

Abstract
We describe a new technology for recruiting specific proteins to RNA through selective recognition of heteroduplexes formed with chemically modified antisense oligonucleotides (ASOs). Typically, ASOs function by hybridizing to their RNA targets and blocking the binding of single-stranded RNA–binding proteins. Unexpectedly, we found that ASOs with 2′-deoxy-2′-fluoro (2′-F) nucleotides, but not with other 2′ chemical modifications, have an additional property: they form heteroduplexes with RNA that are specifically recognized by the interleukin enhancer-binding factor 2 and 3 complex (ILF2/3). 2′-F ASO–directed recruitment of ILF2/3 to RNA can be harnessed to control gene expression by modulating alternative splicing of target transcripts. ILF2/3 recruitment to precursor mRNA near an exon results in omission of the exon from the mature mRNA, both in cell culture and in mice. We discuss the possibility of using chemically engineered ASOs that recruit specific proteins to modulate gene expression for therapeutic intervention.
Introduction
The biogenesis of eukaryotic mRNAs is orchestrated by numerous RNA-binding proteins (RBPs) that control mRNA end processing, splicing, editing, transport, translation, localization and turnover1. This control is accomplished by coordinated expression and recruitment of RBPs to precursor or mature mRNA2, 3, which causes disease when compromised4. RBPs bind their substrates by virtue of domains that recognize single- or double-stranded RNA directly5, 6, 7 or as part of ribonucleoprotein complexes (RNPs) that use RNA as a guide8.
Several antisense-independent or antisense-dependent strategies have been devised to artificially recruit proteins to RNA. The former include engineered proteins composed of an RNA-recognition module fused to a functional module9, 10 and RNA aptamers inserted into transcripts that are recognized by protein ligands11; the latter include bifunctional ASOs with appended RNA or peptide motifs12, 13, 14, 15, 16, RNase H–dependent ASOs, small interfering RNAs (siRNAs) and microRNAs17. In addition, ASOs can also be used to prevent RBP or RNP binding by selectively interfering with RNA-protein or RNA-RNP interactions17, 18.
To improve the potential of ASOs as therapeutic agents, they are chemically modified to increase their metabolic stability, enhance their distribution to cells within tissues and strengthen their binding affinity toward RNA19. Over the last 25–30 years, the search for nucleic acid analogs that confer optimal ASO properties has led to the synthesis and characterization of hundreds of modifications. Some of the most widely used modifications include phosphorothioate linkages in the ASO backbone, replacement of the sugar-phosphate backbone entirely with morpholino or peptide nucleic acids, and modifications of the sugar moiety by substitutions at the 2′ position, such as 2′-O-methyl (2′-OMe), 2′-O-methoxyethyl (2′-MOE), 2′-F and bicyclic sugars, such as locked nucleic acid (LNA) and constrained ethyl (cEt)20, 21. The nuclease resistance, biodistribution and RNA-binding affinity of chemically engineered ASOs have all been intensely studied20, 22. However, one property of modified ASOs that has received little attention is the effect of chemical modifications on the recruitment of proteins to the ASO-RNA duplex. Here we have evaluated whether the chemical composition of ASOs can be exploited to recruit specific proteins to precursor mRNA (pre-mRNA). For functionality, we focused on the ability of ASOs to modulate alternative splicing, a nuclear process controlled by an intricate network of RNA-protein interactions23.
Results
2′-F ASO–directed exon skipping in cell culture
Previously, we identified an 18-mer ASO with 2′-MOE nucleotides (Fig. 1a) that promotes nearly complete inclusion of SMN2 exon 7—an alternative exon—by binding an intronic splicing silencer in intron 724. We transfected this 2′-MOE ASO into HeLa cells and measured splicing of endogenous SMN2 transcripts 24 h later by reverse transcription followed with PCR (RT-PCR). As expected, the 2′-MOE ASO caused almost complete SMN2 exon 7 inclusion compared to untreated cells (Fig. 1b, lane 1 versus lane 3). Using splicing of SMN2 exon 7 as a model system, we evaluated how various chemical modifications of the ribose moiety of the 18-mer ASO affected splicing. Remarkably, an ASO with 2′-F ribose nucleotide residues (Fig. 1a) had the opposite effect to that of the 2′-MOE ASO, resulting in almost complete exon 7 skipping (Fig. 1b, lane 2 versus lane 3). This 2′-F ASO has an identical sequence to that of the 2′-MOE ASO, differing only in the 2′ modification on the sugar (methoxyethoxy versus fluorine). 2′-F ASOs targeted to sites either upstream or downstream of SMN2 exon 7 also caused exon skipping (Supplementary Results, Supplementary Fig. 1). In addition to having SMN2, HeLa cells have the paralogous SMN1 gene with the same ASO target site. SMN1 transcripts normally constitutively include exon 7 (Fig. 1b, lane 3)25. The 2′-F ASO also caused complete SMN1 exon 7 skipping (Fig. 1b, lane 2 versus lane 3).
Figure 1. Modulation of splicing in cell culture as a function of ASO chemistry.

(a) Structure of nucleotides with various 2′ substituents. (b) RT-PCR and DdeI digestion of endogenous HeLa SMN1 and SMN2 mRNA after ASO treatment. The ASOs are complementary to a site 10–27 nucleotides downstream of the 5′ splice site of intron 7. (c) RT-PCR of plasmid-expressed SMN2 mRNA after ASO treatment. The sequence of the ASO target site (same as in b) is shown, and the mutated nucleotides are in lowercase. The WT and MT designations indicate that the ASO is complementary to the wild-type and mutant ASO target sites, respectively. (d) Radioactive RT-PCR of SMN1 and SMN2 mRNAs isolated from fractionated HeLa cells transfected with ASOs. Cyt, cytoplasm; nuc, nuclear; chr, chromatin. Uncropped gels are shown in Supplementary Figure 13. For b–d, the numbered boxes represent specific exons in the SMN1 and SMN2 transcripts.
To ensure that 2′-F ASO–directed exon skipping depended on the ASO hybridizing to the pre-mRNA, we used SMN2 minigene constructs with either a wild-type or a mutated ASO target site. We separately transfected each SMN2 minigene together with an ASO and measured splicing 24 h later by RT-PCR (Fig. 1c). The 2′-F ASO complementary to the wild-type ASO target site (WT 2′-F) caused exon skipping when the target site was wild type, but not when it was mutated (lane 3 versus lane 8). Conversely, the ASO complementary to the mutated ASO target site (MT 2′-F) caused exon skipping when the target site was mutated, but not when it was wild type (lane 8 versus lane 4). 2′-F ASO–directed exon skipping of transcripts from the endogenous SMN2 gene in fibroblasts from patients with spinal muscular atrophy (SMA) was also hybridization dependent (Supplementary Fig. 2).
Splicing typically takes place as RNA polymerase II is generating nascent transcripts on chromatin26. We determined whether ASOs modulate splicing of transcripts on chromatin. The chromatin fraction from HeLa cells was isolated cleanly, as determined by the enrichment of histone H3 (Supplementary Fig. 3a) and intronic RNA (Supplementary Fig. 3b). We found that the inclusion of SMN1 and SMN2 exon 7 is already established on chromatin-associated RNA (Fig. 1d, lane 7 versus lanes 1 and 4), indicating that splicing of these transcripts takes place cotranscriptionally. Treatment of cells with the 2′-MOE ASO caused both SMN1 and SMN2 exon 7 inclusion (lane 8 versus lane 7), whereas the 2′-F ASO caused SMN1 and SMN2 exon skipping on chromatin-associated RNA (lane 9 versus lane 7). Thus, nascent transcripts still associated with chromatin are accessible to ASO-directed splicing modulation.
We next tested whether exon skipping directed by a 2′-F ASO could be extended to other genes for which exon skipping may be therapeutic. The pyruvate kinase M (PKM2) gene has 12 exons, of which exons 9 and 10 are alternatively spliced in a mutually exclusive fashion27. Transcripts that include exon 10 encode an isoform of PKM2 that promotes tumorigenesis28. To determine whether a 2′-F ASO could promote skipping of exon 10 and consequently inclusion of exon 9, we designed a 2′-F ASO complementary to nucleotides 11 through 28 downstream of the 5′ splice site of intron 10. We transfected the 2′-F ASO and the corresponding 2′-MOE ASO into HeLa cells and measured splicing of the endogenous PKM2 transcripts 24 h later by RT-PCR. The 2′-F ASO, but not the 2′-MOE ASO, caused skipping of exon 10 and increased the proportion of transcripts that included exon 9 (Supplementary Fig. 4a).
BCL2L1 (also known as BCL-X) has two alternative 5′ splice sites (XS upstream and XL downstream) that compete for a common 3′ splice site29. Use of the XS or the XL splice site results in the production of proapoptotic or antiapoptotic BCL2L1 protein isoforms, respectively29. We determined whether a 2′-F ASO could inhibit the XL splice site and thereby promote the use of the XS splice site. We designed a 2′-F ASO that binds nucleotides 17 through 34 downstream of the XL splice site, transfected it into HeLa cells and measured splicing 24 h later by RT-PCR. Compared to the 2′-MOE ASO, the 2′-F ASO further decreased the use of the XL splice site and increased the use of the XS splice site (Supplementary Fig. 4b).
MAPT exon 10 can be alternatively spliced to yield tau isoforms with four (4R; with exon 10) or three (3R; without exon 10) microtubule-binding repeats30. Mutations that result in the inclusion of exon 10 lead to frontotemporal dementia and parkinsonism linked to chromosome 1731. We determined whether a 2′-F ASO complementary to nucleotides 17 through 34 downstream of the 5′ splice site of intron 10 could cause the skipping of exon 10. The ASO was transfected into A172 glioblastoma cells, and splicing of exon 10 was measured 24 h later by RT-PCR. The 2′-F ASO caused almost complete skipping of exon 10, whereas the 2′-MOE ASO increased exon 10 inclusion (Supplementary Fig. 4c). These results were confirmed in a separate experiment in which exon 10 inclusion was measured by real-time RT-PCR (Supplementary Fig. 4d).
Proteins recruited to 2′-F ASO–RNA duplexes
The mechanism by which the 2′-F ASO causes exon skipping was determined using SMN2 as a model system. The target site for the 2′-MOE and 2′-F ASOs in SMN2 overlaps ISS-N1, a heterogeneous nuclear ribonucleoprotein (hnRNP) A1– and hnRNP A2–dependent intronic splicing silencer in intron 724,32. We therefore tested whether the opposite effects of these ASOs on exon 7 inclusion could be explained by their differential effects on the binding of hnRNPs A1 and A2 to the pre-mRNA. We conducted RNA pull-down experiments with either of these ASOs duplexed with a 3′-biotinylated 23-mer SMN2 intron 7 RNA fragment (+7 to +29), comprising ISS-N1, attached to streptavidin-coated magnetic beads. We incubated immobilized RNA in the presence or absence of prehybridized ASOs with HeLa cell nuclear extract, washed the beads, eluted the proteins and analyzed them by immunoblotting for the presence of hnRNPs A1 and A2/B1 (Fig. 2a). In the absence of ASOs, hnRNPs A1 and A2/B1 bound the RNA (lane 1).
Figure 2. Identification of proteins that are recruited to a 2′-F ASO–RNA duplex.

(a) Binding of hnRNP A1 and hnRNP A2/B1 to ISS-N1 upon the addition of the 2′-MOE or the 2′-F ASO was determined by western blotting. The ASOs were added before the nuclear extract (before NE), with the nuclear extract (with NE) or after hnRNPs had already bound to the ASO-RNA duplex (after NE). WT, wild-type ISS-N1; MT, mutant ISS-N1. (b) SYPRO Ruby–stained SDS gel of proteins eluted from immobilized duplexes formed with a 2′-MOE or a 2′-F ASO and a complementary biotinylated RNA. B, bound to the duplex; R, released from the duplex. Purified RNase I was loaded in the last lane. Labeled bands were excised and identified by MS as RHA, ILF3, ILF2 and NPM. (c) Western blots of the proteins identified in the released fraction in b. NE, HeLa nuclear extract. Uncropped blots are shown in Supplementary Figure 14.
Mutating two AG dinucleotides critical for hnRNP A1 recognition abrogated binding of hnRNPs A1 and A2/B1, as reported24 (lane 2). Formation of ASO-RNA duplexes with either the 2′-MOE or 2′-F ASOs also prevented hnRNP binding to the RNA (lanes 3 and 4). We then determined whether the ASOs could prevent hnRNP binding when we added them together with the extract and whether they could displace hnRNPs from the RNA when they were added after the hnRNPs had already bound. In both cases, the 2′-MOE and 2′-F ASOs diminished hnRNP binding to RNA (lanes 6 and 7 versus lane 5; lanes 9 and 10 versus lane 8). We conclude that the 2′-MOE ASO causes SMN2 exon 7 inclusion by displacing hnRNP A1/A2 from ISS-N1. Furthermore, the 2′-F ASO does not cause exon 7 skipping by enhancing hnRNP A1/A2 recruitment, as it also displaces these splicing repressors.
Having excluded hnRNPs A1 and A2/B1 as the mediators of 2′-F ASO–directed exon skipping, we asked whether other proteins might be recruited to the ASO-RNA duplex. We sought candidates by doing a pulldown from HeLa nuclear extract with bead-bound ASO-RNA duplexes as before, except that we released protein–ASO–RNA complexes from the beads by RNase I digestion and visualized the proteins by SYPRO Ruby staining. RNase digestion decreased the background owing to nonspecific protein binding to the beads (Fig. 2b, lanes 1 and 3). Several proteins bound both the 2′-MOE ASO–RNA and the 2′-F ASO–RNA duplexes (lanes 2 and 4), whereas others uniquely bound the 2′-F ASO–RNA duplex (lane 4) but not the 2′-MOE ASO–RNA duplex (lane 2). The latter proteins were excised from the gel and identified by MS as RNA helicase A (RHA), ILF2, ILF3 (which forms a heterodimer with ILF233) and nucleophosmin (NPM). Immunoblotting confirmed that these proteins were specifically recruited to the 2′-F ASO–RNA duplex (Fig. 2c).
2′-F ASO–directed exon skipping is mediated by ILF2/3
We next determined whether any of the proteins that preferentially bound the 2′-F ASO–RNA duplex were responsible for mediating 2′-F ASO–directed SMN2 exon 7 skipping. We used siRNAs to decrease the expression of each protein and then determined whether the 2′-F ASO lost its ability to cause exon skipping (Fig. 3a). Specifically, HeLa cells were treated with increasing concentrations of siRNA for 48 h to allow for turnover of the targeted protein. Then we transfected a constant amount of 2′-F ASO, isolated RNA 24 h later and measured SMN splicing by real-time RT-PCR. HeLa cells have both SMN1 and SMN2, so the transcripts that include exon 7 (Fig. 3b) or exclude exon 7 (Fig. 3c) are derived from both genes. In the absence of ILF3 knockdown, the 2′-F ASO caused exon skipping; that is, there were low levels of transcripts including exon 7 (Fig. 3b, blue solid line) and high levels of transcripts lacking exon 7 (Fig. 3c, blue solid line). siRNA treatment dose-dependently reduced ILF3 expression (Fig. 3a). When expression of ILF3 was reduced with increasing concentrations of siRNA, the 2′-F ASO gradually lost its ability to cause exon skipping, giving rise to increasing levels of transcripts including exon 7 (Fig. 3b, blue dashed line) and decreasing levels of transcripts lacking exon 7 (Fig. 3c, blue dashed line). This was also observed when the expression of ILF2 was reduced with siRNA treatment (Supplementary Fig. 5). Thus, ILF2/3 mediates 2′-F ASO–directed SMN1 and SMN2 exon 7 skipping. We obtained consistent results when using a constant amount of siRNA against ILF2 or ILF3 and increasing amounts of the 2′-F ASO (Supplementary Fig. 6). ILF2 and ILF3 heterodimerize and stabilize each other33, explaining why inhibiting the expression of either protein prevented 2′-F ASO–directed exon skipping. In contrast, knockdown of either NPM or RHA had no effect on 2′-F ASO–directed exon 7 skipping (Supplementary Fig. 7). ILF3 knockdown also attenuated the 2′-F ASO–directed conversion of Bcl-XL to Bcl-XS (Supplementary Fig. 8). As expected, none of the siRNA treatments affected the exon-inclusion activity of the 2′-MOE ASO (Fig. 3b,c and Supplementary Figs. 5–8).
Figure 3. 2′-F ASO–directed exon skipping is mediated by ILF2/3.

(a) Real-time RT-PCR analysis following siRNA knockdown of ILF3 in HeLa cells. The data points are the average of duplicate samples. (b) Real-time RT-PCR analysis of SMN transcripts including exon 7 after treating HeLa cells with increasing concentrations of ILF3 siRNA for 48 h and then another 24 h with a fixed concentration of ASO. The data points are the average of duplicate samples. Black solid line, ILF3 knockdown and no ASO; blue solid line, no ILF3 knockdown and 2′-F ASO; blue dashed line, ILF3 knockdown and 2′-F ASO; red solid line, no ILF3 knockdown and 2′-MOE ASO; red dashed line, ILF3 knockdown and 2′-MOE ASO. (c) Same as in b, except that the real-time RT-PCR analysis is for SMN transcripts excluding exon 7.
Determinants for ILF2/3 binding to ASO-RNA duplexes
Having identified the ILF2/3 complex as the mediator of 2′-F ASO–directed exon skipping, we studied the structural requirements of the ASO-RNA duplex for binding the ILF2/3 complex. In pull-down experiments, ILF2/3 bound preferentially to 2′-F ASO–RNA duplexes and not to single-stranded 2′-F ASO (Fig. 4a, lane 6 versus lane 4). In contrast, RHA bound both equally well (lane 6 versus lane 4). Binding of ILF2/3 to 2′-F ASO–RNA duplexes was not sequence specific (Supplementary Fig. 9).
Figure 4. Structural determinants for ILF2/3 binding to ASO–RNA duplexes.

(a) Western blots to measure the binding of ILF2/3 to single-stranded ASOs or to ASO-RNA duplexes. The designation ‘-bio’ indicates a biotin tag. Bead, streptavidin-conjugated beads with no ASO or RNA attached; NE, HeLa nuclear extract. (b) Western blots to measure the binding of ILF2/3 to ASO-RNA duplexes formed with ASOs with various chemical modifications of the ribose moiety. The Tm and the expected helical conformation of each ASO-RNA duplex are shown. NE, HeLa nuclear extract. (c) Western blots to measure the binding of ILF2/3 to ASO-RNA duplexes formed with ASOs with either phosphodiester linkages (P=O) or phosphorothioate linkages (P=S). NE, HeLa nuclear extract. (d) Purified ILF3 binds the 2′-F ASO–RNA (2′-F–RNA) duplex. 2′MOE ASO–RNA is abbreviated 2′-MOE–RNA. The data points are the average of duplicate samples. Uncropped blots are shown in Supplementary Figure 15.
Next, we examined the helical conformational requirements for binding of ILF2/3 to ASO-RNA duplexes (Fig. 4b). As described above, ILF2/3 bound a 2′-F ASO–RNA duplex but not a 2′-MOE–RNA duplex. ILF2/3 also bound an RNA-RNA duplex, as expected34, but not a DNA-RNA duplex. Because RNA-RNA, 2′-F ASO–RNA and 2′-MOE ASO–RNA duplexes are all A-form helices35, 36, whereas DNA-RNA duplexes form helices intermediate between A- and B-form37, the interaction of ILF2/3 with heteroduplexes is more complex than mere recognition of an A-form helix. The 2′-MOE side chain resides in the minor groove of the heteroduplex, which, together with its hydration shell, adds steric bulk36. To further explore the role of bulk in the minor groove, we tested a 2′-OMe (Fig. 1a) ASO–RNA duplex, which is also A-form38 but with less bulk in the minor groove than the 2′-MOE ASO–RNA duplex. The 2′-OMe ASO–RNA duplex bound more ILF2/3, as well as more RHA and NPM, than the 2′-MOE ASO–RNA duplex (Fig. 4b). The 2′-deoxy-2′-fluoroarabinonucleic acid (2′-F-ANA) (Fig. 1a) ASO is a stereoisomer of the 2′-F ASO, with the fluorine at the 2′ position having inverted stereochemistry37, 39. 2′-F-ANA–RNA duplexes form helices intermediate between A- and B-form37. Consistent with the DNA-RNA duplex data, there was a marked decrease in binding of ILF2/3 to the 2′-F-ANA ASO–RNA duplex compared to binding to the 2′-F ASO–RNA duplex (Fig. 4b).
We also explored the binding of ILF2/3 to heteroduplexes formed with ASOs containing bicyclic sugars. These are expected to form A-form helices when duplexed with RNA40. We synthesized ASOs with bicyclic cEt modifications (Fig. 1a), which are analogs of LNA21. ASOs are rarely fully modified with bicyclic sugars41. Therefore, each 2′-F, DNA or 2′-MOE ASO was modified with a cEt nucleotide at every position labeled X in the mixed chemistry structure (mixmer) XFFXFFXFFXFFXFFXFX, with F representing 2′-F, DNA or 2′-MOE nucleotides. Only the 2′-F/cEt mixmer ASO–RNA duplex bound ILF2/3 to a level comparable to that of the 2′-F ASO–RNA duplex (Supplementary Fig. 10). The DNA/cEt mixmer ASO–RNA duplex showed weak binding of ILF2/3, and the 2′-MOE/cEt mixmer ASO–RNA duplex did not bind ILF2/3, presumably because of the steric bulk of the 2′-MOE substitution in the minor groove (Supplementary Fig. 10). These results are in agreement with the results shown in Figure 4b and confirm that an A-form ASO-RNA duplex is not sufficient for ILF2/3 binding. In addition, cEt-modified ASOs allow maximal ILF2/3 binding only when they are combined with 2′-F modifications.
The last parameter we focused on was the type of internucleotide linkage (phosphodiester versus phosphorothioate) (Fig. 4c). As described above, ILF2/3 bound well when the duplex was formed with either RNA ASO or 2′-F ASO with a phosphorothioate backbone (lanes 4 and 6). However, ILF2/3 binding was reduced when the RNA backbone in the heteroduplex was phosphodiester (lane 3 versus lane 4). ILF2/3 still bound well to duplexes formed with 2′-F ASO with a phosphodiester backbone (lane 5 versus lane 6). Therefore, several features are necessary for maximal ILF2/3 binding: an A-form duplex with an ASO that has a phosphorothioate backbone and fluorine at the 2′ position of the ribose.
The experiments described above were performed in HeLa nuclear extracts, and therefore the binding of ILF2/3 to the 2′-F ASO–RNA could have been indirect. To determine whether ILF2/3 binds the 2′-F ASO–RNA duplex directly, we performed an experiment with purified Flag-tagged ILF3 (Supplementary Fig. 11). ILF3 is the subunit of the ILF2/3 complex that has double-stranded RNA-binding domains7. We immobilized Flag-ILF3 on magnetic beads coated with Flag-specific antibodies and then incubated these with 32P-labeled 2′-F ASO–RNA duplexes. After washing the beads, the amount of radioactive duplex that was bound was determined by scintillation counting. The 2′-F ASO–RNA duplex, but not the 2′-MOE ASO–RNA duplex, bound Flag-ILF3 (Fig. 4d). Thus, ILF2/3 binds directly to the 2′-F ASO–RNA duplex.
Exon skipping by 2′-F mixmer ASOs
As only the 2′-F/cEt mixmer ASO–RNA duplex bound ILF2/3 to a similar extent as the 2′-F ASO–RNA duplex (Supplementary Fig. 10), we next determined whether the 2′-F/cEt mixmer ASO and other 2′-F mixmer ASOs could also cause exon skipping. We transfected each ASO at increasing concentrations into fibroblasts from patients with SMA, isolated RNA 24 h later and measured SMN2 splicing by real-time RT-PCR (Fig. 5). As expected, the 2′-F ASO caused exon 7 skipping; that is, low levels of transcripts including exon 7 (Fig. 5b) and high levels of transcripts lacking exon 7 (Fig. 5c). In contrast, the 2′-F-ANA, 2′-OMe and 2′-MOE ASOs caused exon 7 inclusion (Fig. 5b). The 2′-F/cEt mixmer ASO caused exon 7 skipping (Fig. 5c), but the DNA/cEt and 2′-MOE/cEt mixmer ASOs caused exon 7 inclusion. 2′-F/DNA and 2′-F/RNA mixmer ASOs also caused exon 7 skipping, but the skipping decreased with the 2′-F/2′-OMe mixmer ASO and was eliminated with the 2′-F/2′-MOE mixmer ASO (Fig. 5c). We also tested whether siRNAs directed to the same site as the ASOs above could cause SMN2 exon 7 skipping. siRNAs did not cause exon 7 skipping, even when they had 2′-F modifications (Supplementary Fig. 12). Taken together, these results demonstrate that various 2′-F mixmer ASO designs support efficient exon skipping, provided they do not also contain bulky substituents in the minor groove such as 2′-MOE.
Figure 5. Effect of 2′-F mixmer ASOs on exon skipping.

(a) Structure of the mixmer ASOs. F is 2′-F. X is either DNA, RNA, 2′-OMe, 2′-MOE or cEt. For each ASO, every X has the same chemistry. (b) Real-time RT-PCR analysis of SMN2 transcripts including exon 7 following ASO treatment of fibroblasts from patients with SMA. The concentration of transfected ASO is shown above the graph. The data points are the average of duplicate samples. (c) Same as in b, except that the real-time RT-PCR analysis is for SMN2 transcripts excluding exon 7.
2′-F ASO–directed exon skipping in mice
To validate that the observations made in cell culture would translate to a potential therapeutic application, we determined whether the 2′-F ASO caused SMN2 exon 7 skipping in mice transgenic for human SMN2. We administered the 2′-F ASO by intraperitoneal injection at doses of 50 mg per kg body weight every 2 d for a total of four doses, harvested tissues 48 h after the last dose and measured SMN2 splicing by real-time RT-PCR (Fig. 6). As a control, we administered the 2′-MOE ASO, which, as expected, promoted exon 7 inclusion in all tissues examined; this was seen as an increase in transcripts that included exon 7 (Fig. 6a) and a concomitant decrease in transcripts that skipped exon 7 (Fig. 6b). The 2′-F ASO showed completely opposite effects on splicing, as we expected: a decrease in transcripts that included exon 7 (Fig. 6a) and an increase in transcripts that skipped exon 7 (Fig. 6b).
Figure 6. Modulation of splicing in mice as a function of ASO chemistry.

(a) Real-time RT-PCR analysis of SMN2 transcripts including exon 7 in the indicated tissues after treating mice transgenic for human SMN2 with ASOs. PBS, n = 2; 2′-MOE; n = 5; 2′-F, n = 5. For the 2′-MOE and 2′-F groups, the error bars represent the s.d. (b) Same as in a, except that the real-time RT-PCR analysis is for SMN2 transcripts excluding exon 7.
Discussion
Here we have described a new technology for modulating gene expression and demonstrated its underlying mechanism. An ASO can be engineered with specific chemical modifications to direct protein recruitment to RNA for the purpose of modulating alternative splicing in cell culture and in mice. Figure 7 illustrates the proposed mechanism for modulation of SMN2 exon 7 splicing. In the absence of ASOs, hnRNPs are recruited to the pre-mRNA (Fig. 7a). 2′-MOE–modified ASOs bind pre-mRNA and prevent the recruitment of hnRNPs (Fig. 7b). Without the repressive activity of hnRNPs24, SMN2 exon 7 inclusion ensues. 2′-F ASOs also prevent the recruitment of hnRNPs to the pre-mRNA (Fig. 7c). However, the 2′-F ASO–pre-mRNA duplex is recognized by ILF2/3, which causes the skipping of exon 7. A unique way of modulating splicing by using Ago2-loaded antisense RNA to cause exon skipping has also been developed42.
Figure 7. Scheme showing the proposed mechanism for ASO chemistry–dependent modulation of SMN2 exon 7 alternative splicing.
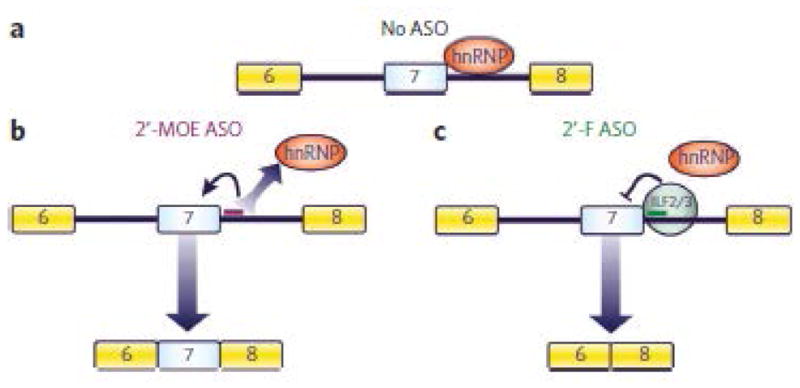
(a) In the absence of ASOs, hnRNPs are recruited to the pre-mRNA. Numbered boxes represent exons. (b) 2′-MOE ASOs bind pre-mRNA and prevent the recruitment of hnRNPs. Without the repressive activity of hnRNPs24, SMN2 exon 7 inclusion ensues. (c) 2′-F ASOs also prevent the recruitment of hnRNPs to the pre-mRNA. However, the 2′-F ASO–pre-mRNA duplex is recognized by ILF2/3, which causes the skipping of exon 7.
Several antisense-dependent strategies have been developed to recruit proteins to pre-mRNA for the purpose of modulating splicing. These strategies use bifunctional ASOs with RNA or peptide motifs appended12, 13, 14, 15. Our technology has several advantages over existing strategies. 2′-F ASOs are specific in that they direct the recruitment of ILF2/3 only after the ASO has hybridized to the pre-mRNA. In contrast, the bifunctional ASOs should be able to interact with their protein ligands even when they have not yet hybridized to pre-mRNA, thereby depleting them, which may increase the chances of unwanted side effects. The 2′-F ASO is an example of an ‘informational drug’ with the unique property of having the information necessary for protein recruitment encoded within its pharmacophore43. This allows for the production of a shorter ASO, with a smaller number of monomers required to synthesize it, which is a key factor in the ultimate cost.
Medicinal chemistry efforts have resulted in the creation of ASOs with more favorable ‘drug-like’ properties20. An important implication of our results is that when chemically modified ASOs are used, it is necessary to take into account the effect that chemistry may have on the recognition of the ASO-RNA duplex by proteins. This is important because it may alter the activity of the ASO for the intended purpose.
2′-F ASO–directed exon skipping may be used to treat diseases for which exon skipping is therapeutically desired4, 18, 44. ASOs uniformly modified with a variety of different chemistries have been used successfully to induce exon skipping18. To cause exon skipping, these ASOs have been carefully designed to target specific motifs within transcripts that are important for exon inclusion. In this sense, the 2′-F ASOs are more versatile because their exon-skipping ability is not restricted to a particular sequence (Fig. 1b and Supplementary Figs. 1 and 4). ILF2/3 is involved in several steps of RNA metabolism45, and these steps may be modulated by harnessing the 2′-F ASO–directed recruitment of ILF2/3. Moreover, our technology may also be useful for synthetic biology applications11, 46. For example, it may be possible to fuse the ILF3 RNA-binding domain to a protein of interest and then recruit the fusion protein to an RNA transcript in a 2′-F ASO–dependent manner. Finally, this study sets the stage for the discovery of other ASO modifications that maintain RNA-hybridization capacity and can recruit specific proteins to RNA for the purpose of modulating transcription, RNA processing, RNA stability, RNA localization or translation.
Methods
Cell transfection
For Figure 1b, HeLa cells in six-well plates were transfected with 30 nM ASO in Opti-MEM (Invitrogen) containing 1.5 μl ml−1 Cytofectin (Genlantis). For Figure 5, fibroblasts from patients with SMA (Coriel; GM03813) in 96-well plates were transfected with increasing concentrations of ASO as above. Four hours later, the transfection medium was replaced with complete medium consisting of DMEM (Invitrogen) supplemented with 10% (v/v) FBS (Invitrogen). Incubation proceeded for another 20 h. For Figure 1c, the transfection medium consisted of Opti-MEM with 1 μg of plasmid DNA, 30 nM ASO and 2.5 μl ml−1 Lipofectamine 2000 (Invitrogen). For Figure 3, HeLa cells in 96-well plates were transfected with increasing concentrations of siRNA in Opti-MEM containing 7.5 μl ml−1 Lipofectamine RNAiMAX (Invitrogen). Four hours later, the transfection medium was replaced with complete medium, as above. Incubation proceeded for 44 h, and then the cells were transfected with 30 nM ASO using Cytofectin, as above. Exposure of ASO to cells was for 24 h.
RT-PCR
For Figure 1b,c, RNA was isolated with an RNeasy Mini Kit (Qiagen). In-column DNA digestion was done with 50 U of DNase I (Invitrogen). With SuperScript II Reverse Transcriptase (Invitrogen), 500 ng of RNA was reverse-transcribed using oligo(dT) as the primer. PCR was performed with Platinum Taq DNA Polymerase (Invitrogen). For Figure 1b, amplification was for 30 cycles (94 °C for 30 s, 55 °C for 30 s and 72 °C for 36 s), and for Figure 1c, amplification was for 28 cycles (94 °C for 30 s, 60 °C for 30 s and 72 °C for 17 s). For Figure 1b, the PCR products were digested with DdeI. All PCR products were separated by agarose gel electrophoresis and stained with ethidium bromide. For Figures 3 and 5, RNA was isolated with an RNeasy 96 Kit (Qiagen). In-column DNA digestion was done with 30 U of DNase I (Invitrogen). Real-time RT-PCR was performed as described47. Total RNA was measured with Quant-iT RiboGreen RNA Reagent (Invitrogen). The ILF3 or SMN1 and SMN2 expression level was normalized to that of total RNA, and this was further normalized to the level in controls that had not been treated with siRNA, ASO or both. For Figure 6 a 3-mm3 piece of tissue was homogenized in a 2-ml tube containing Lysing Matrix D (MP Biomedicals), 500 μl RLT buffer (Qiagen) and 1% (v/v) β-mercaptoethanol. Homogenization was performed for 20 s at 6,000 r.p.m. using a FastPrep Automated Homogenizer (MP Biomedicals). Ten microliters of lysate were used to isolate RNA with the RNeasy 96 Kit (Qiagen), and real-time RT-PCR was performed as described above. The SMN2 expression level was normalized to that of the mouse gene Gapdh, and this was further normalized to the level in PBS-treated mice.
RNA pulldowns
ASOs were annealed to biotinylated RNA oligonucleotides by incubating 3 nmol of ASO with 1 nmol of RNA in 1× siRNA buffer (Thermo Scientific) and heating for 1 min at 90 °C. The solution was allowed to cool to 25 °C and then mixed with 100 μl of prewashed Dynabeads MyOne Streptavidin C1 (Invitrogen). Subsequent washing steps were done according to the manufacturer’s instructions. ASO-RNA bound to beads was then incubated with a 75-μl mixture containing 1.6 mM MgCl2, 20 mM creatine phosphate, 500 μM ATP, 10 mM HEPES-KOH (pH 8), 17% (v/v) Buffer D (20 mM HEPES-KOH, pH 8, 20% (v/v) glycerol, 0.1 M KCl, 0.2 mM EDTA, 1 mM DTT, 0.5 mM phenylmethanesulfonyl fluoride) and 33% (v/v) HeLa nuclear extract24. For lanes 5–7 in Figure 2a, 500 pmol of each ASO were added together with the nuclear extract. Mixtures were incubated with constant rotation for 40 min at 30 °C. Beads were isolated by magnetic selection and washed three times with Buffer D containing 300 mM KCl, except those shown in Figure 2a, for which 150 mM KCl was used instead. For lanes 1–7 in Figure 2a, lanes 1 and 3 in Figure 2b and Figure 4, bound proteins were eluted by resuspending the beads in 50 μl of Laemmli sample buffer (Bio-Rad) containing 5% (v/v) β-mercaptoethanol and were heated for 2 min at 85 °C. Forty percent of each sample was loaded on an SDS polyacrylamide gel. For Figure 2a (lanes 8 and 9), after the three washes the bead-bound RNA was incubated with 500 pmol of ASO, 1.6 mM MgCl2, 10 mM HEPES-KOH (pH 8) and 17% (v/v) Buffer D for another 40 min at 30 °C. After washing the beads once with Buffer D (150 mM KCl), the proteins were eluted and loaded on an SDS polyacrylamide gel as above. For Figure 2b, after washing with buffer D (300 mM KCl), the beads were incubated with a 50-μl mixture containing 1,000 U RNase I (Ambion) and 10 mM Tris-HCl (pH 8) for 40 min at 30 °C with constant rotation. After proteins were eluted from the beads as before, 20% of the released fraction and 10% of the bound fraction were loaded on an SDS polyacrylamide gel. The gel was then stained with SYPRO Ruby Protein Gel Stain (Invitrogen) according to the manufacturer’s instructions. Ten percent of the released fraction was used for western blotting. The list of antibodies used is in Supplementary Table 5.
Dosing of mice
All protocols met ethical standards for animal experimentation and were approved by the Institutional Animal Care and Use Committee (IACUC). Adult SMA type III mice (Smn1−/−; SMN2+/+) were obtained from Jackson Laboratory (FVB.Cg-Tg(SMN2)2HungSMN1tm1Hung/J, stock number 005058). ASOs, dissolved in PBS, were administered by intraperitoneal injection at doses of 50 mg per kg body weight every 2 d for a total of 4 doses. There were five mice per group, and tissues were harvested 48 h after the last dose.
Additional methods
Oligonucleotides, constructs, melting temperature (Tm) measurements, MS, formation of 32P-labeled ASO-RNA duplexes and antibody pulldowns are described in the Supplementary Methods.
Supplementary Material
References
- 1.Glisovic T, Bachorik JL, Yong J, Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008;582:1977–1986. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Licatalosi DD, Darnell RB. RNA processing and its regulation: global insights into biological networks. Nat Rev Genet. 2010;11:75–87. doi: 10.1038/nrg2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calarco JA, Zhen M, Blencowe BJ. Networking in a global world: establishing functional connections between neural splicing regulators and their target transcripts. RNA. 2011;17:775–791. doi: 10.1261/rna.2603911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol. 2007;8:479–490. doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Auweter SD, Oberstrass FC, Allain FH. Sequence-specific binding of single-stranded RNA: is there a code for recognition? Nucleic Acids Res. 2006;34:4943–4959. doi: 10.1093/nar/gkl620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tian B, Bevilacqua PC, Diegelman-Parente A, Mathews MB. The double-stranded-RNA-binding motif: interference and much more. Nat Rev Mol Cell Biol. 2004;5:1013–1023. doi: 10.1038/nrm1528. [DOI] [PubMed] [Google Scholar]
- 8.Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Cheong CG, Hall TM, Wang Z. Engineering splicing factors with designed specificities. Nat Methods. 2009;6:825–830. doi: 10.1038/nmeth.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mackay JP, Font J, Segal DJ. The prospects for designer single-stranded RNA-binding proteins. Nat Struct Mol Biol. 2011;18:256–261. doi: 10.1038/nsmb.2005. [DOI] [PubMed] [Google Scholar]
- 11.Culler SJ, Hoff KG, Smolke CD. Reprogramming cellular behavior with RNA controllers responsive to endogenous proteins. Science. 2010;330:1251–1255. doi: 10.1126/science.1192128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baughan TD, Dickson A, Osman EY, Lorson CL. Delivery of bifunctional RNAs that target an intronic repressor and increase SMN levels in an animal model of spinal muscular atrophy. Hum Mol Genet. 2009;18:1600–1611. doi: 10.1093/hmg/ddp076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cartegni L, Krainer AR. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat Struct Biol. 2003;10:120–125. doi: 10.1038/nsb887. [DOI] [PubMed] [Google Scholar]
- 14.Skordis LA, Dunckley MG, Yue B, Eperon IC, Muntoni F. Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc Natl Acad Sci USA. 2003;100:4114–4119. doi: 10.1073/pnas.0633863100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villemaire J, Dion I, Elela SA, Chabot B. Reprogramming alternative pre-messenger RNA splicing through the use of protein-binding antisense oligonucleotides. J Biol Chem. 2003;278:50031–50039. doi: 10.1074/jbc.M308897200. [DOI] [PubMed] [Google Scholar]
- 16.Goraczniak R, Behlke MA, Gunderson SI. Gene silencing by synthetic U1 adaptors. Nat Biotechnol. 2009;27:257–263. doi: 10.1038/nbt.1525. [DOI] [PubMed] [Google Scholar]
- 17.Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 18.Bauman J, Jearawiriyapaisarn N, Kole R. Therapeutic potential of splice-switching oligonucleotides. Oligonucleotides. 2009;19:1–13. doi: 10.1089/oli.2008.0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennett CF, et al. Antisense oligonucleotide-based therapeutics. In: Templeton NS, editor. Gene and Cell Therapy: Therapeutic Mechanisms and Strategies. Taylor & Francis Group; 2009. pp. 497–522. [Google Scholar]
- 20.Swayze EE, Balkrishen B. The medicinal chemistry of oligonucleotides. In: Crooke ST, editor. Antisense Drug Technology: Principles, Strategies and Applications. Taylor & Francis Group; 2008. pp. 143–182. [Google Scholar]
- 21.Seth PP, et al. Synthesis and biophysical evaluation of 2′,4′-constrained 2′O-methoxyethyl and 2′,4′-constrained 2′O-ethyl nucleic acid analogues. J Org Chem. 2010;75:1569–1581. doi: 10.1021/jo902560f. [DOI] [PubMed] [Google Scholar]
- 22.Levin AA, Yu RZ, Geary RS. Basic principles of the pharmacokinetics of antisense oligonucleotide drugs. In: Crooke ST, editor. Antisense Drug Technology: Principles, Strategies and Applications. Taylor & Francis Group; 2008. pp. 183–215. [Google Scholar]
- 23.Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009;10:741–754. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82:834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pandya-Jones A, Black DL. Co-transcriptional splicing of constitutive and alternative exons. RNA. 2009;15:1896–1908. doi: 10.1261/rna.1714509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noguchi T, Inoue H, Tanaka T. The M1- and M2-type isozymes of rat pyruvate kinase are produced from the same gene by alternative RNA splicing. J Biol Chem. 1986;261:13807–13812. [PubMed] [Google Scholar]
- 28.Christofk HR, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 29.Boise LH, et al. bcl-x, a bcl-2–related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 30.Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 1989;8:393–399. doi: 10.1002/j.1460-2075.1989.tb03390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rademakers R, Cruts M, van Broeckhoven C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum Mutat. 2004;24:277–295. doi: 10.1002/humu.20086. [DOI] [PubMed] [Google Scholar]
- 32.Singh NK, Singh NN, Androphy EJ, Singh RN. Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol. 2006;26:1333–1346. doi: 10.1128/MCB.26.4.1333-1346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guan D, et al. Nuclear factor 45 (NF45) is a regulatory subunit of complexes with NF90/110 involved in mitotic control. Mol Cell Biol. 2008;28:4629–4641. doi: 10.1128/MCB.00120-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liao HJ, Kobayashi R, Mathews MB. Activities of adenovirus virus-associated RNAs: purification and characterization of RNA binding proteins. Proc Natl Acad Sci USA. 1998;95:8514–8519. doi: 10.1073/pnas.95.15.8514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawasaki AM, et al. Uniformly modified 2′-deoxy-2′-fluoro phosphorothioate oligonucleotides as nuclease-resistant antisense compounds with high affinity and specificity for RNA targets. J Med Chem. 1993;36:831–841. doi: 10.1021/jm00059a007. [DOI] [PubMed] [Google Scholar]
- 36.Teplova M, et al. Crystal structure and improved antisense properties of 2′-O-(2-methoxyethyl)-RNA. Nat Struct Biol. 1999;6:535–539. doi: 10.1038/9304. [DOI] [PubMed] [Google Scholar]
- 37.Li F, et al. 2′-Fluoroarabino- and arabinonucleic acid show different conformations, resulting in deviating RNA affinities and processing of their heteroduplexes with RNA by RNase H. Biochemistry. 2006;45:4141–4152. doi: 10.1021/bi052322r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inoue H, et al. Synthesis and hybridization studies on two complementary nona(2′-O-methyl)ribonucleotides. Nucleic Acids Res. 1987;15:6131–6148. doi: 10.1093/nar/15.15.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilds CJ, Damha MJ. 2′-Deoxy-2′-fluoro-β-D-arabinonucleosides and oligonucleotides (2′F-ANA): synthesis and physicochemical studies. Nucleic Acids Res. 2000;28:3625–3635. doi: 10.1093/nar/28.18.3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vester B, Wengel J. LNA (locked nucleic acid): high-affinity targeting of complementary RNA and DNA. Biochemistry. 2004;43:13233–13241. doi: 10.1021/bi0485732. [DOI] [PubMed] [Google Scholar]
- 41.Jepsen JS, Sorensen MD, Wengel J. Locked nucleic acid: a potent nucleic acid analog in therapeutics and biotechnology. Oligonucleotides. 2004;14:130–146. doi: 10.1089/1545457041526317. [DOI] [PubMed] [Google Scholar]
- 42.Liu J, Hu J, Corey DR. Expanding the action of duplex RNAs into the nucleus: redirecting alternative splicing. Nucleic Acids Res. 2011;40:1240–1250. doi: 10.1093/nar/gkr780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cohen JS. Informational drugs: a new concept in pharmacology. Antisense Res Dev. 1991;1:191–193. doi: 10.1089/ard.1991.1.191. [DOI] [PubMed] [Google Scholar]
- 44.Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007;8:749–761. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- 45.Barber GN. The NFAR’s (nuclear factors associated with dsRNA): evolutionarily conserved members of the dsRNA binding protein family. RNA Biol. 2009;6:35–39. doi: 10.4161/rna.6.1.7565. [DOI] [PubMed] [Google Scholar]
- 46.Win MN, Liang JC, Smolke CD. Frameworks for programming biological function through RNA parts and devices. Chem Biol. 2009;16:298–310. doi: 10.1016/j.chembiol.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hua Y, et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010;24:1634–1644. doi: 10.1101/gad.1941310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.