the fluorescent signals coming from each individual fluorescent transcript at the locus