

Abstract
DesII is a member of the radical SAM family of enzymes that catalyzes radical-mediated transformations of TDP-4-amino-4,6-didexoy-D-glucose as well as other sugar nucleotide diphosphates. Like nearly all radical SAM enzymes, the reactions begin with the reductive homolysis of SAM to produce a 5′-deoxyadenosyl radical which is followed by regiospecific hydrogen atom abstraction from the substrate. What happens next, however, depends on the nature of the substrate radical so produced. In the case of the biosynthetically relevant substrate, a radical-mediated deamination ensues; however, when this amino group is replaced with a hydroxyl, one instead observes dehydrogenation. The factors that govern the fate of the initially generated substrate radical as well as the mechanistic details underlying these transformations have been a key focus of research into the chemistry of DesII. This review will discuss recent discoveries pertaining to the enzymology of DesII, how it may relate to understanding other radical-mediated lyases and dehydrogenases and the working hypotheses currently being investigated regarding the mechanism of DesII catalysis.
Keywords: Enzyme Catalysis, Radical Reactions, Deamination, Dehydrogenation, Carbohydrates
1. Introduction
DesII is a S-adenosyl-L-methionine (SAM, 6)-dependent lyase that catalyzes the radical-mediated deamination of TDP-4-amino-4,6-dideoxy-D-glucose to TDP-4,6-dideoxy-3-keto-D-glucose (1→2, see Fig. 1A).1,2 This transformation represents the key step in the biosynthesis of TDP-desosamine (3) in Streptomyces venezuelae, which is a required component for the biological construction of various macrolide antibiotics such as erythromycin and pikromycin.3,4 DesII catalysis involves the reductive homolysis of SAM (6) via electron transfer from an active site [4Fe-4S]1+ cluster to form L-methionine (8) and a 5′-deoxyadenosyl radical (7), the latter of which is responsible for initiating the radical-mediated elimination reaction as shown in Fig. 1B.1,2 This mechanism of 5′-deoxyadenosyl radical generation places DesII among the rapidly expanding class of radical SAM enzymes.5,6,7
Figure 1.

A, Reactions catalyzed by DesII. B, The reductive homolysis of S-adenosyl-L-methionine (6, Ade: adenine) by an active site [4Fe-4S]1+ cluster is the principle step that characterizes nearly all radical SAM enzymes currently under study including DesII.6,7,12 This results in the formation of L-methionine (Met, 8) and a 5′-deoxyadenosyl radical (7). The latter can then generate additional radical intermediates such as 21/28 in the case of DesII via H-atom abstraction.
One aspect of radical SAM enzymology that is of particular interest is its mechanistic diversity. Whereas almost all radical SAM catalyzed reactions characteristically begin with the reductive homolysis of SAM to form 7 (Dph2 and B12-independent glycerol dehydrogenase being notable exceptions8 with their own peculiarities), what happens next to actually effect the radical-mediated transformation of substrate to product is frequently found to be highly case-specific.7 This is in contrast to most other enzyme classes where the catalytic principles understood from one enzyme (e.g., an NAD-dependent dehydrogenase or PLP-dependent enzyme) are often directly applicable to understanding the mechanism of a separate, related enzyme. The radical SAM enzymes as a whole can thus be considered an ideal system for studying how enzymes utilize radicals to catalyze diverse chemical transformations.
DesII is a system of particular interest due to its dual functionality. In addition to the biosynthetically relevant deamination of TDP-4-amino-4,6-dideoxy-D-glucose (1→2), DesII also catalyzes the dehydrogenation of TDP-D-quinovose to form TDP-6-deoxy-3-keto-D-glucose (4→5).2 The only difference between the substrates 1 and 4 is whether there is an amino or hydroxyl group at the C4 position. Furthermore, both of these reactions begin in essentially the same way with C3-hydrogen atom abstraction from the TDP-sugar substrate by the 5′-deoxyadenosyl radical (see Fig. 1B).2,9 This has led to a number of questions. For example, why is 1 not also oxidized (the C3 position is oxidized to a carbonyl, but the C4 position is simultaneously reduced to a methylene)? Likewise, the precedence set by adenosylcobalamin-dependent diol dehydratase (see Fig. 2B)10 and nonenzymatic systems involving α,β-dihydroxyalkyl radicals11 indicates that dehydration of TDP-D-quinovose (4) should not only be possible but even rapid. So, what keeps this from happening not only with respect to 4 but also the C2 hydroxyl group during deamination? These questions in addition to simply trying to understand how the various transformations proceed have been a driving principle in the study of DesII as a model of radical enzymology in general.
Figure 2.

Examples of radical-mediated lyase and dehydrogenation reactions. A, Dehydrogenation reactions catalyzed by the radical SAM enzyme BtrN and the anaerobic sulfatase maturating enzymes (anSMEs). B, Adenosylcobalamin-dependent lyase reactions catalyzed by ethanolamine ammonia lyase (EAL) and diol dehydratase.
A number of comprehensive and detailed reviews on the radical SAM class of enzymes are already available.6,7,12 In particular, the recent review by Broderick and coworkers7 provides a detailed discussion of not only the radical SAM enzymes as a group but also the current working models for generation of the 5′-deoxyadenosyl radical. Likewise, an extensive review on DesII together with BtrN was published in 2012 and detailed the biosynthetic role of DesII, its place among other radical SAM enzymes and our understanding of its catalytic cycle at that time.13 Therefore, readers interested in these aspects of radical SAM catalysis and DesII enzymology are directed to these reviews. The present discussion will focus on current models of DesII catalysis that attempt to explain how the fate of the substrate radical is directed once generated.
2. Deamination vs. Dehydrogenation
The DesII deamination reaction is redox-neutral in so far as there is no net flux of reducing equivalents either into or out of the sugar ring during turnover. For this reason, DesII should in principle be able to regenerate SAM with each turnover as has been reported for other redox-neutral radical SAM transformations such as those catalyzed by lysine aminomutase (LAM),14–16 sporephotoproduct lyase17,18 and QueE19. However, this is not the case with DesII. Instead, SAM undergoes a net reduction in vitro by an external source of reducing equivalents such as sodium dithionite or the more biologically relevant NADPH / flavodoxin / flavodoxin reductase coupled reducing system.20 Furthermore, the stoichiometry of SAM reduction versus deamination of 1 is 1:1 irrespective of the reducing system.20 This suggests that the reduction of SAM is not a consequence of uncoupling, whereby the 5′-deoxyadenosyl radical (7) is formed but is reduced to 5′-deoxyadenosine (9) without concomitant conversion of the substrate to product. It remains unclear whether the net reduction of SAM during the DesII-catalyzed deamination is indeed an inherent property of catalysis or is merely an artifact of the in vitro system studied so far.
In contrast to the deamination of 1, dehydrogenation of 4 results in the net oxidation of the TDP-sugar substrate. SAM is again consumed in a 1:1 ratio with TDP-D-quinovose. Notably, multiple dehydrogenation turnovers are still possible when the external source of reducing equivalents (i.e., Na2S2O4) is allowed to decompose leaving only the reduced enzyme prior to substrate addition.20 This is consistent with a model where SAM, as opposed to an external electron acceptor, is acting as the formal two-electron oxidant. The dehydrogenation of TDP-D-quinovose by DesII is similar in overall appearance to the dehydrogenation of 2-deoxy-scyllo-inosamine to amino-dideoxy-scyllo-inosose (10→11, see Fig. 2A) catalyzed by BtrN during the biosynthesis of deoxystreptamine in Bacillus circulans.21 Both enzymes also appear to exhibit ordered sequential kinetic mechanisms (SAM binds first) with similar steady state kinetic parameters.21,22 In this sense, DesII can be considered a well-controlled and efficient radical-mediated dehydrogenase even though it acts as a lyase in its biosynthetic context.
3. The Substrate Radical
Significant insight into the mechanism of DesII catalysis was obtained when a radical intermediate was freeze-trapped during steady state dehydrogenation of TDP-D-quinovose (4) and characterized by electron paramagnetic resonance (EPR) spectroscopy.9 The X-band EPR spectrum of this intermediate exhibits an isotropic doublet of doublets splitting pattern centered at g = 2.003 with hyperfine splitting constants of 33.6 G for both doublets. This implied a C3-centered substrate radical (21) that is equally coupled to the H-atom nuclei at both C2 and C4. Furthermore, when TDP-D-[4-2H]quinovose was employed in the experiment, the spectrum collapsed to a broad doublet that was best simulated when one of the two 33.6 G doublet couplings is replaced with a 5.2 G triplet splitting.9 This 6.5-fold reduction in splitting constant is consistent with the ratio of magnetogyric ratios for protium versus deuterium (gH/gD) and further substantiated the assignment of a C3-centered radical.
These results provided direct evidence that the dehydrogenation reaction begins with H-atom abstraction from the C3-position of TDP-D-quinovose (see 20→21, Fig. 4). While it has not yet been possible to trap an EPR active species during the deamination reaction, previous experiments showed that deuterium is transferred from TDP-4-amino-4,6-dideoxy-D-[3-2H]glucose to 5′-deoxyadenosine.2 Based on these observations, the deamination reaction also likely commences with the formation of an analogous C3-centered substrate radical (see 27→28), albeit one with a much reduced half-life. The deuterium tracer experiment also supported the 5′-deoxyadenosyl radical itself as the agent responsible for the H-atom abstraction event rather than an intermediary glycyl or cysteinyl protein radical generated by the initially formed 5′-deoxyadenosyl radical.7
Figure 4.

Working model for the dehydrogenation of TDP-D-quinovose (4) by DesII. The substrate radical 21 assumes a configuration that does not allow optimal hyperconjugation between the partially filled p-orbital at C3 and C–OH bonds at either C2 or C4.9 This impedes elimination of the hydroxyl groups such that the dehydration products 24 and 25 are not observed. Transformations are depicted with only single arrows for simplicity; however, steps may in fact be freely reversible.
The EPR characterization of the dehydrogenation substrate radical provided two additional and mechanistically important pieces of information. The first is that the linewidth of the EPR spectra using both natural abundance and C4-deuterated TDP-D-quinovose decreased from 7.2 G in H2O to 5.0 G in D2O.9 This isotope effect on the linewidth of the radical arises from a change in inhomogeneous broadening23 due to unresolved hyperfine coupling between the unpaired electron at C3 and a solvent exchangeable hydron. When the solvent protons are exchanged for deuterons, this hyperfine coupling is reduced due to the smaller magnetogyric ratio of deuterium. This is observed as a reduction in the inhomogeneous broadening and sharper EPR lineshapes. Based on this reasoning, the EPR-active species was assigned as a C3 α-hydroxyalkyl radical (•C–OH) rather than a ketyl radical (•C–O−) indicating that deprotonation of the C3 hydroxyl is not required for H-atom abstraction (20→21). Had the C3 hydroxyl been deprotonated in the EPR active species (i.e., a ketyl radical), then a change in the linewideth would not have been expected.9 It is nevertheless still arguable that the isotope effect is due to more long range hyperfine couplings; however, subsequent measurements of solvent deuterium kinetic isotope effects further supported the assignment of an α-hydroxyalkyl radical and are discussed below.22
The other key structural insight available from the EPR results is the stereochemical configuration of the substrate radical 21. Using the Heller-McConnell equation applied to the magnitude of the observed coupling constants,24 it is possible to estimate the dihedral angle between the p-orbital harboring the unpaired electron at C3 and the adjacent C–H bonds at C2 and C4.9 This dihedral angle was determined to be approximately 15° in both cases indicating a more clinal configuration of the corresponding C–OH bonds (see Fig. 3). Therefore, the C–OH σ-bonds at C2 and C4 of 21 are more orthogonally oriented with respect to the p-orbital at C3 than would be expected for a radical-mediated elimination reaction.
Figure 3.
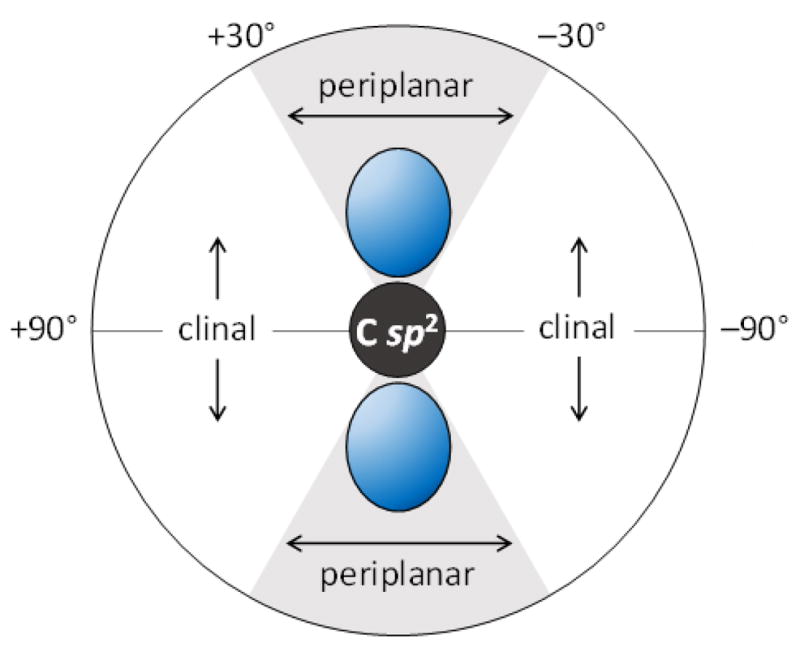
Periplanar and clinal regions of the dihedral angle between the partially occupied p-orbital (blue) at the sp2-hybridized radical center and an adjacent sp3-hybridized carbon. In the radical lyases EAL and diol dehydratase26,27 characterized by EPR, the adjacent C–OH/NH3+ bond lies in the grey, periplanar region. In the case of the radical dehydrogenases DesII and BtrN,9,25 the adjacent C–OH bonds lie in the white, clinal regions.
Indeed, substrate radical intermediates have also been trapped and characterized by EPR in the cases of BtrN,25 ethanolamine ammonia lyase (EAL)26 and diol dehydratase27. Like DesII, BtrN is also a radical SAM dehydrogenase involving a cyclitol substrate (10) with a β-hydroxy functionality adjacent to the carbon that is oxidized.21 The orientation between the partially filled p-orbital and the single adjacent C–OH bond was also observed to be clinal in the case of BtrN. In contrast, EAL and diol dehydratase are radical-mediated lyases that employ adenosylcobalamin rather than radical SAM chemistry to generate the 5′-deoxyadenosyl radical (Fig. 1B). In both cases, a periplanar configuration has been observed between the p-orbital at the radical center and the adjacent amino or hydroxyl functionality to be eliminated.
While the substrate radical intermediate generated during the DesII-catalyzed deamination reaction has yet to be completely characterized, the EPR results for the dehydrogenation reaction together with the observations reported for other radical-mediated dehydrogenases and lyases have offered a model to account for how the radical intermediate is controlled during DesII catalysis.13 This model proposes that the substrate radical of TDP-4-amino-4,6-dideoxy-D-glucose (see 28, Fig. 6) assumes a different configuration within the DesII active site compared to the substrate radical of TDP-D-quinovose (21, Fig. 4). Such a configuration would reduce the dihedral angle between the C–NH3+ σ-system and the p-orbital at C3, resulting in improved hyperconjugation and facilitating either an elimination or migration reaction (see below). Lyase activity is thus observed as a consequence. In contrast, if hyperconjugation is insufficient due to poor overlap, then elimination is impeded to the extent that an oxidative side reaction can take place instead. This would result in the observation of dehydrogenase activity. This model of radical control forms the basis of the more detailed discussions of the dehydrogenation and deamination mechanisms that follow.
Figure 6.

Working model for the deamination of TDP-4-amino-4,6-dideoxy-D-glucose (1) by DesII. In contrast to TDP-D-quinovose (4, see Fig. 4), 1 is proposed to bind within the DesII active site in a configuration that optimizes the overlap between the partially filled p-orbital at C3 and the C–NH3+ bond at C4, but not the C–OH bond at C2 (see 28). The substrate radical is thus primed for elimination via either direct elimination or migration, and neither dehydrogenation nor dehydration take place to generate ei ther 33 or 34, respectively. Transformations are depicted with only single arrows for simplicity; however, steps may in fact be freely reversible.
4. Mechanism of Dehydrogenation
4.1. Movement of Reducing Equivalents
Dehydrogenation of TDP-D-quinovose (4) requires the net transfer of two reducing equivalents from the substrate to SAM. This transfer, however, is not direct and likely involves three separate single electron reduction reactions (see Fig. 4). The first of these is the electron transfer event between the [4Fe-4S]1+ cluster and SAM (19→20). The result is the formation of 5′-deoxyadenosyl radical 7 and an oxidized [4Fe-4S]2+ cluster. The second redox reaction, which may happen concerted with the first such that a discrete 5′-deoxyadenosyl radical never actually forms, is the H-atom abstraction from C3 of TDP-D-quinovose resulting in the C3-centered α-hydroxyalkyl radical observed by EPR (20→21).9 The H-atom abstraction is equivalent to a single electron transfer event and accomplishes the reduction of SAM to L-methionine and 5′-deoxyadenosine. In order to complete the dehydrogenation of TDP-D-quinovose, a second one-electron oxidation of the substrate radical must subsequently take place. The ability of the dehydrogenation reaction to proceed for multiple turnovers in the absence of an external reducing agent20 implies that the substrate radical itself is responsible for the reduction of the [4Fe-4S]2+ cluster back to the [4Fe-4S]1+ redox state (21→22→23). This regenerates the active form of the enzyme and concludes the catalytic cycle.
This mechanism of DesII-catalyzed dehydrogenation is at least aesthetically appealing, because it minimizes the required number of electron transfers and would permit the enzyme to function as a dehydrogenase in the absence of an external redox system. In other words, once in the activated [4Fe-4S]1+ state, the enzyme would remain an active dehydrogenase utilizing SAM as an oxidant just as an NAD(P)-dependent dehydrogenase would utilize NAD(P). Therefore, one might expect that the radical SAM enzymes which have evolved to catalyze dehydrogenation reactions would follow a similar mechanistic paradigm. It is therefore of some interest that mounting evidence continues to suggest that this expectation may actually be misguided.
The mechanistic difference between DesII and other radical SAM dehydrogenases can first be appreciated by comparing the number of [4Fe-4S] clusters present per protein monomer. The catalytic iron-sulfur cluster of DesII is coordinated by the three residues: Cys141, Cys145 and Cys148 in Streptomyces venezuelae.3 These three cysteine residues constitute the CxxxCxxC motif that is common to most known radical SAM enzymes and is responsible for coordinating the catalytic [4Fe-4S] cluster (see Fig. 1B).5,28,29 DesII has only one additional cysteine residue (Cys15 in Streptmyces venezuelae3) making the presence of a second cluster highly unlikely, and iron-sulfur titrations of the reconstituted enzyme are consistent with this conclusion.2
In contrast, the radical SAM dehydrogenases BtrN and the anaerobic sulfatase maturating enzymes (anSMEs) have all been shown to possess multiple iron-sulfur clusters. The primary sequence of BtrN from Bacillus circulans contains eight cysteine residues, though only three of these make up the characteristic CxxxCxxC motif.30 Subsequent studies including X-ray crystallography confirmed that four of the remaining cysteine residues are involved in the coordination of a second, auxiliary [4Fe-4S] cluster.31,32 The anSMEs activate sulfatase zymogens by catalyzing the dehydrogenation of cysteine or serine residues to form the catalytic Cα-formylglycine residues (e.g., 12→13 & 14→13 in Fig. 2A).33–35 These radical SAM enzymes such as AtsB,36,37 anSMEcpe38–40 and anSMEbt41 contain between 13 and 18 cysteine residues and have been shown to coordinate three [4Fe-4S] clusters each. Furthermore, these enzymes exhibit all or part of a common domain architecture that has been named the SPASM motif and is responsible for coordinating the auxiliary clusters.31,40,42 This subfamily has already turned out to be quite large and growing,40 though not all of these enzymes are dehydrogenases.
Before structural data became available, these observations suggested a provocative hypothesis regarding the catalytic mechanism of BtrN and the anSMEs. It was proposed that one of the auxiliary [4Fe-4S] clusters is also site differentiated thereby allowing the hydroxyl (or sulfhydral) moiety undergoing oxidation to coordinate one of the iron centers.32 Prior to H-atom abstraction, the auxiliary cluster serves as a Lewis acid in this model to promote deprotonation of the hydroxyl rendering it a better reductant. This in turn facilitates the initial H-atom abstraction to produce the substrate radical. Following H-atom abstraction, the auxiliary cluster could then serve as an electron sink to catch the second reducing equivalent via an inner-sphere electron transfer. This results in the complete oxidation of the hydroxyl group to a carbonyl and is reminiscent of the working model for galactose oxidase, which utilizes a CuII coordinated 3′-(S-cysteinyl)tyrosyl radical to dehydrogenate the C6 hydroxyl of galactose.43,44 A mechanism would then be needed to facilitate net electron transfer back to the primary [4Fe-4S] cluster and complete the catalytic cycle.
Later structural studies of BtrN31 and anSMEcpe,40 however, revealed that the auxiliary clusters are neither site-differentiated nor situated close enough to the bound substrate to permit direct coordination. In fact, in both cases the carbon undergoing oxidation is roughly equidistant (ca. 9 Å) between the catalytic and nearest auxiliary cluster (see Fig. 5).31,40 It is therefore suggested that the auxiliary clusters effectively constitute a conduit by which the second reducing equivalent is transferred from the substrate radical to an external oxidant at the protein surface.31,40 Direct biochemical evidence supporting this hypothesis has been provided in the cases of BtrN32 and anSMEcpe39 in so far as the auxiliary clusters are required for full activity and anSMEcpe is able to remain largely oxidized during steady state turnover in the presence of flavodoxin. Therefore, the additional clusters do appear to be direct participants in catalysis rather than vestigial remnants common to all the SPASM-type enzymes.
Figure 5.

Crystal structure31 of the B. circulans BtrN active site emphasizing the relative positioning of the substrate 2-deoxy-scyllo-inosamine (10, DOIA) versus the auxiliary and catalytic [4Fe-4S] clusters (PDB ID code 4M7T). The carbon of the substrate that is oxidized as well as the 5′-carbon of SAM are highlighted in dark grey. The guanidinium η-nitrogens of Arg152 (blue) are within 4 Å of the C3-hydroxyl of the substrate, and the residue may serve as a catalytic base.31 Phe188 (green) is situated between the substrate and the auxiliary cluster. The four and three cysteines that coordinate the auxiliary and catalytic clusters, respectively, are also illustrated.
While the mechanisms proposed for BtrN and the anSMEs might seem to be unnecessarily complicated in light of the working model for DesII dehydrogenation, they may actually be more efficient especially in vivo where the naturally selected oxidation-reduction system would operate. Under these conditions, the catalytic cluster of radical SAM enzymes may be significantly more susceptible to reduction by the physiological redox system. If this were to happen such that the [4Fe-4S]2+ cluster were reduced back to the [4Fe-4S]1+ state before the substrate radical is oxidized, then the latter would essentially be trapped.13 The auxiliary clusters may thus serve as a “backdoor” to let the second electron out from the active site in the case of such a scenario.
The apparent differences between DesII, BtrN and the anSMEs highlight the mechanistic diversity of the radical SAM enzymes. Although these enzymes all use radical chemistry initiated by the reductive homolysis of SAM, the substrate radical intermediates generated in each case may be manipulated in different ways to effect the same overall transformation. Therefore, it remains an interesting research topic as to how and why the multiple auxiliary clusters are necessary for catalysis among the biosynthetic radical SAM dehydrogenases.
4.2. Oxidation of the Substrate Radical
The defining step of the dehydrogenation reaction is the one-electron oxidation of the substrate radical. It has been argued that the rate of intraprotein electron tunneling is largely dependent on the distance between the donor and acceptor sites with exergonic electron transfer taking place rapidly over distances of 14 Å or less.45,46 Therefore, the roughly 9 Å distances observed between the substrate radicals and [4Fe-4S] clusters in the structures of BtrN and anSMEcpe are well within the range necessary to facilitate the oxidation.31,40 A similar distance likely also exists between the substrate radical and the catalytic [4Fe-4S] cluster in the DesII active site. However, a facile outersphere electron transfer would not be desirable in the case of DesII, because it may poison the biologically important deamination reaction and yield 33 instead of 2 as the product. Hence, the oxidation of the substrate radical in the DesII active site is likely slowed in some manner, and a mechanism is in place to accelerate electron transfer only during the dehydrogenation reaction.
As opposed to exergonic electron transfer between redox centers, endergonic transfer can be significantly slower, all else being equal.45 The reduction potential of the catalytic [4Fe-4S]2+ cluster has been measured in the case of LAM and found to have a value of approximately −0.5 V, which is sensitive to the coordination environment.47,48 In contrast, free thiolate-coordinated [4Fe-4S]2+ clusters exhibit reduction potentials of about −1 V in aprotic solvents,49 further demonstrating the variability in these potentials. This is to be compared with the oxidation potential of an α-hydroxyalkyl radical, which is approximately +0.9 V.50 Therefore, slow, endergonic oxidation of the substrate radical by the catalytic cluster cannot be entirely ruled out. Likewise, if the pathway between the redox centers is poorly optimized for electron transfer, then the combined effect could help to prevent an oxidative side reaction during deamination.
If correct, one way to increase the rate of electron transfer during the dehydrogenation reaction is to convert the substrate radical into a stronger reductant. The α-hydroxyalkyl radicals are typically 105-fold more acidic than their reduced counterparts (i.e., primary and secondary alcohols),51 such that deprotonation of the TDP-D-quinovose substrate radical (21) by an active site base is within the capability of some protein residues. The result would be the production of a ketyl radical (22) having an oxidation potential of approximately +1.5 V51 and thus sufficient reducing power to accelerate outersphere electron transfer to the catalytic [4Fe-4S]2+ cluster (22→23).
Two observations made previously support the hypothesis that deprotonation of the α-hydroxyalkyl substrate radical is necessary for dehydrogenation to proceed. The first is the pH-dependence of steady state parameters for dehydrogenation.22 A decrease in V/K is seen under both acidic and basic conditions, though the latter loss in activity is believed to be due to titration of the N3 nitrogen of the TDP moiety in the free substrate. In contrast, V plateaus at alkaline pH with a pKaV of approximately 9, consistent with the presence of an active site general base. The second observation is the measurement of a significant solvent deuterium isotope effect on the net rate constant for decomposition of the substrate radical observed by EPR.22 This latter finding represents a key piece of evidence supporting the general base hypothesis; however, a brief review of the theory of net rate constants first described by W.W. Cleland in 1975 may be worthwhile before discussing the isotope effect on the net rate constant in detail.52
The assumption of steady state kinetics implies that the net concentration of each intermediary species can be treated as a constant for small changes in substrate concentration. If we consider an arbitrary serial reaction with four intermediates, then we can write
| (1) |
where Ei is the ith intermediate or enzyme form, S is the free substrate and P is the product with concentrations ei, s and p, respectively. Cleland’s insight52 was to map such a reaction to a kinetically more tractable system which exhibits the same steady state intermediate concentrations but is composed of only irreversible steps, i.e.,
| (2) |
In order to maintain the steady state for this hypothetical irreversible system, the flux through each step must equal the overall steady state rate, v. In other words, the rate equation
| (3) |
must be true for each intermediate. a In this formulation, the conceptualized rate constants k′i are referred to as net rate constants.
In these expressions, it is important to recognize that the steady state concentration of a given intermediate (i.e., ei in Eqn. (3)) depends on the steady state rate v. It will thus take on different values depending on the degree to which the enzyme is saturated with substrate. In contrast, the net rate constants are fixed and determined entirely by the microscopic rate constants describing the overall system (1). It can be shown52,53 that this later relationship can be formulated using a simple rule of thumb: the net rate constant for a given intermediate is equal to the forward rate constant for that intermediate modulated by the probability that the subsequent intermediate advances on completely to product versus returning via the reverse reaction. For example, this probability will be equal to k′3 / (k′3 + k32) for intermediate E3 such that the net rate constant for E2 will be given by
| (4) |
Note that if k32 = 0, such that E3 must advance entirely to product, then we would have k′2 = k23. In other words, if a step is indeed irreversible, then the net rate constant is simply equal to the true microscopic forward rate constant. This permits the inductive formulation of the net rate constant for any intermediate upstream of an irreversible step.52
As a consequence of these rules, net rate constants depend only on the sequence of steps between the intermediate of interest and the first irreversible downstream step inclusive. All other steps do not contribute. The analysis of net rate constants can thus provide a considerable reduction in the kinetic complexity of a steady state system and overcome some of the restrictions due to rate limitation. For example, such an approach has been used by Tittmann and coworkers to study the detailed kinetics of thiamin-dependent enzymes.54 In the case of DesII, the concentrations of the TDP-D-quinovose radical, eradH2O and eradD2O, under near saturating conditions in H2O and D2O, respectively, were measured by EPR. The ratio of these concentrations, D2Oεrad = eradH2O/eradD2O, was thus measured to be 0.22 ± 0.03 indicating a roughly five-fold greater accumulation of the radical in D2O versus H2O under saturating conditions.22 In light of Eqn. (3), the solvent deuterium isotope effect on the saturating rate of turnover, i.e., D2OV = 1.8 ± 0.2, can be divided by D2Oεrad to provide the isotope effect on the net rate constant for the α-hydroxyalkyl radical intermediate, i.e., D2Ok′rad = 8 ± 2.
An intuitive interpretation of this isotope effect is available by considering three possible idealized mechanisms for decomposition of the substrate radical, Erad (i.e., 21 in Fig. 4), via oxidation. The first mechanism involves electron transfer followed by rapid deprotonation of the protonated ketone, ECOH, (i.e., conjugate acid of 23) to produce the enzyme-bound ketone product, ECO (i.e., 23 in Fig. 4),
| (5) |
where kPT, kFET and kRET are the first order rate constants for proton transfer, forward electron transfer and reverse electron transfer, respectively. The net rate constant k′rad is then given by
| (6) |
In these expressions, deprotonation of a protonated ketone, which has a negative pKa, is expected to be effectively irreversible within an enzyme active site. Now, if this deprotonation is much faster than electron transfer from the cluster back to the protonated ketone (i.e., kPT ≫ kRET), as would also be the case if the electron transfer itself is irreversible, then k′rad will essentially be equal to kFET and independent of proton transfer. In other words, one would expect a unit solvent deuterium isotope effect on k′rad, which is inconsistent with the measured value.
Of the remaining two possibilities, the first involves deprotonation of the α-hydroxyalkyl radical (Erad, 21) to form a ketyl radical, Eket (i.e., 22 in Fig. 4), followed by electron transfer,
| (7) |
where kET, kFPT and kRPT denote the first order rate constants for electron transfer, forward proton transfer and reverse proton transfer, respectively. This is the mechanism depicted by the sequence 21→22→23 in Fig. 4. The second possibility is concerted deprotonation and electron transfer,
| (8) |
where kEPT is the first order rate constant for the concerted process. In both cases the net rate constant associated with Erad (21) will be influenced by the rate of proton transfer and thus exhibit a solvent deuterium kinetic isotope effect. The observed isotope effect of approximately 8, which approaches the semiclassical limit for a primary deuterium kinetic isotope effect,55 is consistent with these expectations. It was therefore concluded that deprotonation of the α-hydroxyalkyl radical plays an important role in its decomposition.22 It remains unclear, however, whether a discreet, transient ketyl radical (i.e., 22) ever actually forms or not.
5. Mechanism of Deamination
In contrast to the dehydrogenation reaction, much less information is available regarding the deamination reaction catalyzed by DesII. Nevertheless, possible mechanisms for deamination can be broadly classified into two types as shown in Fig. 6. The first possible mechanism is similar to the working models of EAL56,57 and diol dehydratase10 catalysis and involves a radical-mediated 1,2-migration of the protonated amino group (28→29). The result would be a radical carbinolamine (29). Completion of the reaction would then require reduction of the radical intermediate and elimination of ammonia to yield the C3 ketone (e.g., 29→30→31). One interesting aspect of this mechanism is that there is no clear need for the general base proposed based on the observations of the dehydrogenation reaction. It is possible that the putative general base does not operate as such during deamination, which would be consistent with its elevated pKaV. However, it should be kept in mind that the acidity constants for steady state kinetic parameters are apparent and can be altered both upon binding substrate and by the degree to which the pH-sensitive step is rate limiting.53 For example, the pKa associated with the acidic limb of V/K during dehydrogenation is approximately one log-unit lower than that for V.22
The alternative mechanism for deamination is a direct elimination of the amino group (28→32). In this case, deprotonation of a substrate α-hydroxyalkyl radical would yield a ketyl radical that could serve as the conjugate base in an E1cb-type elimination. The carbonyl conjugated (i.e., enol) product radical (32) would then need to be reduced to complete the reaction. A similar type of chemistry has been proposed for ribonucleotide reductase and 2-hydroxyacyl-CoA dehydrogenase,58,59 though neither of these systems involve deamination. A direct elimination mechanism would be consistent with the presence of an active site general base, but would seemingly leave the ketyl radical open to oxidation and production of a 4-amino-3-keto byproduct (33) which has not been observed. Furthermore, computational results for the EAL system suggest that direct elimination of NH3 would be enthalpically unfavorable unless it simultaneously accepts the proton from the adjacent α-hydroxy radical.57 This would obviate the need for an active site general base, especially if deprotonation and elimination proceed in a concerted manner in order to avoid production of a discrete ketyl radical as shown in Fig. 6. Nevertheless, both the elimination and migration mechanisms remain largely speculative at this point and neither can be definitively ruled out.
6. Summary: A Working Model
Despite a far from complete understanding of DesII catalysis, a working model can nevertheless be proposed. The catalytic cycle begins with the formation of a ternary complex involving a TDP-sugar substrate, SAM and DesII having its iron-sulfur cluster in the reduced [4Fe-4S]1+ state (19 or 26). Radical initiation ensues with reductive homolysis of SAM concomitant with H-atom abstraction from the C3 position of the substrate. Deprotonation of the C3 hydroxyl group is not a prerequisite of this step, which may take place in either a single, concerted phase or sequentially with the transient production of a discrete 5′-deoxyadensyl radical. In either case, the result is a C3-centered α-hydroxyalkyl radical along with the reduction products of SAM (i.e., 5′-deoxyadenosine and L-methionine) bound to the active site in the presence of an oxidized [4Fe-4S]2+ cluster (21 or 28).
What happens next is hypothesized to depend in large part on the binding configuration of the substrate. When TDP-D-quinovose (4) serves as the substrate, the substrate radical is expected to assume a chair-like conformation that results in poor hyperconjugation between the sp2-hybridized C3 and the adjacent C–OH σ-bonds at both C2 and C4. As a consequence, a dehydration reaction involving either the C2 or C4 hydroxyl groups is avoided and products such as 24 and 25 do not form. This results in a stalling of the catalytic cycle and the accumulation of the substrate radical amenable to EPR analysis. At this point an active site residue, which likely coordinates the C3 hydroxyl as an H-bond acceptor, can serve as a general base to deprotonate the α-hydroxylalkyl radical (21→22) due to the decreased pKa of the latter. This deprotonation step leads to the formation of a ketyl radical facilitating outersphere electron transfer to the [4Fe-4S]2+ cluster (22→23). Following electron transfer, the C3-oxidation of TDP-D-quinovose would be complete and the enzyme returns to the active [4Fe-4S]1+ state, finishing the catalytic cycle.
In contrast, the biosynthetically relevant TDP-4-amino-4,6-dideoxy-D-glucose substrate (1) is hypothesized to bind in a different conformation compared to TDP-D-quinovose due to the presence of the protonated C4 amino group. Following H-atom abstraction, this alternative conformation is expected to facilitate overlap between the p-orbital at C3 and the C–NH3+ σ-system at C4 while at the same time maintaining the more orthogonal configuration between C3 and the C–OH bond at C2 observed with TDP-D-quinovose. The C3 substrate radical (28) is also likely to be an α-hydroxyalkyl radical making oxidation of the C3 hydroxyl unfavorable. In this way, slower partitioning of the substrate radical along either a dehydrogenation (to form 33) or a dehydration (to form 34) pathway is prevented due to a more rapid elimination (28→32) or migration (28→29) reaction for which the system is primed. Depending on the mechanism of elimination, the result is one of two possible product radicals (29 or 32) that needs to be reduced. While there is currently little information regarding the mechanism of these latter steps (only one of several possibilities is shown for each in Fig. 6), the reduction does not result in the regeneration of SAM and the reduced [4Fe-4S]1+ cluster in vitro. Therefore, the system must rely on an external source of reducing equivalents (such as dithionite in vitro) to obtain the two additional electrons required to complete the reaction.
The study of radical SAM mechanistic enzymology can be regarded as the investigation of how enzymes control highly labile radical intermediates. With its dual activities and recognized subtleties underlying the catalytic processes, DesII in particular has proven to be a particularly fertile ground for the development and testing of principles regarding the enzymology of radical-mediated reactions. While this system has already provided significant insights into radical catalysis, primarily with respect to the dehydrogenation of alcohols, a large number of questions remain and represent an active area of further research.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (GM035906) and the Welch Foundation (F-1511).
Biographies

Hung-wen (Ben) Liu was born in Taipei (Taiwan). He received a PhD under Koji Nakanishi from Columbia University and did his postdoctoral training under Chris Walsh at MIT. He joined the faculty of Chemistry at the University of Minnesota in 1984. He was promoted through the ranks becoming the Distinguished McKnight University Professor in 1999. In 2000, he moved to the University of Texas at Austin, where he now holds the George H. Hitchings Regents Chair in Drug Design and is Professor of Medicinal Chemistry and Chemistry. His research lies at the crossroads of organic and biological chemistry, with particular emphasis on enzymatic reaction mechanisms, natural-product biosynthesis, carbohydrate chemistry, protein function regulation, inhibitor design and synthesis, and metabolic pathway engineering.

Mark W. Ruszczycky received his PhD and MD through the Medical Scientist Training Program at Case Western Reserve University in Cleveland, Ohio. His PhD work was completed under the guidance of Vernon E. Anderson with a focus on mechanistic enzymology and isotope effects. Upon completing his studies in 2008, he realized he enjoyed being a scientist much more than a physician and decided to forego residency to continue studying enzymes with Professor Liu.
Footnotes
In the case of the first step, the substrate concentration s must also be included, i.e., v = s k′1 e1.
Contributor Information
Mark W. Ruszczycky, Email: mwr8@utexas.edu.
Hung-wen Liu, Email: h.w.liu@mail.utexas.edu.
References
- 1.Szu P-h, He X, Zhao L, Liu H-w. Angew Chem Int Ed. 2005;44:6742–6746. doi: 10.1002/anie.200501998. [DOI] [PubMed] [Google Scholar]
- 2.Szu P-h, Ruszczycky MW, Choi S-h, Liu H-w. J Am Chem Soc. 2009;131:14030–14042. doi: 10.1021/ja903354k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xue Y, Zhao L, Liu H-w, Sherman DH. Proc Natl Acad Sci USA. 1998;95:12111–12116. doi: 10.1073/pnas.95.21.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thibodeaux CJ, Melancon CE, III, Liu H-w. Angew Chem Int Ed. 2008;47:9814–9859. doi: 10.1002/anie.200801204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sofia HJ, Chen G, Hetzler BH, Reyes-Spindola JF, Miller NE. Nuc Acids Res. 2001;29:1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frey PA. Acc Chem Res. 2014;47:540–549. doi: 10.1021/ar400194k. [DOI] [PubMed] [Google Scholar]
- 7.Broderick JB, Duffus BR, Duschene KS, Shepard EM. Chem Rev. 2014;114:4229–4317. doi: 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Zhang Y, Zhu X, Torelli AT, Lee M, Dzikovski B, Koralewski RM, Wang E, Freed J, Krebs C, Ealick SE, Lin H. Nature. 2010;465:891–898. doi: 10.1038/nature09138. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Demick JM, Lanzilotta WN. Biochemistry. 2011;50:440–442. doi: 10.1021/bi101255e. [DOI] [PubMed] [Google Scholar]
- 9.Ruszczycky MW, Choi S-h, Mansoorabadi SO, Liu H-w. J Am Chem Soc. 2011;133:7292–7295. doi: 10.1021/ja201212f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toraya T. Chem Rev. 2003;103:2095–2127. doi: 10.1021/cr020428b. [DOI] [PubMed] [Google Scholar]
- 11.a) Bansal KM, Gratzel M, Henglein A, Janata E. J Phys Chem. 1973;77:16–19. [Google Scholar]; b) Steenken S. J Phys Chem. 1979;83:595–599. [Google Scholar]; c) Steenken S, Davies MJ, Gilbert BC. J Chem Soc, Perkin Trans 2. 1986:1003–1010. [Google Scholar]
- 12.a) Frey PA, Hegeman AD, Ruzicka FJ. Crit Rev Biochem Mol Biol. 2008;43:63–88. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]; b) Duschene KS, Veneziano SE, Silver SC, Broderick JB. Curr Opin Chem Biol. 2009;13:74–83. doi: 10.1016/j.cbpa.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruszczycky MW, Ogasawara Y, Liu H-w. Biochim Biophys Acta. 2012;1824:1231–1244. doi: 10.1016/j.bbapap.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chirpich TP, Zappia V, Costilow RN, Barker HA. J Biol Chem. 1970;245:1778–1789. [PubMed] [Google Scholar]
- 15.Moss M, Frey PA. J Biol Chem. 1987;262:14859–14862. [PubMed] [Google Scholar]
- 16.Baraniak J, Moss ML, Frey PA. J Biol Chem. 1989;264:1357–1360. [PubMed] [Google Scholar]
- 17.Cheek J, Broderick JB. J Am Chem Soc. 2002;124:2860–2861. doi: 10.1021/ja017784g. [DOI] [PubMed] [Google Scholar]
- 18.Buis JM, Cheek J, Kalliri E, Broderick JB. J Biol Chem. 2006;281:25994–26003. doi: 10.1074/jbc.M603931200. [DOI] [PubMed] [Google Scholar]
- 19.McCarty RM, Krebs C, Bandarian V. Biochemistry. 2013;52:188–198. doi: 10.1021/bi301156w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruszczycky MW, Choi S-h, Liu H-w. J Am Chem Soc. 2010;132:2359–2369. doi: 10.1021/ja909451a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yokoyama K, Numakura M, Kudo F, Ohmori D, Eguchi T. J Am Chem Soc. 2007;129:15147–15155. doi: 10.1021/ja072481t. [DOI] [PubMed] [Google Scholar]
- 22.Ruszczycky MW, Choi S-h, Liu H-w. Proc Nat Acad Sci USA. 2013;110:2088–2093. doi: 10.1073/pnas.1209446110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weil JA, Bolton JR, Wertz JE. Electron paramagnetic resonance: Elementary theory and practical applications. John Wiley & Sons; New York: 1994. [Google Scholar]
- 24.Heller C, McConnell HM. J Chem Phys. 1960;32:1535–1539. [Google Scholar]
- 25.Yokoyama K, Ohmori D, Kudo F, Eguchi T. Biochemistry. 2008;47:8950–8960. doi: 10.1021/bi800509x. [DOI] [PubMed] [Google Scholar]
- 26.Bandarian V, Reed GH. Biochemistry. 2002;41:8580–8588. doi: 10.1021/bi0201217. [DOI] [PubMed] [Google Scholar]
- 27.Yamanishi M, Ide H, Murakami Y, Toraya T. Biochemistry. 2005;44:2113–2118. doi: 10.1021/bi0481850. [DOI] [PubMed] [Google Scholar]
- 28.Zhao L, Borisova SA, Yeung S-M, Liu H-w. J Am Chem Soc. 2001;123:7909–7910. doi: 10.1021/ja010587x. [DOI] [PubMed] [Google Scholar]
- 29.a) Walsby CJ, Ortillo D, Broderick WE, Broderick JB, Hoffman BM. J Am Chem Soc. 2002;124:11270–11271. doi: 10.1021/ja027078v. [DOI] [PubMed] [Google Scholar]; b) Chen D, Walsby C, Hoffman BM, Frey PA. J Am Chem Soc. 2003;125:11788–11789. doi: 10.1021/ja036120z. [DOI] [PubMed] [Google Scholar]
- 30.Ota Y, Tamegai H, Kudo F, Kuriki H, Koike-Takeshita A, Eguchi T. J Antibiot. 2000;53:1158–1167. doi: 10.7164/antibiotics.53.1158. [DOI] [PubMed] [Google Scholar]
- 31.Goldman PJ, Grove TL, Booker SJ, Drennan CL. Proc Natl Acad Sci USA. 2013;110:15949–15954. doi: 10.1073/pnas.1312228110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grove TL, Ahlum JH, Sharma P, Krebs C, Booker SJ. Biochemistry. 2010;49:3783–3785. doi: 10.1021/bi9022126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fang Q, Peng J, Dierks T. J Biol Chem. 2004;279:14570–14578. doi: 10.1074/jbc.M313855200. [DOI] [PubMed] [Google Scholar]
- 34.Benjdia A, Subramanian S, Leprince J, Vaudry H, Johnson MK, Berteau O. J Biol Chem. 2008;283:17815–17826. doi: 10.1074/jbc.M710074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benjdia A, Leprince J, Guillot A, Vaudry H, Rabot S, Berteau O. J Am Chem Soc. 2007;129:3462–3463. doi: 10.1021/ja067175e. [DOI] [PubMed] [Google Scholar]
- 36.Szameit C, Miech C, Balleininger M, Schmidt B, von Figura K, Dierks T. J Biol Chem. 1999;274:15375–15381. doi: 10.1074/jbc.274.22.15375. [DOI] [PubMed] [Google Scholar]
- 37.Grove TL, Lee K-H, St Clair J, Krebs C, Booker SJ. Biochemistry. 2008;47:7523–7538. doi: 10.1021/bi8004297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berteau O, Guillot A, Benjdia A, Rabot S. J Biol Chem. 2006;281:22464–22470. doi: 10.1074/jbc.M602504200. [DOI] [PubMed] [Google Scholar]
- 39.Grove TL, Ahlum JH, Qin RM, Lanz ND, Radle MI, Krebs C, Booker SJ. Biochemistry. 2013;52:2874–2887. doi: 10.1021/bi400136u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goldman PJ, Grove TL, Sites LA, McLaughlin MI, Booker SJ, Drennan CL. Proc Natl Acad Sci USA. 2013;110:8519–8524. doi: 10.1073/pnas.1302417110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng Q, Hwa V, Salyers AA. J Bacteriol. 1992;174:7185–7193. doi: 10.1128/jb.174.22.7185-7193.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haft DH, Kumar BM. J Bateriol. 2011;193:2745–2755. [Google Scholar]
- 43.Whittaker JW. Arch Biochem Biophys. 2005;433:227–239. doi: 10.1016/j.abb.2004.08.034. [DOI] [PubMed] [Google Scholar]
- 44.Whittaker JW. Chem Rev. 2003;103:2347–2363. doi: 10.1021/cr020425z. [DOI] [PubMed] [Google Scholar]
- 45.Moser CC, Anderson JLR, Dutton PL. Biochim Biophys Acta. 2010;1797:1573–1586. doi: 10.1016/j.bbabio.2010.04.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Page CC, Moser CC, Chen X, Dutton PL. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 47.Hinckley GT, Frey PA. Biochemistry. 2006;45:3219–3225. doi: 10.1021/bi0519497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang SC, Frey PA. Biochemistry. 2007;46:12889–12895. doi: 10.1021/bi701745h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.a) Daley CJA, Holm RH. Inorg Chem. 2001;40:2785–2793. doi: 10.1021/ic010039k. [DOI] [PubMed] [Google Scholar]; b) Daley CJA, Holm RH. J Inorg Biochem. 2003;97:287–298. doi: 10.1016/s0162-0134(03)00280-0. [DOI] [PubMed] [Google Scholar]; c) Rao PV, Holm RH. Chem Rev. 2004;104:527–559. doi: 10.1021/cr020615+. [DOI] [PubMed] [Google Scholar]
- 50.a) Rao PS, Hayon E. J Am Chem Soc. 1974;96:1287–1294. doi: 10.1021/ja00812a005. [DOI] [PubMed] [Google Scholar]; b) Rao PS, Hayon E. Anal Chem. 1976;48:564–568. [Google Scholar]
- 51.Hayon E, Simic M. Acc Chem Res. 1974;7:114–121. [Google Scholar]
- 52.Cleland WW. Biochemistry. 1975;14:3220–3224. doi: 10.1021/bi00685a029. [DOI] [PubMed] [Google Scholar]
- 53.Cook PF, Cleland WW. Enzyme Kinetics and Mechanism. Taylor and Francis; New York: 2007. [Google Scholar]
- 54.a) Tittmann K, Golbik R, Uhlemann K, Khailova L, Schneider G, Patel M, Jordan F, Chipman DM, Duggleby RG, Hubner G. Biochemistry. 2003;42:7885–7891. doi: 10.1021/bi034465o. [DOI] [PubMed] [Google Scholar]; b) Tittmann K, Vyazmensky M, Hubner G, Barak Z, Chipman DM. Proc Natl Acad Sci USA. 2005;102:553–558. doi: 10.1073/pnas.0408210101. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tittmann K, Neef H, Golbik R, Hubner G, Kern D. Biochemistry. 2005;44:8697–8700. doi: 10.1021/bi050522x. [DOI] [PubMed] [Google Scholar]; d) Schutz A, Golbik R, Konig S, Hubner G, Tittmann K. Biochemistry. 2005;44:6164–6179. doi: 10.1021/bi0473354. [DOI] [PubMed] [Google Scholar]
- 55.Wiberg KB. Chem Rev. 1955;55:713–743. [Google Scholar]
- 56.a) Babior BM. J Biol Chem. 1969;244:449–456. [PubMed] [Google Scholar]; b) Bandarian V, Reed GH. In: Chemistry and Biochemistry of B12. Banerjee R, editor. John Wiley & Sons; New York: 1999. pp. 811–833. [Google Scholar]; c) Warncke K, Canfield JM. J Am Chem Soc. 2004;126:5930–5931. doi: 10.1021/ja031569d. [DOI] [PubMed] [Google Scholar]
- 57.Semialjac M, Schwarz H. J Am Chem Soc. 2002;124:8974–8983. doi: 10.1021/ja020101s. [DOI] [PubMed] [Google Scholar]
- 58.Stubbe J, van der Donk WA. Chem Biol. 1995;2:793–801. doi: 10.1016/1074-5521(95)90084-5. [DOI] [PubMed] [Google Scholar]
- 59.Buckel W. Angew Chem Int Ed. 2009;48:6779–6787. doi: 10.1002/anie.200900771. [DOI] [PubMed] [Google Scholar]