

Abstract
Bacterial persistence represents a simple of phenotypic heterogeneity, whereby a proportion of cells in an isogenic bacterial population can survive exposure to lethal stresses such as antibiotics. In contrast, genetically based antibiotic resistance allows for continued growth in the presence of antibiotics. It is unclear, however, whether resistance and persistence are complementary or alternative evolutionary adaptations to antibiotics. Here, we investigate the co‐evolution of resistance and persistence across the genus Pseudomonas using comparative methods that correct for phylogenetic nonindependence. We find that strains of Pseudomonas vary extensively in both their intrinsic resistance to antibiotics (ciprofloxacin and rifampicin) and persistence following exposure to these antibiotics. Crucially, we find that persistence correlates positively to antibiotic resistance across strains. However, we find that different genes control resistance and persistence implying that they are independent traits. Specifically, we find that the number of type II toxin–antitoxin systems (TAs) in the genome of a strain is correlated to persistence, but not resistance. Our study shows that persistence and antibiotic resistance are complementary, but independent, evolutionary adaptations to stress and it highlights the key role played by TAs in the evolution of persistence.
Keywords: antibiotic resistance, bacteria, comparative studies, life‐history trade‐offs, persistence
Introduction
It is often assumed that populations of isogenic bacteria are phenotypically and physiologically identical. However, even under laboratory conditions specifically designed to generate homogeneous conditions, bacteria almost always show considerable phenotypic variation within a population (Dubnau & Losick, 2006; Graumann, 2006; Veening et al., 2008; Lidstrom & Konopka, 2010). For example, a small fraction of cells in clonal bacterial populations are phenotypically resistant to sudden exposure to stress, a phenomenon that has been termed ‘persistence’. Although first described in the 1940s (Hobby et al., 1942; Bigger, 1944a, b), the interest in bacterial persistence has increased in the last 15 years (Lewis, 2007; Allison et al., 2011; Balaban, 2011; Fauvart et al., 2011). This is because persistence protects bacteria against a number of clinically relevant stressors including high doses of antibiotics, but also phagocytosis by macrophages (Ramage et al., 2009; Helaine et al., 2014), and consequently, persistence is thought to contribute to many chronic and recurrent infections (Lewis, 2007, 2008; Allison et al., 2011; Balaban, 2011). Whether or not persister cells are dormant remains an ongoing discussion (Balaban et al., 2013; Wood et al., 2013), but mechanistically persisters represent a subset of cells recalcitrant to sudden stress. Although persisters are known to be induced by certain stimuli, many persister cells are formed prior to the onset of any stress as a result of stochastic processes operating at a cellular level (Balaban et al., 2004). Persisters therefore represent a form of evolutionary bet‐hedging (Slatkin, 1974; Philippi & Seger, 1989); although most cells of a given genotype will grow in favourable conditions, a subset of cells differentiate into a stress tolerant state to insure against sudden environmental change.
Persistence is an example of phenotypic plasticity, and represents one evolutionary strategy for coping with stressful environments, and is often referred to as antibiotic tolerance (Lewis, 2008; Kint et al., 2012; Fridman et al., 2014). Alternatively, bacterial populations can adapt to stress by evolving more conventional nonplastic adaptations which allow continued population growth in the presence of the stress, which in the context of antibiotics is referred to as resistance. Genetically elevated levels of resistance typically evolve by chromosomal mutations that modify the targets of antibiotics or by acquiring mobile elements carrying genes that chemically modify or degrade antibiotics (reviewed in Blair et al., 2015). Whereas persistence allows a small fraction of a bacterial population to escape from stress in a nonreplicative state, genetic resistance allows bacteria to continue to reproduce under stressful conditions. Very broadly, theory predicts that the constancy of exposure to stress is crucial in determining whether selection favours the evolution of tolerance or resistance (Kussell et al., 2005). If a selection pressure is constant, there is no advantage to a plastic trait such as tolerance. Indeed, in the case of persistence, increased levels could not evolve in response to continuous exposure to stress as cells exiting from a persistent to a replicative state would be killed by the stressor. In contrast, intermittent selection could favour either elevated persistence or resistance.
As some environments can favour the evolution of either resistance or tolerance, whether persistence and antibiotic resistance represent complementary or alternative adaptations is unclear. Intuitively, a population which has evolved a high level of antibiotic resistance will experience less selection for high‐level persistence, as persistence is not required to survive novel stresses. Similarly, in a population with high‐level persistence, a resistant mutant will have a smaller selective benefit. Conversely, as both are fundamentally adaptations to the same environmental stress, both may evolve in response to the same selection pressure. Empirical support for the relationship between persistence and resistance is mixed. For example, several papers have shown that there is no association between antibiotic susceptibility and persistence between clones from the same species (Stewart & Rozen, 2012; Hofsteenge et al., 2013), suggesting that resistance and persistence may be unrelated. However, these studies did not control for the potentially confounding effects of phylogenetic nonindependence, so their results should be treated with caution. In contrast, analysis of bacteria from chronic infections suggests the two traits may have a more complicated relationship. The evolution of antibiotic resistance during chronic infections is a problem of significant medical importance (Hogardt & Heesemann, 2013; Smith et al., 2013; Cullen & McClean, 2015). However, despite repeated exposures to antibiotics, not all chronic infections evolve antibiotic resistance, which has sometimes been referred to as the paradox of chronic infections (Lewis, 2010a, b). Instead, longitudinal analysis of isolates from chronic infections suggests that some populations evolve persistence but not resistance (LaFleur et al., 2010; Mulcahy et al., 2010). This suggests that in some circumstances, resistance and persistence may represent alternative adaptations to the same selection pressure.
Although data on the relationship between resistance and persistence at a microevolutionary scale are limited, there is even less evidence at larger evolutionary scales. Therefore, to further explore the relationship between persistence and resistance, we tested for a correlation between the two types of defence strategies from bacteria from different strains of Pseudomonas. We wished to test whether different strains adopted fundamentally different strategies with coping with stress and whether some strains evolve high resistance but low persistence, whereas some favoured vice versa. If stressful environments generate selection for both resistance and persistence, then these traits should be positively correlated across strains. On the other hand, if persistence and resistance are alternative evolutionary strategies, then a negative correlation should exist between these traits. Finally, it is possible that there is no general relationship between persistence and resistance. Phylogenetic nonindependence can lead to spurious correlations between traits across strains that are driven by common ancestry. To account for this problem, we tested for associations between persistence and resistance using comparative methods that explicitly account for the phylogeny of Pseudomonas (Vogwill et al., 2014). Specifically, trait‐association studies can be biased by unequal phylogenetic relatedness. In other words, traits can appear associated because closely related strains are more likely than average to have similar phenotypes, and thereby obscure any actual relationship between the traits from differing branches of a phylogeny.
The second aim of this study was to explore the genetic mechanisms underpinning this variation in persistence. Although the mechanistic basis of persistence was long seen as highly complicated and elusive, recent work has shown that type II toxin–antitoxin systems (TAs) play a central role in persister formation. Type II TAs encode a toxin protein and corresponding antitoxin protein (Schuster & Bertram, 2013). The antitoxin neutralizes the toxin, preventing the activity of the toxin when both are present. The antitoxins in these systems are either less stress tolerant or have a shorter half‐life than the toxins. Therefore, certain stresses (Maisonneuve et al., 2013), or simply stochastic gene expression (Fasani & Savageau, 2013), can result in the toxin becoming active. Several different lines of evidence implicate type II TAs in persister formation (Lewis & Hansen, 2013; Schuster & Bertram, 2013; Holden, 2015). For example, the first mutant isolated with a higher persister fraction was an Escherichia coli which had a mutation in the HIPA toxin gene (Moyed & Bertrand, 1983). Moreover, by sequentially knocking out type II TAs, it has been experimentally demonstrated the number of TAs in a genome is proportional to the number of persisters within that population (Maisonneuve et al., 2011). However, to our knowledge, there are no comparative tests of this link between type II TA frequency and persister formation. We therefore used our experimental data to test the hypothesis that the number of TAs within a genome would predict the level of persistence but not resistance. This would allow us to separate whether the relationship between resistance and persistence was a result of a direct mechanistic link.
Materials and methods
Strains
The eight strains of Pseudomonas are listed in Fig. 1. Prior to experimentation, all strains were stored in 20% glycerol at −80 °C. Strains were cultured in liquid King's B media (KB), at 30 °C with constant shaking at 250 rpm.
Figure 1.
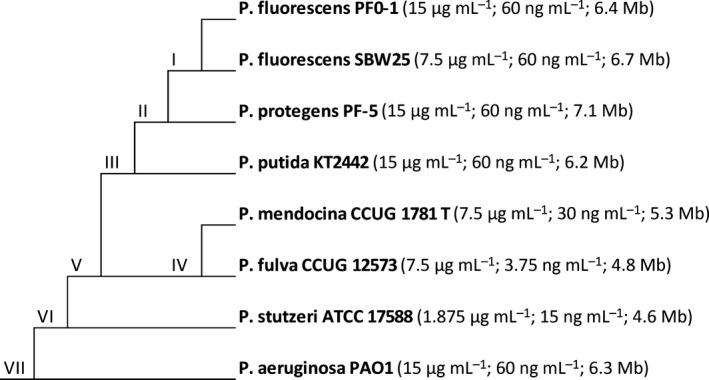
Schematic of the study system showing the phylogenetic relationship of the strains. The numbers following each strain's name represent the minimum inhibitory concentrations to rifampicin and ciprofloxacin, respectively, followed by the strain's genome size.
Kill curves
We define persistence as the number of cells surviving 4 h of antibiotic exposure. To assay the level of persistence of our bacterial isolates, we performed survival curves for each strain of bacteria for two antibiotics. We use the DNA gyrase inhibitor, ciprofloxacin, and the RNA polymerase inhibitor, rifampicin. These antibiotics have different cellular targets and are chemical distinct. Each bacterial isolate was initially cultured overnight in liquid King's B media (KB) at 30 °C with constant shaking at 250 rpm. Samples were then diluted 20‐fold into fresh KB media and recultured for 2 h to standardize all culture to exponential phase growth. After 2 h, an aliquot of this culture was plated to determine its density, after which the remaining culture had the relevant antibiotic added to it. After 4 h, another aliquot of this culture was plated to redetermine its density, and the level of persistence was calculated as the proportion or bacteria surviving antibiotic exposure. This assay was performed for both antibiotics, at the MIC (minimum inhibitory concentration, see below), 2× and 4× MIC for the strain with highest MIC for that antibiotic. Conveniently, this range of doses included 4× the MIC for seven of the strains to each antibiotic. For the strains with 4× MIC outside this range, we performed additional kill curves, specifically P. fulva CCUG 12573 to rifampicin and P. stutzeri ATCC 17588 to ciprofloxacin. Pilot kill curves were initially performed in which the density of bacteria was assayed every hour for 6 h post‐antibiotic exposure (Fig. 2). Four hours was found to representative to the level of persistence regardless of time period for the antibiotics included here.
Figure 2.
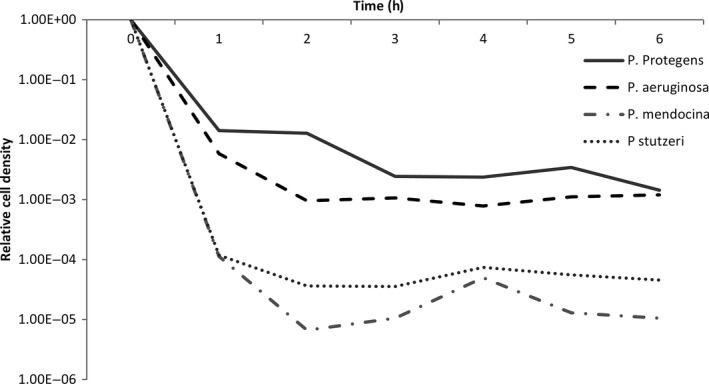
Example of the kill curves used to assay persistence, for four bacterial strains at one concentration of an antibiotic. An initial sample is plated at 0 h, after which antibiotics are immediately added to each tube. Subsequent samples are then plated every hour for 6 h. Cell densities are expressed relative to the density at 0 h.
Resistance profiles
The resistance profiles for these strains were taken from a manuscript in preparation by Furio and Maclean. Briefly, to assay the resistance of our bacterial isolates, the MICs were determined by EUCAST standard methods for each of 24 antibiotics. Five to ten morphologically similar colonies of each strain were suspended in sterile saline solution (NaCl 0.9%). A sample of this suspension was then used to inoculate approximately 105 cells into each well of a microtitre plate contacting a two‐fold dilution series of a particular antibiotic. The plate was then incubated for 24 h at 30 °C, after which the optical density at 595 nm was measured using a Bio‐Tek Synergy 2 microplate reader (Bio‐Tek, Winooski, VT, USA). The lowest antibiotic concentration at which growth had been completely inhibited was considered the MIC. This was repeated three times for each antibiotic by strain combination. This was performed for a total of 24 antibiotics: ceftazidime, cefotaxime, ceftriaxone, meropenem, piperacillin, carbencillin, azteronam, kanamycin, streptomycin, gentamycin, amikacin, tobramycin, ciprofloxacin, levofloxacin, feroxacin, colistin, polymixin B, sulfamethoxazole, mafenide, chloramphenicol, rifampicin, fosfomycin, spectinomycin and tetracycline. All antibiotics were ordered from Sigma, and stocks were prepared according to the manufacturer's instructions and stored at −20 °C.
The MICs of different antibiotics can vary across several magnitudes of scale, rendering a raw mean across different antibiotics largely meaningless. For example, some antibiotics are active at ng mL−1, whereas others require mg mL−1. Therefore, to provide a general measure of resistance, we standardize the differing antibiotics by first calculating a z‐score for each strain to each antibiotic. A z‐score is a dimensionless standardized variable, calculated as the difference between a variable and its mean, standardized by dividing by the standard deviation of the mean. For each antibiotic, MICs were first log‐transformed so that they would be on a linear scale, then all three replicates for each strain–antibiotic combination were averaged prior to z‐scores being calculated. As some of the antibiotics used are from the same family, we averaged z‐scores for antibiotics of the same family together. This reduced our 24 scores to just 10: beta‐lactams, aminoglycosides, fluoroquinolones, polymyxins, sulphonamides, chloramphenicol, rifampicin, fosfomycin, spectinomycin and tetracycline. Finally, we took the average of those 10 z‐scores as our resistance measurement for each strain.
Phylogenetic analysis
Trait‐association studies can be biased by unequal phylogenetic relatedness. In other words, traits can appear associated because closely related strains are more likely than average to have similar phenotypes, and thereby obscure any actual relationship between the traits from differing branches of a phylogeny. Here, we control for this using phylogenetic independent contrasts (PICs) performed using the method of Felsenstein (Felsenstein, 1985), calculated using the Pseudomonas phylogeny from Vogwill et al. (2014). In short, this method compares the change in a trait between neighbouring nodes or tips and then standardizes for the distance between these nodes/tips, thereby controlling for some strains being more closely related than others. When this is performed for more than one trait, the size or direction of these contrasts can then be analysed to look for correlated evolution of different traits. For the eight strains in this study system, we can calculate seven contrasts. However, as it is customary to constrain the correlation of these traits through the origin, there is no loss of statistical power.
Bioinformatic analysis of toxin–antitoxin systems
The number of toxin–antitoxin pairs per strain was downloaded from the Toxin‐Antitoxin Database (TADB). This database provides a confidence score for each TA loci as a percentage. We use anything predicted to be a TAs with at least 50% confidence from the TADB. Note that this value correlates with both anything reported as a TAs in the TADB, as well as with the number of TAs reported with 90% confidence.
Statistical analysis
All statistical tests were performed in SPSS 16 (SPSS Inc., Chicago, IL, USA). Variation in persistence was analysed using general linear models and associates between different traits using phylogenetically corrected correlations. Proportions of variances explained were calculated using standard methods.
Results
Persistence varies between strains, antibiotic and doses of antibiotic
Using kill curves, we measured levels of persistence for eight isolates of Pseudomonas (Fig. 1) to three doses of the antibiotics ciprofloxacin and rifampicin (Fig. 3). We find that persistence is significantly affected by both the bacterial strain (full statistical results in Table 1; strain: F 7,239 = 115.89, P < 0.001) and the dose of the antibiotic (dose: F 2,239 = 89.68, P < 0.001), but not directly by the identity antibiotic itself (antibiotic: F 1,239 = 1.11, P = 0.293). Specifically, the strain explained 46.3% of the variance in persistence, whereas the dose of antibiotic explained 10.2% of the variance, demonstrating that the majority of variation in persistence is between strains.
Figure 3.
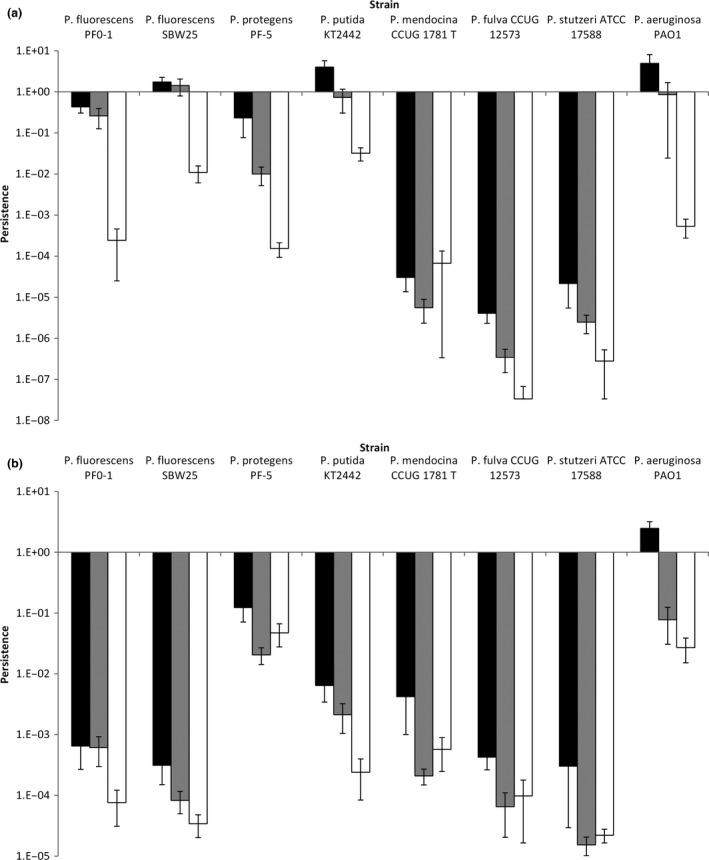
Persistence varies between strains, antibiotics and doses. Bars represent mean proportion surviving cells (± SE) after 4 h exposure to either ciprofloxacin (panel a) or rifampicin (panel b). For panel a, black, grey and white bars represent 60, 120 and 240 ng L−1, respectively. For panel b, black, grey and white bars represent 15, 30 and 60 μg L−1, respectively.
Table 1.
Results of a fully factorial general mixed model on the mean level of persistence across different strains of Pseudomonas for the antibiotics rifampicin and ciprofloxacin
| Factor | d.f. | F‐statistic | P‐value | Proportion variance explained (eta‐squared) |
|---|---|---|---|---|
| Strain | 7,239 | 98.14 | > 0.001 | 46.32 |
| Dose | 2,239 | 75.95 | > 0.001 | 10.24 |
| Antibiotic | 1,239 | 00.94 | 0.293 | 00.06 |
| Strain × dose | 14,239 | 01.96 | > 0.01 | 01.85 |
| Strain × antibiotic | 7,239 | 50.29 | > 0.001 | 23.74 |
| Antibiotic × dose | 2,239 | 21.13 | > 0.001 | 02.85 |
| Antibiotic × strain × dose | 14,239 | 01.10 | 0.208 | 01.04 |
As antibiotic dose significantly affected persistence, we repeated the analysis using just persistence at 4× MIC for each strain. Bacterial strain was again found to significantly affect persistence (strain: F 7,80 = 13.44, P < 0.001) and explained 36.6% of the variance. At this dose, there was statistically significant variation in persistence between antibiotics (antibiotic: F 1,80 = 7.48, P < 0.01), but the strength of this effect was weak, accounting for only 2.9% of the variance in persistence. The interaction of strain and antibiotic, on the other hand, accounted for 28.2% of the variance in persistence (antibiotic by strain interaction: F 7,80 = 10.39, P < 0.001). As strain is found to be the strongest predictor of persistence, we therefore define persistence for the rest of this manuscript as the mean antibiotic survival at four times a strain's MIC, averaged across both antibiotics. However, it should be noted that the results of the analysis do not change if a mean across all doses is used, or if persistence at any particular dose is also used.
Persistence and resistance: complimentary or alternative adaptations
To provide a broad measure of antibiotic susceptibility, we first measured the MIC for 24 antibiotics for each strain. We then converted this into a z‐score to provide a relative measure of antibiotic resistance. We find that mean antibiotic susceptibility does significantly correlate with mean persistence at 4× MIC (Fig. 4a; r = 0.787, d.f. = 6, P < 0.05). This is also true if we limit the measure of antibiotic resistance to just the mean of the two antibiotics used to assay persistence (r = 0.769, d.f. = 6, P < 0.05), or if we treat the two antibiotics as independent measures of resistance and persistence (r = 0.594, d.f. = 14, P < 0.05). However, this analysis does not correct for phylogenetic nonindependence, which is the tendency for traits to be similar between closely related strains solely due to phylogenetic history. If not controlled for, traits can be incorrectly concluded to be selected by the same ecological pressures, when in fact they are correlated due to so‐called phylogenetic inertia. Therefore, to control for the effects of phylogeny, we repeated the analysis using PICs via the method of Felsenstein. This analysis reveals that mean persistence at 4× MIC and mean resistance are still significantly correlated after correcting for phylogeny (Fig. 4b; r = 0.731, d.f. = 6, P < 0.05).
Figure 4.
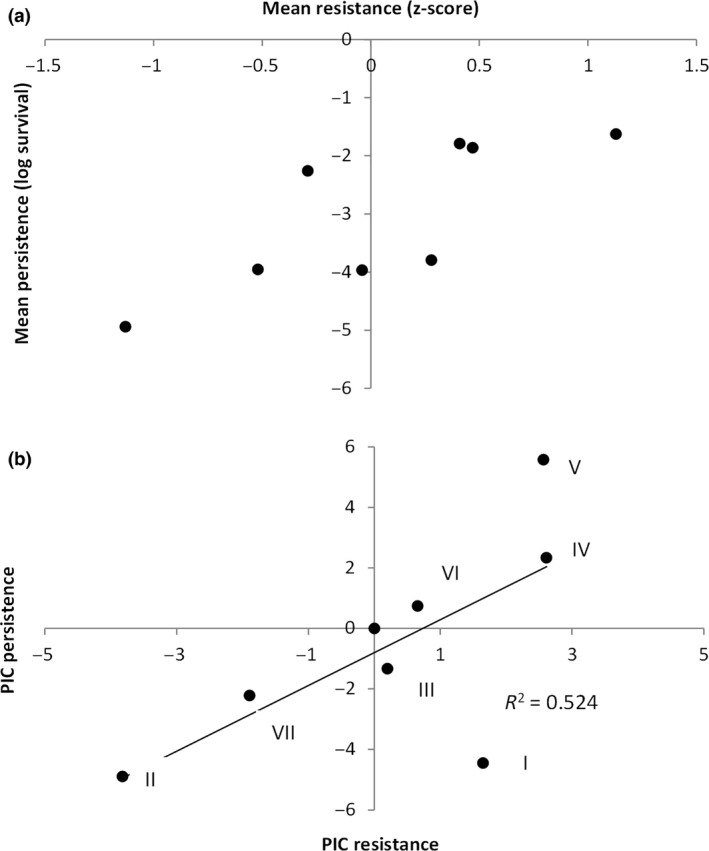
(a) mean resistance and mean persistence correlate across different strains of Pseudomonas. Each point represents a different strain. (b) Resistance and persistence still correlate after correcting for phylogeny. Each point represents a different phylogenetic independent contrast (PIC, numbered as in Fig. 1).
To explore the mechanistic basis of variation in persistence, we compared our phenotypic data with the number of TAs in the genomes of our isolates. Although type II TAs are known to play a major role in persister formation, it is unclear to what extent variation in the copy number of type II TAs between isolates can explain variation in persistence. Strikingly, the number of predicted TAs in a genome significantly correlates with the mean level of persistence (Fig. 5a, b; phylogenetically independent correlation: r = 0.793, d.f. = 6, P < 0.05). In other words, the greater the number of TAs in a genome the higher the mean level of persistence. In contrast, there is no significant relationship between the number of TAs and antimicrobial resistance (Fig. 6a, b; phylogenetically independent correlation: r = 0.478, d.f. = 6, P = 0.231). In other words, TAs do not appear to contribute to antimicrobial resistance. Therefore, although persistence and resistance are phenotypically correlated, they appear to be mechanistically unrelated.
Figure 5.
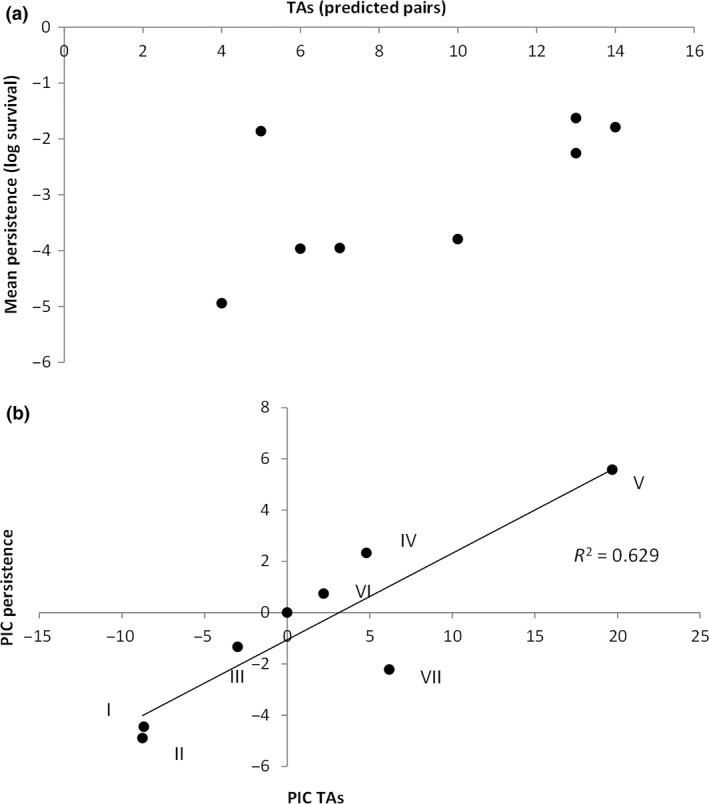
(a) the number of type II toxin–antitoxin (TAs) pairs predicts the mean level of persistence across strains. Each point represents a separate strain. (b) The number of TAs still significantly correlates with persistence after correcting for phylogeny. Each point represents a different phylogenetic independent contrast (PIC, numbered as in Fig. 1).
Figure 6.
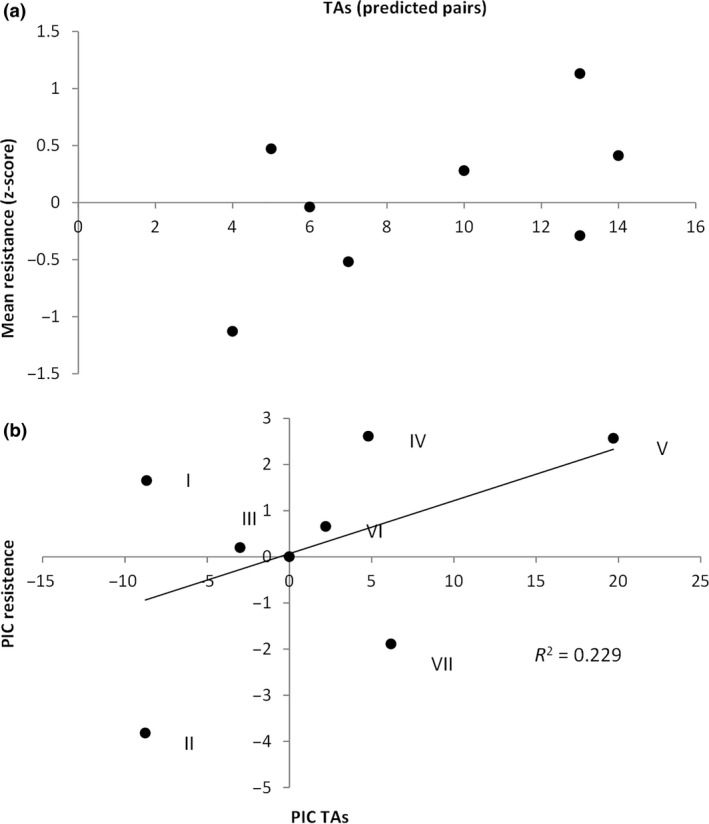
(a) the number of type II toxin–antitoxin (TAs) pairs has no significant relationship with the mean level of resistance across strains. Each point represents a separate strain. (b) The number of TAs still has no significant relationship with resistance after correcting for phylogeny. Each point represents a different phylogenetic independent contrast (PIC, numbered as in Fig. 1).
One feature which may underlie this pattern is variation in genome size, as the isolates used in our system vary by over 2.5 MB in genome size (Özen & Ussery, 2012). It has been previously demonstrated that the number of TAs roughly correlates with genome size (Makarova et al., 2009). We therefore reanalysed the relationship between persistence and resistance using partial correlations to control for the effect of genome size. Partial correlations are a technique whereby the correlation between two variables is tested for whether it could be caused by both variables independently correlating with a third variable. This analysis reveals that the correlation between persistence and resistance is no stronger than would be expected if both are unrelated to each other but both independently correlated with genome size (phylogenetically corrected partial correlation: r = 0.223, d.f. = 5, P = 0.630). In contrast, the relationship between persistence and the number of TAs is stronger than expected from both being simply properties of genome size (phylogenetically corrected partial correlation: r = 0.894, d.f. = 5, P < 0.01). This suggests that the positive correlation between resistance and persistence simply reflects the fact that strains of Pseudomonas with large genomes have independently evolved high levels of both persistence and resistance.
Discussion
In this manuscript, we report a phenotypic association between antibiotic susceptibility and bacterial persistence. This suggests that at a broad evolutionary scale, they represent complimentary adaptations to stressful ecological niches. However, as resistance and persistence are known to have different mechanistic causes, we unsurprisingly find that the relationship appears to not be causal. Specifically, we present evidence that the level of persistence of a bacterial strain can be predicted from the number of type II TAs in its genome. Admittedly, this is only from a sample of eight isolates, so requires additional genomewide association studies (GWAS) to be fully verified. However, with the increasing volumes of genomic data available for pathogenic bacteria, there is increasing interest in predicting bacterial phenotypes from sequence data. So‐called GWAS have had some success predicting virulence and antimicrobial resistance (reviewed in Read & Massey, 2014). From our relatively small study, it appears persistence may be predicted from sequence data. This is somewhat surprising when one considers how only a few years ago the molecular basis of persistence was still obscure. Although many genes were known to influence persister formation, they seemed so disparate and general that predicting persistence solely from genomic data would have appeared impossible. However, given the emerging role of TAs in persistence phenotypes, it seems tentatively more promising to relate persistence to sequence data. A caveat to this argument is that persisters are thought to be a universal property of bacteria (Lewis, 2010a, b), whereas TAs are not (Pandey & Gerdes, 2005; Makarova et al., 2009). Aside from whether improved knowledge of TAs will reveal their presence in these genomes, the predictive power of TAs is limited to strains where TAs are a major mechanism of persister formation.
Across a diverse range of isolates, we find strain to be the best predictor of persistence. Although persistence did significantly differ between antibiotics, this effect was much smaller than the effect of strain. It has been previously demonstrated that persistence varies between genotypes and antibiotics (Stewart & Rozen, 2012; Hofsteenge et al., 2013); however, neither previous work analysed the effect of strain and antibiotic in the same statistical test, which would allow the partitioning of variance between the two factors. Although both papers do show that persistence does not linearly correlate between different antibiotics, this does not preclude strain having the greatest predictive power for persistence. Indeed, in the data presented here although there is no significant correlation in persistence between the two antibiotics, strain is still found to explain a greater proportion of variance than antibiotic. It is important to understand whether persistence is mainly due to particular genotypes or particular antibiotics (or indeed other factors), as it could directly inform clinical antibiotic choices particular with regard to chronic or potentially chronic infections. For example, if certain antibiotics always yield high fractions of persisters, then this antibiotic should be avoided for the treatment of potentially chronic or recurrent infections. In contrast, if certain genotypes always have higher persister fractions, then greater effort could be made to understand the epidemiology and spread of these lineages.
The study system used here encompasses the entire breadth of a genus, and it is therefore questionable whether our findings can be applied at smaller evolutionary scales. At a within‐species scale, there is still considerable variation in both antibiotic susceptibility and persistence (Stewart & Rozen, 2012; Hofsteenge et al., 2013), making a correlation between resistance and persistence at least possible. Previous work has generally focussed on strains within species with the same level of resistance, resulting in a lack of empirical insight on this topic. There is still considerable variation in genome size within a bacterial species (for example Bergthorsson & Ochman, 1998; Thompson et al., 2005; Loper et al., 2012), although admittedly not as much variation as across a genus. However, if similar patterns of genomic content are observed in other taxa as in the Pseudomonas genus, then a correlation between persistence and resistance is quite likely, if perhaps weaker than found here. It is also interesting to speculate whether the number of TAs will still be predictive of persistence at a smaller phylogenetic scale. Although there is still considerable variation in persistence within a species (Stewart & Rozen, 2012; Hofsteenge et al., 2013), it is still unclear whether there is also variation in TAs per genome at this scale. However, variation in TAs is not the only source of variation in persistence. Although TAs have a key role in persister formation, many other factors can also influence the number of persisters found within a population (Lewis, 2007; Allison et al., 2011; Balaban, 2011; Balaban et al., 2013). Variation in persistence within a species could be a combination of variation of TA number, as well as other factors such as the precise identity of the TAs and also these other cellular systems which influence persister formation.
We find that isolates with greater genome sizes possess higher persistence but also higher resistance. If high levels of both persistence and resistance are found in the same isolate, it raises the question of what if anything limits the evolution of these traits. Crucially, there is growing evidence of trade‐offs associated with both persistence and resistance. Although the presence of costs from high‐level antibiotic resistance is well established (Melnyk et al., 2015; Vogwill & MacLean, 2015), there is also emerging evidence that higher proportions of persisters are also more costly in terms of growth rate and competitive ability (Stewart & Rozen, 2012; Stepanyan et al., 2015). However, if both traits are costly, and both traits are adaptation to similar selection pressures, it seems surprising that there is not some sort of trade‐off or inverse relationship between the two. Therefore, persistence and resistance are perhaps best viewed as adaptations to different properties of the same selection pressure, resulting in the traits being linked but not necessarily driven by the exact same properties of an environment of niche. Or in other words, in an environment which does not require a high level of antibiotic resistance, it also does not require a high level of persistence, but not for precisely the same reason.
In addition to the variation between strains, we found a significant effect of antibiotic dose and significant interactions with antibiotic identity on persistence. Finding that persistence is a highly variable phenotype is consistent with previous work from a range of systems and illustrates that persister cells do not represent a homogenous phenotype (Allison et al., 2011; Fasani & Savageau, 2013). Instead, the ‘persistence’ phenotype is actually a diverse composite of many different cellular mechanisms, each of which subtly differs in how it affects cellular growth and survival, with some mechanisms resulting in high recalcitrance to some stresses but less to others. Underlying this is the diversity of TAs within most bacterial genomes, as well as the complex up‐stream regulation of TAs. For example, if a genome contains ten TAs, then it has potentially ten distinct mechanisms of persister formation, or more if the differing TAs can work in combinations. Although the molecular basis of most TAs is still unclear, known mechanisms include inhibition of DNA gyrase, degradation of mRNA or inhibition of translation (reviewed in Gerdes et al., 2005). These differing mechanisms may represent physiological states which are tolerant of different types of cellular stress. Additionally, whether these separate routes to persistence represent alternative or synergistic approaches to cellular plasticity remains unclear. Therefore, having multiple TAs may permit differing levels of persistence depending on how many TAs are simultaneously active, as well as their precise molecular mechanism.
Our study system contains isolates predominately drawn from environmental samples, as opposed to clinical samples, although P. aeruginosa PAO1 is a clinical specimen from 1955. Therefore, the selection pressures shaping this system are likely to be low‐level selection of resistance and tolerance from naturally produced antibiotics and other toxins. Clinical systems will in general exert far greater selection pressures from antibiotic because of the higher concentrations of antibiotics required to treat infections. Furthermore, samples from chronic infections are likely to have been exposed to even greater selection for both resistance and persistence. However, whether within each of these groups persistence and resistance will also be correlated remains to be tested. Perhaps when these traits start to get extremely high, trade‐offs start to become more important.
Acknowledgments
The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007–2013)/ERC grant agreement no. 281591 and from the Royal Society. VF was supported by an MEC Postdoctoral Fellowship from the Spanish Government (EX‐2010‐0958).
Data deposited at Dryad: doi: 10.5061/dryad.s7j42
References
- Allison, K.R. , Brynildsen, M.P. & Collins, J.J. 2011. Heterogeneous bacterial persisters and engineering approaches to eliminate them. Curr. Opin. Microbiol. 14: 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban, N.Q. 2011. Persistence: mechanisms for triggering and enhancing phenotypic variability. Curr. Opin. Genet. Dev. 21: 768–775. [DOI] [PubMed] [Google Scholar]
- Balaban, N.Q. , Merrin, J. , Chait, R. , Kowalik, L. & Leibler, S. 2004. Bacterial persistence as a phenotypic switch. Science 305: 1622–1625. [DOI] [PubMed] [Google Scholar]
- Balaban, N.Q. , Gerdes, K. , Lewis, K. & McKinney, J.D. 2013. A problem of persistence: still more questions than answers? Nat. Rev. Microbiol. 11: 587–591. [DOI] [PubMed] [Google Scholar]
- Bergthorsson, U. & Ochman, H. 1998. Distribution of chromosome length variation in natural isolates of Escherichia coli . Mol. Biol. Evol. 15: 6–16. [DOI] [PubMed] [Google Scholar]
- Bigger, J. 1944a. The bactericidal action of penicillin on Staphylococcus pyogenes . Ir. J. Med. Sci. 19: 585–595. [Google Scholar]
- Bigger, J. 1944b. Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet 244: 497–500. [Google Scholar]
- Blair, J.M.A. , Webber, M.A. , Baylay, A.J. , Ogbolu, D.O. & Piddock, L.J.V. 2015. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 13: 42–51. [DOI] [PubMed] [Google Scholar]
- Cullen, L. & McClean, S. 2015. Bacterial adaptation during chronic respiratory infections. Pathogens 4: 66–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubnau, D. & Losick, R. 2006. Bistability in bacteria. Mol. Microbiol. 61: 564–572. [DOI] [PubMed] [Google Scholar]
- Fasani, R.A. & Savageau, M.A. 2013. Molecular mechanisms of multiple toxin–antitoxin systems are coordinated to govern the persister phenotype. Proc. Natl. Acad. Sci. U.S.A. 110: E2528–E2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauvart, M. , De Groote, V.N. & Michiels, J. 2011. Role of persister cells in chronic infections: clinical relevance and perspectives on anti‐persister therapies. J. Med. Microbiol. 60: 699–709. [DOI] [PubMed] [Google Scholar]
- Felsenstein, J. 1985. Phylogenies and the comparative method. Am. Nat. 125: 1–15. [Google Scholar]
- Fridman, O. , Goldberg, A. , Ronin, I. , Shoresh, N. & Balaban, N.Q. 2014. Optimization of lag time underlies antibiotic tolerance in evolved bacterial populations. Nature 513: 418–421. [DOI] [PubMed] [Google Scholar]
- Gerdes, K. , Christensen, S.K. & Lobner‐Olesen, A. 2005. Prokaryotic toxin‐antitoxin stress response loci. Nat. Rev. Microbiol. 3: 371–382. [DOI] [PubMed] [Google Scholar]
- Graumann, P.L. 2006. Different genetic programmes within identical bacteria under identical conditions: the phenomenon of bistability greatly modifies our view on bacterial populations. Mol. Microbiol. 61: 560–563. [DOI] [PubMed] [Google Scholar]
- Helaine, S. , Cheverton, A.M. , Watson, K.G. , Faure, L.M. , Matthews, S.A. & Holden, D.W. 2014. Internalization of salmonella by macrophages induces formation of nonreplicating persisters. Science 343: 204–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobby, G.L. , Meyer, K. & Chaffee, E. 1942. Observations on the mechanism of action of penicillin. Exp. Biol. Med. 50: 281–285. [Google Scholar]
- Hofsteenge, N. , van Nimwegen, E. & Silander, O. 2013. Quantitative analysis of persister fractions suggests different mechanisms of formation among environmental isolates of E. coli . BMC Microbiol. 13: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogardt, M. & Heesemann, J. 2013. Microevolution of Pseudomonas aeruginosa to a chronic pathogen of the cystic fibrosis lung In: Between Pathogenicity and Commensalism, 358 (Dobrindt U., Hacker J.H., Svanborg C., eds), pp. 91–118. Springer, Berlin Heidelberg. [DOI] [PubMed] [Google Scholar]
- Holden, D.W. 2015. Persisters unmasked. Science 347: 30–32. [DOI] [PubMed] [Google Scholar]
- Kint, C.I. , Verstraeten, N. , Fauvart, M. & Michiels, J. 2012. New‐found fundamentals of bacterial persistence. Trends Microbiol. 20: 577–585. [DOI] [PubMed] [Google Scholar]
- Kussell, E. , Kishony, R. , Balaban, N.Q. & Leibler, S. 2005. Bacterial persistence: a model of survival in changing environments. Genetics 169: 1807–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFleur, M.D. , Qi, Q. & Lewis, K. 2010. Patients with long‐term oral carriage harbor high‐persister mutants of Candida albicans . Antimicrob. Agents Chemother. 54: 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, K. 2007. Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 5: 48–56. [DOI] [PubMed] [Google Scholar]
- Lewis, K. 2008. Multidrug tolerance of biofilms and persister cells In: Bacterial Biofilms, 322 (Romeo T., ed), pp. 107–131. Springer, Berlin Heidelberg. [DOI] [PubMed] [Google Scholar]
- Lewis, K. 2010a. Persister cells. Annu. Rev. Microbiol. 64: 357–372. [DOI] [PubMed] [Google Scholar]
- Lewis, K. 2010b. Persister cells and the paradox of chronic infections. Microbe 5: 429–437. [Google Scholar]
- Lewis, K. & Hansen, S. 2013. Type II toxin‐antitoxin loci, hipBA and persisters In: Prokaryotic Toxin‐Antitoxins (Gerdes K., ed), pp. 189–203. Springer, Berlin Heidelberg. [Google Scholar]
- Lidstrom, M.E. & Konopka, M.C. 2010. The role of physiological heterogeneity in microbial population behavior. Nat. Chem. Biol. 6: 705–712. [DOI] [PubMed] [Google Scholar]
- Loper, J.E. , Hassan, K.A. , Mavrodi, D.V. , Davis, E.W. II , Lim, C.K. , Shaffer, B.T. et al 2012. Comparative genomics of plant‐associated Pseudomonas spp.: insights into diversity and inheritance of traits involved in multitrophic interactions. PLoS Genet. 8: e1002784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonneuve, E. , Shakespeare, L. , Jorgensen, M. & Gerdes, K. 2011. Bacterial persistence by RNA endonucleases. Proc. Natl. Acad. Sci. U.S.A. 108: 13206–13211. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Maisonneuve, E. , Castro‐Camargo, M. & Gerdes, K. 2013. (p)ppGpp controls bacterial persistence by stochastic induction of toxin‐antitoxin activity. Cell 154: 1140–1150. [DOI] [PubMed] [Google Scholar]
- Makarova, K.S. , Wolf, Y.I. & Koonin, E.V. 2009. Comprehensive comparative‐genomic analysis of Type 2 toxin‐antitoxin systems and related mobile stress response systems in prokaryotes. Biol. Direct 4: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnyk, A.H. , Wong, A. & Kassen, R. 2015. The fitness costs of antibiotic resistance mutations. Evol. Appl. 8: 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyed, H.S. & Bertrand, K.P. 1983. hipA, a newly recognized gene of Escherichia coli K‐12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 155: 768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy, L. , Burns, J. , Lory, S. & Lewis, K. 2010. Emergence of pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J. Bacteriol. 192: 6191–6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özen, A. & Ussery, D. 2012. Defining the Pseudomonas genus: where do we draw the line with azotobacter? Microb. Ecol. 63: 239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey, D.P. & Gerdes, K. 2005. Toxin–antitoxin loci are highly abundant in free‐living but lost from host‐associated prokaryotes. Nucleic Acids Res. 33: 966–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippi, T. & Seger, J. 1989. Hedging one's evolutionary bets, revisited. Trends Ecol. Evol. 4: 41–44. [DOI] [PubMed] [Google Scholar]
- Ramage, H.R. , Connolly, L.E. & Cox, J.S. 2009. Comprehensive functional analysis of Mycobacterium tuberculosis toxin‐antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS Genet. 5: e1000767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read, T. & Massey, R. 2014. Characterizing the genetic basis of bacterial phenotypes using genome‐wide association studies: a new direction for bacteriology. Genome Med. 6: 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster, C.F. & Bertram, R. 2013. Toxin–antitoxin systems are ubiquitous and versatile modulators of prokaryotic cell fate. FEMS Microbiol. Lett. 340: 73–85. [DOI] [PubMed] [Google Scholar]
- Slatkin, M. 1974. Hedging one's evolutionary bets. Nature 250: 704–705. [Google Scholar]
- Smith, T. , Wolff, K.A. & Nguyen, L. 2013. Molecular biology of drug resistance in Mycobacterium tuberculosis . Curr. Top. Microbiol. Immunol. 374: 53–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanyan, K. , Wenseleers, T. , Duéñez‐Guzmán, E.A. , Muratori, F. , Van den Bergh, B. , Verstraeten, N. et al 2015. Fitness trade‐offs explain low levels of persister cells in the opportunistic pathogen Pseudomonas aeruginosa . Mol. Ecol. 24: 1572–1583. [DOI] [PubMed] [Google Scholar]
- Stewart, B. & Rozen, D. 2012. Genetic variation for antibiotic persistence in Escherichia coli . Evolution 66: 933–939. [DOI] [PubMed] [Google Scholar]
- Thompson, J.R. , Pacocha, S. , Pharino, C. , Klepac‐Ceraj, V. , Hunt, D.E. , Benoit, J. et al 2005. Genotypic diversity within a natural coastal bacterioplankton population. Science 307: 1311–1313. [DOI] [PubMed] [Google Scholar]
- Veening, J.‐W. , Smits, W.K. & Kuipers, O.P. 2008. Bistability, epigenetics, and bet‐hedging in bacteria. Annu. Rev. Microbiol. 62: 193–210. [DOI] [PubMed] [Google Scholar]
- Vogwill, T. & MacLean, R.C. 2015. The genetic basis of the fitness costs of antimicrobial resistance: a meta‐analysis approach. Evol. Appl. 8: 284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogwill, T. , Kojadinovic, M. , Furió, V. & MacLean, R.C. 2014. Testing the role of genetic background in parallel evolution using the comparative experimental evolution of antibiotic resistance. Mol. Biol. Evol. 31: 3314–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood, T.K. , Knabel, S.J. & Kwan, B.W. 2013. Bacterial persister cell formation and dormancy. Appl. Environ. Microbiol. 79: 7116–7121. [DOI] [PMC free article] [PubMed] [Google Scholar]