

Abstract
Alteration/deficiency in activation 3 (ADA3) is an essential component of specific histone acetyltransferase (HAT) complexes. We have previously shown that ADA3 is required for establishing global histone acetylation patterns and for normal cell cycle progression (S. Mohibi et al., J Biol Chem 287:29442–29456, 2012, http://dx.doi.org/10.1074/jbc.M112.378901). Here, we report that these functional roles of ADA3 require its acetylation. We show that ADA3 acetylation, which is dynamically regulated in a cell cycle-dependent manner, reflects a balance of coordinated actions of its associated HATs, GCN5, PCAF, and p300, and a new partner that we define, the deacetylase SIRT1. We use mass spectrometry and site-directed mutagenesis to identify major sites of ADA3 acetylated by GCN5 and p300. Acetylation-defective mutants are capable of interacting with HATs and other components of HAT complexes but are deficient in their ability to restore ADA3-dependent global or locus-specific histone acetylation marks and cell proliferation in Ada3-deleted murine embryonic fibroblasts (MEFs). Given the key importance of ADA3-containing HAT complexes in the regulation of various biological processes, including the cell cycle, our study presents a novel mechanism to regulate the function of these complexes through dynamic ADA3 acetylation.
INTRODUCTION
Alteration/deficiency in activation 3 (ADA3) protein is a conserved component of key chromatin-modifying complexes that contain either GCN5 or PCAF histone acetyl transferases (HATs), such as SAGA (Spt/Ada/Gcn5) in yeast, ATAC (ADA2a-containing complex), STAGA (SPT3/TAFII31/GCN5 acetyltransferase), and TFTC (TATA binding protein free-TAF containing complex) in metazoans (1–7). Within these complexes, ADA3 associates with GCN5 and ADA2 to form the HAT module. ADA3 has also been shown to associate with p300, the most well-defined HAT of mammalian systems (8, 9). ADA3 is essential for the HAT activity of p300- and GCN5-containing HAT complexes toward histones (10–14) as well as of nonhistone proteins, such as p53 and β-catenin (15, 16).
Although strongly implicated in the regulation of HAT activity of ADA3-containing complexes, additional functions for ADA3 have been reported. For example, we identified ADA3 as a novel human papillomavirus E6 oncoprotein-binding protein (17), and additional studies revealed that ADA3 binds to nuclear hormone receptors, such as estrogen receptor and retinoid acid receptor, and enhances their transcriptional activation function (8, 18–21). Recent studies have identified an essential role of ADA3 in normal cell cycle progression and maintenance of genomic stability (5, 13, 22, 23).
Whether ADA3's role in these processes is merely a passive structural one or is actively regulated is unknown. Posttranslational modification represents one potential mechanism to regulate ADA3 function, and in fact yeast ADA3 was found to be modified by acetylation (24). Consistent with this idea, we observed that human ADA3 is also acetylated in vitro by its interacting HAT p300 (13). Here, we present evidence that, in addition to p300, mammalian ADA3 is acetylated by GCN5 and PCAF, and that ADA3 acetylation is balanced by deacetylation by the histone deacetylase (HDAC) SIRT1. Mass spectrometry analyses identified seven p300 and one GCN5 acetylation site on ADA3. ADA3 acetylation is cell cycle dependent, and acetylation-defective mutants of ADA3 fail to restore global histone acetylation patterns or H3K9 acetylation at the c-Myc enhancer and failed to rescue cell cycle progression block caused by endogenous Ada3 deletion. These results demonstrate that acetylation plays an important role in ADA3 function in histone modification and cell cycle progression. Taken together, our findings demonstrate that acetylation of ADA3 by its associated HATs is essential for its key role in histone acetylation and cell cycle progression.
MATERIALS AND METHODS
Plasmids, siRNA, and transient transfection.
Construction of FLAG-ADA3 has been described previously (13). Various FLAG-ADA3 point mutants were generated using an Invitrogen GeneArt site-directed mutagenesis kit and then verified by DNA sequencing. Primers for site-directed mutagenesis were designed using the GeneArt Primer design tool on the manufacturer's website (http://www.thermofisher.com/order/oligoDesigner/). Primer sequences are available upon request. The His-GCN5L2 HAT domain was a gift from Cheryl Arrowsmith (plasmid 25482; Addgene). Wild-type (WT) p300, p300ΔHAT mutant, FLAG-HDACs, FLAG-SIRT1, FLAG-SIRT2, and FLAG-SIRT3 were generous gifts from Kishor Bhakat. FLAG-SIRT4, FLAG-SIRT5, FLAG-SIRT6, and FLAG-SIRT7 were purchased from Addgene (plasmids 13815, 13816, 13817, and 13818, respectively). FLAG-SIRT1-H363Y was generated using an Invitrogen GeneArt site-directed mutagenesis system kit and verified by DNA sequencing. For transient-transfection experiments, the indicated plasmids were transfected using X-tremeGene HP transfection reagent (06366236001; Roche) according to the manufacturer's protocol. Control and SIRT1 (sc-40986) short interfering RNA (siRNA) were purchased from Santa Cruz Biotechnology. For cotransfection of FLAG-ADA3 and control or SIRT1 siRNA, 3 μg FLAG-ADA3 and 20 nM siRNA were cotransfected using X-tremeGENE siRNA transfection reagent (04476093001) by following the manufacturer's protocol.
Cell culture, viral infections, and cell cycle analysis.
76NTERT cells were cultured in DFCI medium as described before (25). A549, HEK293T, and Ada3FL/FL murine embryonic fibroblasts (MEFs) were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. Ada3FL/FL MEFs stably expressing wild-type FLAG-ADA3 or acetylation-defective mutants were generated as previously described (13). Adenoviruses expressing enhanced green fluorescent protein (EGFP)-Cre or EGFP alone (Adeno-EGFP) were purchased from the University of Iowa (Gene Transfer Vector Core). Cre-mediated deletion of Ada3 was performed as described previously (13). Cell cycle analysis by fluorescence-activated cell sorting (FACS) in 76NTERT cells was performed as described previously (26).
Antibodies.
FLAG-horseradish peroxidase (HRP) (A8592), β-actin (A2228), and α-tubulin (T-5168) antibodies were purchased from Sigma. Anti-acetyl-H3K56 (04-1135), anti-acetyl-H3K9 (07-352), and histone H3 (06-755) were purchased from Millipore. ADA2a (ab-57489) and ADA2b (ab-57953) antibodies were purchased from Abcam. p300 (sc-584 and sc-585), PCAF (sc-13124), TRRAP (sc-5405), and HSC-70 (sc-7298) antibodies were from Santa Cruz Biotechnology, and anti-acetyl-lysine (9681), anti-acetyl-lysine–HRP (6952), GCN5 (3305), SIRT1 (9475), and hemagglutinin (HA)-HRP (2999) were from Cell Signaling. Generation of mouse monoclonal anti-ADA3 was described previously (13). ADA3 antibody was labeled with HRP using Lightning-Link HRP kit from Novus Biologicals (701-0030) by following the manufacturer's protocol.
Reagents.
Trichostatin A (TSA; T8552), nicotinamide (NAM; N0636), β-NAD sodium salt (NAD+; N0632), and acetyl coenzyme A sodium salt (A2056) were purchased from Sigma. EX-527 was purchased from Selleckchem (S1541). Recombinant p300 HAT domain was purchased from Active Motif (31205).
Immunoprecipitation and immunoblotting.
For immunoprecipitation (IP), cells were harvested in lysis buffer (20 mm Tris-HCl [pH 7.5], 150 mm NaCl, 0.5% Nonidet P-40, 0.1 mm Na4VO3, 1 mm NaF, and protease inhibitor mixture, containing 2 μM TSA and 10 mM NAM for acetylation experiments) and whole-cell extracts were subjected to anti-FLAG M2 affinity gel (Sigma) overnight at 4°C, and then beads were washed five times at 5,000 rpm for 1 min with lysis buffer. Unless otherwise indicated, the immunoprecipitated FLAG tag proteins were eluted with 0.25 μg/μl 3× FLAG peptide (Sigma) into lysis buffer. The elutes were subjected to SDS-PAGE and analyzed by immunoblotting as indicated. For FLAG-ADA3 and p300 coimmunoprecipitation, HEK293T cells were cross-linked by dithiobis(succinimidyl propionate) (DSP; 22585; Thermo Scientific) before immunoprecipitation. In brief, cells were incubated with 1.5 mM DSP in 1× phosphate-buffered saline (PBS) for 15 min at room temperature, followed by quenching with excess Tris, pH 7.4. Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer, and FLAG immunoprecipitation remained the same as mentioned above, except for washing the beads with RIPA buffer. For endogenous ADA3 immunoprecipitation, equal amounts of lysates were incubated with 5 μg anti-ADA3 mouse monoclonal antibody overnight at 4°C, followed by incubation with protein A/G-agarose (sc-2003; Santa Cruz Biotechnology) for 2 h. Beads were washed with lysis buffer 5 times as mentioned above and eluted in 2× SDS sample buffer. Elutes were then subjected to SDS-PAGE analysis and immunoblotted with the indicated antibodies.
Immunofluorescence.
Ada3FL/FL MEFs stably expressing wild-type FLAG-ADA3 or acetylation-defective mutants were infected with Adeno-EGFP or Adeno-EGFP-Cre in P-100 dishes as described above. One day after infection, 2,000 cells were replated on glass coverslips in 12-well plates, and 4 days later cells were fixed in 4% paraformaldehyde for 20 min. Staining was performed with anti-ADA3 antibody. The secondary antibody used was Alexa Fluor 594, from Life Technologies. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). The coverslips were then placed on slides using the mounting medium. Fluorescent images were captured using an LSM 510 META confocal fluorescence microscope (Zeiss).
In vitro acetylation and deacetylation assays.
Purified glutathione S-transferase (GST)–ADA3 or various GST-ADA3 mutants were acetylated by recombinant HAT GCN5 or p300. Briefly, 1 μg GST-ADA3 WT or mutants were incubated with 25 ng p300 HAT domain or 50 ng GCN5 HAT domain in HAT buffer (50 mM Tris-HCl, pH 8.0, 50 mM KCl, 5% glycerol, 0.1 mM EDTA, 1 mM dithiothreitol [DTT], 2 μM TSA, 50 μM acetyl coenzyme A sodium salt, and 1 mM phenylmethylsulfonyl fluoride [PMSF]) at 30°C for 30 min. The reaction was stopped by adding 3× SDS sample buffer, and products were subjected to SDS-PAGE analysis and immunoblotted with the indicated antibodies. For deacetylation reactions, the products of GST-ADA3 WT acetylation reactions (acetylated with p300 or GCN5) were then incubated with 20 ng FLAG-SIRT1, wild type (WT) or H363Y, in HDAC buffer (50 mM Tris-HCl, pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 3 mM NAD+, 200 nM TSA) at 37°C for 1 h. The deacetylation reaction was then stopped by 6× SDS sample buffer and analyzed by immunoblotting with pan-anti-acetyl-lysine antibody. FLAG-SIRT1, WT or H363Y, used in these deacetylation reactions was ectopically expressed and immunoprecipitated from HEK293T cells. The immunoprecipitates were eluted with 3×FLAG peptide, and a fraction was analyzed on SDS-PAGE gel by Coomassie brilliant blue staining along with various amounts of bovine serum albumin (BSA) for quantification.
GST pulldown assays.
GST pulldown assay was performed as described previously (23). Briefly, 1 μg of bacterially purified GST or GST-ADA3 protein bound to beads was used as bait and incubated with 300 ng of baculovirally purified SIRT1 protein, purchased from R&D Systems (catalog no. 7714-DA). Following incubation for 2 h at 4°C, the beads were washed 5 times with lysis buffer and samples were subjected to SDS-PAGE, followed by immunoblotting with anti-SIRT1 antibody.
Identification of acetylation sites on ADA3 by mass spectrometry.
For identification of acetylation sites in vivo, 76NTERT cells stably overexpressing FLAG-ADA3 or HEK293T cells transiently transfected with FLAG-ADA3 were treated with 1 μM TSA and 5 mM NAM for 10 h. Following this procedure, whole-cell lysates were immunoprecipitated with anti-FLAG conjugated agarose beads. Immunoprecipitates were then eluted with 3×FLAG peptide, run on SDS-PAGE, and stained with Coomassie stain. A band corresponding to FLAG-ADA3 was cut from the gel (about 1.5 μg) and then subjected to either chymotrypsin or trypsin digestion. The samples were cleaned up using a Millipore μC18 ZipTip, resuspended in 0.1% formic acid, and injected through an Eksigent cHiPLC column onto a 5600 TripleTOF. Database searching was performed using PEAKS studio 6 software, and peptides were identified with 95% confidence levels. For identification of lysines specifically acetylated by either GCN5 or p300, 1.5 μg of bacterially purified GST-ADA3 was incubated with 50 ng of GCN5 catalytic domain or with 25 ng of p300 catalytic domain (amino acids 965 to 1810) for 30 min at 30°C. The samples were eluted on SDS-PAGE, stained with Coomassie stain, and subjected to mass spectrometric analysis, as indicated above.
Examining ADA3 acetylation during cell cycle progression.
76NTERT cells were synchronized in G1 phase by growth factor deprivation and released into the cell cycle by stimulation with growth factor containing medium as described previously (26). Following stimulation, cells were harvested at various time points for FACS analysis by propidium iodide staining, and lysates were prepared in the IP lysis buffer (described above). Four hundred fifty micrograms of lysates was used to perform endogenous ADA3 IP using anti-ADA3 monoclonal antibody, followed by immunoblotting with anti-acetyl-lysine–HRP antibody and anti-ADA3–HRP antibody. Ten micrograms of lysates (∼2%) was loaded as the input to determine the total levels of ADA3 in various cell cycle phases.
Rescue proliferation and rescue colony formation assays.
Rescue proliferation assays for various acetylation-defective mutants were performed as previously described (23). Briefly, Ada3FL/FL MEFs stably overexpressing empty vector, ADA3 WT or the K418R, 7KR, or 8KR mutant were plated in triplicates in 96-well plates (150 cells/well) 24 h after infection with adenovirus EGFP or EGFP-Cre. Cell proliferation was measured at various days using CellTiter Glo assay (Promega) according to the manufacturer's protocol. For calculating percent rescue for each cell line, two levels of normalization were used. First, CellTiter Glo values for each day for Cre-infected cells were divided by their corresponding control infected cell values. Subsequently, the data obtained were normalized to day 1 for each cell line. Rescue colony formation assays were performed as previously described (13), and colony numbers were counted followed by calculation of percent rescue. Percent rescues from CellTiter Glo assay or colony formation assay represent means ± standard deviations (SD) from three independent experiments, and P values were computed using Student's t test.
ChIP.
Chromatin immunoprecipitation (ChIP) assay was performed using a ChIP-IT express kit from Active Motif according to the manufacturer's protocol, with slight modifications in fixation and sonication conditions. Stably overexpressing empty vector, ADA3 WT, or K418R, 7KR, or 8KR mutant Ada3 FL/FL MEFs were infected with adenovirus expressing GFP-Cre in order to delete endogenous Ada3. Forty-eight hours after infection, cells were switched to 0.1% serum-containing medium for 72 h followed by stimulation with 10% serum containing media for 40 min. Cells were then fixed in ethylene glycol bis(succinimidyl succinate) (EGS; Thermo Scientific; and formaldehyde at room temperature. In particular, cells were incubated in 1.5 mM EGS in 1× PBS on a shaking platform for 15 min. To this, formaldehyde (1% working concentration) was added for another 15 min. The fixation reaction was then stopped by 1× glycine at room temperature for 5 min. Chromatin was isolated and sonicated for 12 min in a Bioruptor UCD-200 (Diagenode) attached to a NESLAB RTE7 water bath circulator (Thermo Scientific). Remaining steps for ChIP were followed exactly per the manufacturer's protocol. Eluted chromatin from immunoprecipitation and input was then ethanol precipitated and used as templates in reverse transcription-PCRs (RT-PCRs) performed in four replicates. ChIP RT-PCR data were analyzed using the percent input method and normalized against the vector. In brief, the percent input was calculated as 100 × 2(adjusted input − CT of IP), where adjusted input is the CT (threshold cycle) of the input minus the log2(dilution factor).
RESULTS
ADA3 is subject to acetylation and deacetylation across different cell types.
Acetylation of proteins is a reversible biochemical reaction, and the level of acetylation is governed by the opposing actions of lysine acetyltransferases and deacetylases (27). To examine whether ADA3 acetylation is also dynamically regulated, we expressed FLAG epitope-tagged ADA3 (FLAG-ADA3) in HEK293T cells, treated the cultures with trichostatin A (TSA) and nicotinamide (NAM) to inhibit cellular deacetylases (28, 29), performed anti-FLAG immunoprecipitations for FLAG-ADA3, followed by Western blotting with pan-anti-acetyl-lysine antibodies. Compared to basal levels of ADA3 acetylation in untreated cells, acetylation of ADA3 was markedly increased upon treatment of cells with TSA and NAM (Fig. 1A). We next treated A549, a lung cancer cell line, with TSA and NAM as described above and performed anti-ADA3 immunoprecipitation followed by blotting for acetylation signals to demonstrate the acetylation of endogenous ADA3 (Fig. 1B). ADA3 acetylation was also observed in other cell lines, including 76NTERT, an immortal human mammary epithelial cell line (25), and MCF-7, a breast cancer cell line (see Fig. S1A and B in the supplemental material). Furthermore, a reciprocal immunoprecipitation with anti-acetyl-lysine antibody and immunoblotting with anti-FLAG–HRP antibody also demonstrated ADA3 acetylation in 76NTERT cells (see Fig. S1C). These results demonstrate that ADA3 is acetylated across different cell types and that the level of acetylation is actively regulated by acetylation and deacetylation.
FIG 1.

ADA3 is subject to acetylation and deacetylation across different cell types, and acetylation is mediated by GCN5 and p300. (A) HEK293T cells were transfected with FLAG-ADA3, and 30 h posttransfection cells were treated with 1 μM trichostatin A (TSA) and 5 mM nicotinamide (NAM) or vehicle, as indicated, for an additional 10 h. Cell lysates were immunoprecipitated with anti-M2 FLAG–agarose beads. Immunoprecipitates were then eluted with 3×FLAG peptide and analyzed on SDS-PAGE using anti-acetyl-lysine (Ac-lysine) antibody. The same blot was stripped and probed with HRP-labeled FLAG antibody. (B) A549 cells were treated with TSA (1 μM) and NAM (5 mM) or vehicle for 10 h. Whole-cell extracts were then immunoprecipitated with normal mouse IgG or anti-ADA3 antibodies and immunoblotted (IB) with the indicated antibodies. (C and D) Cell lysates from vector- or FLAG-ADA3-transfected HEK293T cells were subjected to immunoprecipitation with M2 FLAG-agarose beads, and interaction with p300 and GCN5 was assessed by immunoblotting with anti-p300 (C) and anti-GCN5 (D) antibodies. (E and F) In vitro HAT assay using GST or GST-ADA3 as the substrate and catalytic domains of p300 (E) or GCN5 (F) as enzymes. The assay was followed by immunoblotting with anti-acetyl-lysine antibody. (G and H) HEK293T cells were transfected with FLAG-ADA3 along with empty vector, p300 WT, or p300 ΔHAT mutant (G) or with GCN5 WT or GCN5 ΔHAT mutant (H). Forty-eight hours after transfection, cell lysates were subjected to immunoprecipitation by M2 agarose beads and immunoblotted with the indicated antibodies.
ADA3 acetylation is mediated by GCN5, PCAF, and p300.
Since ADA3 is in a complex with the HAT GCN5, PCAF, or p300 (8–10), we reasoned that these HATs may mediate ADA3 acetylation. We first validated the association of ADA3 with these HATs by probing anti-FLAG immunoprecipitates from FLAG-ADA3-transfected HEK293T cells for p300 (Fig. 1C), GCN5 (Fig. 1D), or PCAF (see Fig. S2A in the supplemental material). These experiments showed efficient coimmunoprecipitation of endogenous p300, GCN5, or PCAF with FLAG-ADA3. We next used in vitro lysine acetyltransferase assays to assess the acetylation of bacterially purified ADA3 by the catalytic domains of recombinant p300 or GCN5. As shown in Fig. 1E and F, both p300 and GCN5 were able to efficiently acetylate ADA3 in vitro. We corroborated these findings by coexpressing FLAG-ADA3 with wild-type p300 or GCN5 or their respective HAT activity-deficient mutants (p300ΔHAT or GCN5ΔHAT). Consistent with the in vitro analyses, we observed that acetylation of FLAG-ADA3 was increased with coexpression of wild-type p300 or GCN5 but not when their HAT activity-deficient mutants were coexpressed (Fig. 1G and H). Expression of PCAF also led to increased ADA3 acetylation in HEK293T cells (see Fig. S2B). Taken together, these results demonstrate that ADA3-interacting lysine acetyl transferases GCN5, PCAF, and p300 can acetylate ADA3.
Acetylation of ADA3 is counteracted by class III HDACs (SIRTs).
The marked increase in the levels of ADA3 acetylation upon global inhibition of deacetylation supported the likelihood that ADA3 is dynamically regulated by deacetylation. Histone deacetylases (HDACs), enzymes that remove the acetyl group from lysine, have been categorized into three classes: class I, consisting of HDACs 1, 2 and 3; class II, consisting of HDACs 4, 5, and 6; and class III, consisting of sirtuins (SIRTs) 1 to 7 (30). TSA has been widely used as an inhibitor of class I and II HDAC activities (28), whereas NAM has been demonstrated to be a broad inhibitor of SIRTs (29). In order to determine the class of HDACs responsible for ADA3 deacetylation, we ectopically expressed FLAG-ADA3 in HEK293T cells and then treated the cells with either TSA or NAM. We observed that ADA3 acetylation was markedly enhanced in the presence of NAM, whereas TSA had little or no effect on ADA3 acetylation (Fig. 2A and B), suggesting that one or more SIRTs are likely to function in ADA3 deacetylation.
FIG 2.
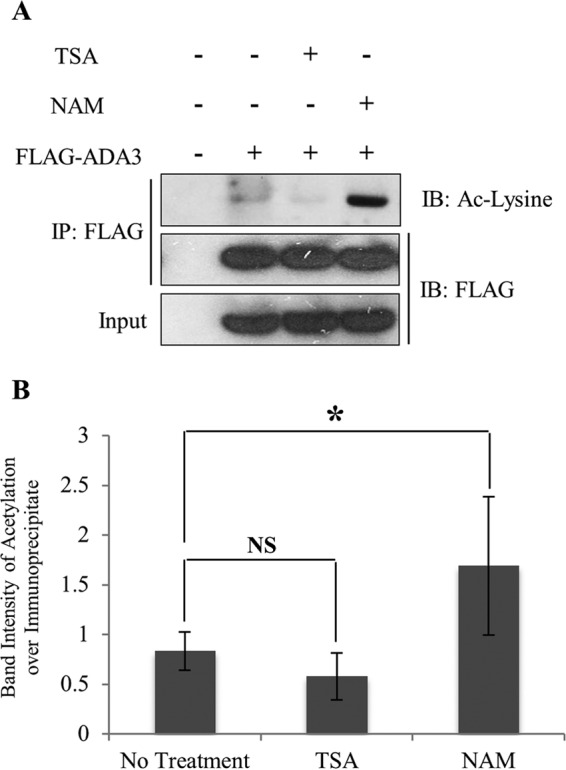
Acetylation of ADA3 is counteracted by class III HDACs (SIRTs). (A) HEK293T cells were transfected with FLAG-ADA3 or empty vector. Thirty hours after transfection, cells were treated either with vehicle or 1 μM HDAC class I and II inhibitor trichostatin A (TSA) or 5 mM HDAC class III inhibitor nicotinamide (NAM) for 10 h. Cell lysates were then subjected to immunoprecipitation by M2 FLAG-agarose beads and immunoblotted with either anti-acetyl-lysine or FLAG-HRP antibodies. (B) The band intensities of acetylated FLAG-ADA3 over immunoprecipitated FLAG-ADA3 were quantified using ImageJ software and averaged from four independent experiments. The statistical significance between different groups was computed using Student's t test. NS, not significant; *, P ≤ 0.05.
ADA3 exclusively interacts with SIRT1 among class I, II, and III deacetylases.
Based on increased ADA3 acetylation specifically with a SIRT inhibitor, we further explored if one or more of the SIRTs are involved in deacetylation of ADA3. We first examined the interaction of endogenous ADA3 with ectopically expressed FLAG-tagged HDACs or SIRTs in HEK239T cells. Coimmunoprecipitation analyses demonstrated that, out of 13 HDACs (HDAC1 through HDAC6 and SIRT1 through SIRT7) tested, only SIRT1 specifically interacted with ADA3 (Fig. 3A). A reciprocal coimmunoprecipitation further verified the strong association of ectopically expressed FLAG-ADA3 with endogenous SIRT1 (Fig. 3B). More importantly, immunoprecipitation with anti-SIRT1 antibody followed by Western blotting with anti-ADA3 antibody confirmed the association of endogenous ADA3 and SIRT1 (Fig. 3C).
FIG 3.

ADA3 exclusively interacts with SIRT1 among class I, II, and III deacetylases. (A) HEK293T cells were transfected with empty vector or various FLAG-tagged HDAC and SIRT constructs. Forty-eight hours after transfection, cell lysates were subjected to immunoprecipitation by M2 FLAG-agarose beads, eluted with 3×FLAG peptide, and immunoblotted with anti-ADA3 or FLAG-HRP antibody. (B) HEK293T cells were transfected either with empty vector or FLAG-ADA3. Forty-eight hours after transfection, cell lysates were subjected to immunoprecipitation by M2 FLAG-agarose beads, and immunoprecipitates were immunoblotted with anti-SIRT1 or FLAG-HRP antibody. (C) HEK293T cell lysates were subjected to immunoprecipitation using anti-SIRT1 antibody, and immunoprecipitates were analyzed by Western blotting using anti-ADA3–HRP and SIRT1 antibodies. (D) In vitro GST pulldown assay in which 300 ng recombinant SIRT1 was incubated with 1 μg glutathione-bound GST or GST-FLAG-ADA3, followed by immunoblotting with anti-SIRT1 antibody. (E) Lysates of HEK293T cells transfected with FLAG-SIRT1 wild type or its catalytically inactive mutant, H363Y, were immunoprecipitated as described for panel A, followed by immunoblotting with anti-ADA3 and FLAG-HRP antibodies. The values represent the ratios of signal intensities of ADA3 bands over that of total SIRT1 immunoprecipitated as calculated using ImageJ.
We next assessed whether the interaction of ADA3 with SIRT1 is direct using purified recombinant GST-ADA3 and recombinant SIRT1 proteins. GST-ADA3, but not GST alone, was able to bind to recombinant SIRT1, indicating a direct interaction (Fig. 3D). We next tested if the catalytic activity of SIRT1 is required for its interaction with ADA3 by expressing either wild-type or catalytically inactive (H363Y) SIRT1 in HEK293T cells. Notably, while wild-type SIRT1 was able to coimmunoprecipitate ADA3, the interaction with SIRT1 (H363Y) mutant was markedly lower (Fig. 3E), suggesting that SIRT1 activity is facilitating its physical interaction with ADA3.
SIRT1 deacetylates ADA3 in cells and in vitro.
To assess if SIRT1 functions as a deacetylase toward ADA3, we examined the acetylation of ADA3 in HEK293T cells in the presence or absence of SIRT1-specific inhibitor EX-527, which inhibits SIRT1 activity by occupying the binding site for its cofactor, NAD+ (31). Immunoprecipitation of FLAG-ADA3 followed by immunoblotting for acetylated lysine showed an increase in ADA3 acetylation in the presence of EX-527 (Fig. 4A). Interestingly, SIRT1 levels did not change significantly after EX-527 treatment, which strongly suggests that increased ADA3 acetylation with the inhibitor treatment reflects inhibition of SIRT1 activity (Fig. 4A). In a complementary approach, we cotransfected HEK293T cells with FLAG-ADA3 along with scrambled or SIRT1-specific siRNA and then examined ADA3 acetylation. Similar to the effect of EX-527, we observed enhanced ADA3 acetylation upon SIRT1 knockdown (Fig. 4B). Finally, to directly test the ability of SIRT1 to deacetylate ADA3, we subjected recombinant purified ADA3 to in vitro acetylation using GCN5 or p300 as HAT and then subjected this acetylated ADA3 to an in vitro deacetylation in the presence of purified wild-type SIRT1 or its catalytic inactive mutant (H363Y). Consistent with observations in cells, a dramatic decrease in ADA3 acetylation was observed upon incubation with wild-type SIRT1 but not with catalytically inactive SIRT1-H363Y mutant (Fig. 4C and D). Taken together, these results establish SIRT1 as a deacetylase for acetylated ADA3. The ability of SIRT1 to deacetylate ADA3 that was acetylated by either GCN5 or p300 suggests that SIRT1 does not discriminate among ADA3 acetylations imparted by different HATs.
FIG 4.

SIRT1 deacetylates ADA3 in cells and in vitro. (A) HEK293T cells were transfected with FLAG-ADA3 or empty vector. Thirty hours after transfection, cells were treated either with DMSO or with 10 μM EX-527, a SIRT1-specific inhibitor, for 16 h. Cell lysates were then subjected to immunoprecipitation by M2 FLAG-agarose beads and immunoblotted with either anti-acetyl-lysine or FLAG-HRP antibody. (B) HEK293T cells were cotransfected together with FLAG-ADA3 and control (Ctrl) vector or siRNA against SIRT1. Forty-eight hours after transfection, ADA3 acetylation was analyzed by immunoprecipitation followed by immunoblotting as described for panel A. Whole-cell extracts were also immunoblotted with anti-SIRT1 and anti-β-actin antibodies to examine SIRT1 knockdown. (C) Schematic depicting the strategy used for in vitro deacetylation assay of ADA3. (D) Bacterially purified ADA3 (1 μg) was in vitro acetylated by either recombinant p300 HAT domain (25 ng) or recombinant GCN5 HAT domain (50 ng). Following this, acetylated ADA3, as a substrate, was subjected to in vitro deacetylation assay. FLAG-tagged wild-type SIRT1 or catalytic inactive mutant SIRT1 H363Y was overexpressed in HEK293T cells, immunoprecipitated by FLAG M2-agarose beads, and eluted by 3×FLAG peptide. Twenty-nanogram aliquots of these elutes were used as enzymes in the presence or absence of SIRT1 cofactor NAD+ or SIRT1 inhibitor EX-527. Following the enzymatic reaction, the reaction mixtures were subjected to SDS-PAGE and immunoblotted with anti-acetyl-lysine antibody.
Mass spectrometry-based identification of lysine residues in ADA3 acetylated by GCN5 and p300.
To delineate specific lysine residues in ADA3 that are modified by acetylation by GCN5 or p300 in intact cells, we utilized a mass spectrometry approach. HEK293T or 76NTERT cells transiently or stably overexpressing FLAG-ADA3 were treated with TSA and NAM, and FLAG-ADA3 was purified using anti-FLAG beads. The anti-FLAG bead-bound FLAG-ADA3 was eluted using a tandem triple FLAG peptide and resolved by SDS-PAGE, and the band corresponding to FLAG-ADA3 was excised and subjected to mass spectrometry analysis after digestion with trypsin or chymotrypsin. This analysis revealed three ADA3 acetylation sites in HEK293T cells (K109, K194, and K418) and four sites in 76NTERT cells (K109, K122, K124, and K222) with the K109 site common to both cell lines (Fig. 5A; see also Fig. S3 in the supplemental material). To determine if any of these lysine residues were specifically acetylated by GCN5 or p300, we performed in vitro acetylation of purified recombinant GST-ADA3 with either GCN5 or p300, followed by mass spectrometric analysis after trypsin or chymotrypsin digestion. This analysis demonstrated that GCN5 acetylated a single lysine residue on ADA3 (K418), whereas p300 acetylated seven distinct lysine residues (K109, K122, K124, K147, K194, K222, and K312) (Fig. 5A; see also Fig. S3). Notably, all but two (K147 and K312) acetylation sites identified on in vitro-acetylated ADA3 were observed in one of the two cell types examined, supporting the idea that the identified ADA3 lysine residues are bona fide acetylation sites. Notably, the identified lysine residues are preserved in all mammals, and a majority are conserved among vertebrates (Fig. 5B), supporting a likely functional role of ADA3 acetylation.
FIG 5.

Mass spectrometry-based identification and validation of lysine residues in ADA3 acetylated by GCN5 and p300. (A) Summary of various acetylation sites identified on ADA3 in various experimental settings, as indicated, using mass spectrometry. (B) Sequence alignment of various ADA3 vertebrate sequences reveals high conservation of acetylated lysine residues. (C and D) In vitro HAT assay was performed using GCN5 (C) or p300 (D) catalytic domains as acetyltransferases and recombinant GST-FLAG-ADA3, GST-ADA3-K418R, GST-ADA3-5KR, or GST-ADA3-7KR as substrates. The assay was followed by immunoblotting with anti-acetyl-lysine antibody. (E and F) HEK293T cells were transfected with FLAG-ADA3 wild-type, ADA3-K418R, ADA3-5KR, or ADA3-7KR with or without GCN5 (E) and p300 (F). Forty-eight hours after transfection, cell lysates were subjected to immunoprecipitation with M2 agarose-FLAG beads and immunoblotted with the indicated antibodies. (G) HEK293T cells were transfected with the indicated plasmids. Forty-two hours after transfection, cells were treated with TSA (1 μM) and NAM (5 mM) for 6 h. Whole-cell extracts were subjected to immunoprecipitation with M2 agarose. Immunoprecipitates were then eluted with 3×FLAG peptide and immunoblotted with the indicated antibodies.
Validation of GCN5- and p300-mediated acetylation sites on ADA3 in vitro and in intact cells.
To further validate the GCN5- and p300-mediated acetylation sites on ADA3, we used site-directed mutagenesis to mutate various lysine (K) residues to arginine (R) residues to preserve their positive charge but render them acetylation incompetent. Based on results presented above, a single K418R substitution was made to assess the GCN5-mediated ADA3 acetylation, while we made two overlapping mutants to assess the p300-mediated acetylation: (i) K→R mutation of K109, K122, K124, K194, and K222 (termed 5KR) or (ii) K→R mutation of all seven lysine residues identified above (K109, K122, K124, K147, K194, K222, and K312; denoted 7KR). These mutants were expressed as recombinant protein in vitro or in cells to assess their ability to serve as substrates of acetylation by GCN5 or p300. The in vitro acetylation assay demonstrated that K418 was the major site of acetylation by GCN5, as the level of acetylation upon incubation with GCN5 was greatly reduced compared to that of wild-type ADA3 (Fig. 5C). When cotransfected in HEK293T cells, GCN5 efficiently acetylated the wild-type ADA3 but not the K418R mutant (Fig. 5E). A similar defect in K418R mutant acetylation was observed with PCAF (see Fig. S4 in the supplemental material). Taken together, these results demonstrate that both GCN5 and PCAF acetylate ADA3 at lysine 418. We next analyzed the 5KR and 7KR mutants of ADA3 in vitro to assess the impact on p300-mediated acetylation. The 5KR mutation of ADA3 greatly reduced its p300-mediated acetylation, whereas the 7KR mutation almost completely abrogated ADA3 acetylation (Fig. 5D). These results were confirmed by coexpressing wild-type ADA3, ADA3-5KR, or ADA3-7KR with HA-tagged p300 in HEK293T cells (Fig. 5F). We next generated an ADA3 mutant with K→R substitutions of all eight lysine residues (those acetylated by either GCN5 or p300; referred to as 8KR) and compared the level of acetylation of wild-type ADA3 to that of its K418R, 7KR, and 8KR mutants expressed in HEK293T cells upon treatment with TSA and NAM. Under these conditions, the 8KR mutant showed complete abrogation of acetylation (Fig. 5G). Together, these results validate the eight lysine residues identified by mass spectrometry as the major acetylation sites on ADA3 that are modified by its interacting HATs.
ADA3 acetylation levels change during cell cycle progression.
Having demonstrated that ADA3 acetylation is dynamically regulated by its associated HATs (GCN5, PCAF, and p300) and the deacetylase SIRT1, we asked if the levels of ADA3 acetylation are regulated under particular physiological conditions. Since we have established that ADA3 plays an important role in cell cycle progression (13, 23), we reasoned that its acetylation would vary with cell cycle progression. To assess if this is the case, we arrested 76NTERT immortal mammary epithelial cells in the G1 phase of the cell cycle by growth factor deprivation, released the cells from G1 block by adding growth factor-containing medium, and analyzed cells at various time points during cell cycle progression (based on FACS analysis) for levels of acetylation on immunoprecipitated ADA3 (26, 32). FACS analysis confirmed that a majority of cells were G1 arrested upon growth factor deprivation followed by release from synchrony and entry into S and G2/M phase by 16 h and 20 h, respectively, after culture in growth factor-rich medium (Fig. 6A). Western blotting of whole-cell extracts showed that total ADA3 levels remained relatively invariant during cell cycle progression (Fig. 6B). As antibodies recognizing specific ADA3 acetylation sites are not available, ADA3 was immunoprecipitated from cell lysates followed by immunoblotting with pan-acetyl-lysine-reactive antibody. Compared to acetylation levels at time zero (G1-arrested cells), we observed an initial peak of ADA3 acetylation at 4 h followed by a second peak that persisted throughout S and G2/M phases. Notably, these dynamic changes in ADA3 acetylation are consistent with previous reports of increases in GCN5 protein levels as cells enter the S phase (33) and a requirement of the HAT activity of p300 for G1-S transition (34–38). Taken together, our results demonstrate that ADA3 acetylation is cell cycle dependent and raised the possibility that acetylation of ADA3 regulates its function in cell cycle progression.
FIG 6.

ADA3 acetylation levels change during cell cycle progression. 76NTERT cells were growth factor deprived in DFCI-3 medium for 72 h and then stimulated with growth factors containing DFCI-1 medium for the indicated times. (A) Cells were fixed in 70% ethanol, stained with propidium iodide, and then subjected to FACS analysis. (B) Whole-cell extracts (input) were immunoblotted either with anti-ADA3 or anti-HSC70 antibody. (C) Equal amounts of whole-cell extracts from each time point were immunoprecipitated with normal mouse IgG or anti-ADA3 antibody. Immunoprecipitates were then immunoblotted with HRP-labeled pan-acetylated or anti-ADA3 antibody. ADA3 acetylation band intensities were quantified using ImageJ software and normalized against 0 h and are represented underneath the blot. GFS, growth factor stimulation; IP, immunoprecipitation.
Acetylation-defective mutants of ADA3 fail to rescue the block in cell proliferation imposed by Ada3 deletion in Ada3FL/FL MEFs.
To investigate the functional importance of ADA3 acetylation, we assessed the ability of acetylation-defective ADA3 mutants versus the WT ADA3 to rescue the block in cell proliferation and/or the defective histone acetylation observed upon induced Ada3 deletion in Ada3FL/FL MEFs (13). For these experiments, we generated stable Ada3FL/FL MEF cell lines expressing either vector, WT FLAG-ADA3, or one of its acetylation-defective mutants, K418R, 7KR, or 8KR. Western blotting confirmed that the expression of ectopic WT ADA3 or its mutants was comparable (Fig. 7N). As expected (13), deletion of endogenous Ada3 by adenovirus-Cre infection led to severe proliferation defects in vector-expressing Ada3FL/FL MEFs, whereas cells expressing wild-type FLAG-ADA3 showed unperturbed cell proliferation, demonstrating a functional rescue (Fig. 7A and B). Notably, partial proliferation defects were seen upon endogenous Ada3 deletion in cells expressing K418R or 7KR mutants (Fig. 7C and D). More significantly, a severe proliferative block was seen upon Ada3 deletion in MEFs expressing the 8KR mutant, suggesting that both GCN5- and p300-mediated acetylation is important for ADA3 function in cell proliferation (Fig. 7E).
FIG 7.


Acetylation-defective mutants of ADA3 fail to rescue the block in cell proliferation imposed by Ada3 deletion in Ada3FL/FL MEFs. Ada3FL/FL MEFs stably expressing either empty vector, ADA3 WT, or acetylation-defective mutants were infected either with GFP only or with GFP-Cre expressing adenovirus, and then CellTiter-Glo, Western blotting, or colony formation assays were performed in these cells. (A to E) Relative luminescence units of various Ada3FL/FL MEFs at various days after the infection are shown. The RLU shown here are means ± SD from three independent experiments, each done in three replicates, and P values were computed using Student's t test. (F to J) Western blots showing the expression of mADA3 and hADA3 WT or mutant proteins. HSC70 was used as a loading control. (K) A composite computation of percent rescue with respect to day 1. Error bars show means ± SEM from three independent experiments (*, P < 0.05; **, P < 0.01, computed using Student's t test). (L to N) Colony formation assay was performed 7 days after the infection, and percent rescue was calculated as described in Materials and Methods. Expression of mADA3 and hADA3 WT or mutants was examined by Western blotting with HSC70 as a loading control.
We confirmed the deletion of endogenous mouse Ada3 in the Adeno-Cre-transduced MEFs by immunoblotting at various time points (Fig. 7F to J). While a significant endogenous ADA3 depletion was seen in each cell line until day 7, the recovery in the expression of endogenous mouse ADA3 at day 9 reflects the outgrowth of cells in which Ada3 was not deleted. We calculated the percent rescue in proliferation (see Materials and Methods) for WT ADA3 or each mutant at various time points after Adeno-Cre transduction (Fig. 7K). A significant defect in the rescue of cells from proliferation block was observed with each acetylation-defective ADA3 mutant compared to WT ADA3 at day 7 after Ada3 deletion (Fig. 7K). At day 9, the defect in rescue with K418R or 7KR was not significant but was significant with the 8KR mutant as well as for the vector-alone-expressing cells (Fig. 7K), likely due to the outgrowth of MEFs in which endogenous Ada3 was not deleted, as suggested by the results of Western blotting (Fig. 7H and I).
We also observed similar defects in the ability of mutant ADA3 proteins to rescue proliferation defects in Ada3-deleted MEFs using an independent colony formation assay (Fig. 7L and M). In this assay, while vector-expressing cells showed about 90% defect in rescue, both ADA3 K418R- and 7KR-expressing cells showed about 40% defect, whereas 8KR showed about 70% defect in rescue. Taken together, our results demonstrate that both GCN5- and p300-mediated acetylation of ADA3 is required for its function in cell proliferation.
To test the possibility that the observed defect in cell cycle rescue is due to the mislocalization of mutants, we analyzed the localization of WT and acetylation-defective mutants 5 days after control or Cre adenovirus infection of Ada3FL/FL MEFs expressing these mutants. As expected, the endogenous ADA3 in vector control cells was exclusively localized to the nucleus; however, the overexpressed WT ADA3 was localized to both nucleus and cytoplasm even after deletion of endogenous ADA3 (see Fig. S5 in the supplemental material). Similar to WT ADA3, all acetylation-defective mutants of ADA3 were present in both the nucleus and cytoplasm (see Fig. S5). Thus, the acetylation-defective mutants are competent at nuclear entry, excluding the possibility that their defective ability to rescue ADA3-depleted MEFs from a proliferation block is due to exclusion from the nucleus. Taken together, our results conclusively show that acetylation of ADA3 is pivotal for its function to promote cell cycle progression.
Acetylation-defective mutants retain the ability to interact with various HATs and other HAT complex components yet fail to rescue the histone acetylation defects globally and at c-Myc enhancer.
Endogenous Ada3 deletion in Ada3FL/FL MEFs leads to defects in global histone acetylation and a reduction in the levels of various HATs, and these defects can be rescued by ectopic WT human ADA3 (13). As defective histone acetylation is likely the basis for functional defects associated with ADA3 depletion, we reasoned that the functional impairment of acetylation-defective ADA3 mutants arises from impaired histone acetylation due to their inability to help assemble the HAT modules or to promote HAT activity. To test these possibilities, WT FLAG-ADA3 or its acetylation-defective mutants expressed in HEK293T cells were examined for their associations with endogenous HATs and other HAT complex components by coimmunoprecipitation. Notably, all three acetylation-defective mutants were as efficient as wild-type ADA3 in their ability to coimmunoprecipitate endogenous GCN5, PCAF, or p300 (Fig. 8A and B). In addition, these mutants retained a strong association with other STAGA complex subunits, TRRAP and ADA2b, and the ATAC-specific subunit ADA2a (Fig. 8A). Importantly, while the vector cells showed a dramatic decrease in the levels of various HATs upon Ada3 deletion, the HAT levels remained unaltered in cells reconstituted with acetylation-defective ADA3 mutants, similar to those expressing the WT ADA3 (see Fig. S6 in the supplemental material). These results ruled out the possibility that acetylation-defective mutants are defective in their assembly into HAT complexes or have diminished interaction with HATs.
FIG 8.

Acetylation-defective mutants retain the ability to interact with various HATs and other HAT complex components yet fail to rescue the histone acetylation defects globally and at the c-Myc enhancer. (A) HEK293T cells were transfected with empty vector, ADA3 WT, or various acetylation-defective mutants. Forty-eight hours after transfection, whole-cell extracts were subjected to immunoprecipitation by M2 agarose-FLAG beads, eluted with 3×FLAG peptide, and then immunoblotted with the indicated antibodies. (B) HEK293T cells were transfected with empty vector, ADA3 WT, or various acetylation-defective mutants. Forty-eight hours after transfection, whole-cell extracts were subjected to immunoprecipitation by M2 agarose-FLAG beads and immunoblotted with the indicated antibodies. (C) Cell lysates of day 7 from cell cycle rescue experiments were immunoblotted with the indicated antibodies. The numbers underneath the blots indicate the band intensities computed from ImageJ normalized over total H3 with respect to the Ctrl. (D) A ChIP quantitative PCR of H3K9(Ac) signals at the c-Myc enhancer in Ada3-deleted MEFs overexpressing ADA3 WT or various acetylation-defective mutants. The y axis shows enrichment as a percentage of input normalized over signals in vector cells. Data represent the means ± SD from three different experiments. (E) Model showing the role of ADA3 acetylation in cell proliferation.
We next examined if the HAT complexes formed by the acetylation-defective ADA3 mutants are functionally active by assessing global chromatin histone acetylation. As expected from our previous study (13), a dramatic decrease in H3K9 and H3K56 acetylation was observed upon Ada3 deletion in Ada3FL/FL cells. However, this defect was rescued by exogenous WT ADA3 (Fig. 8C). Notably, the acetylation-defective mutants ADA3-K418R and ADA3-7KR showed only approximately 50% rescue of global H3K9 acetylation compared to wild-type ADA3 (Fig. 8C), similar to the level of rescue of the proliferation block (about 60%) with these mutants. While ADA3 K418R and 7KR mutants did not show any significant defect in the rescue of global H3K56 acetylation, the 8KR mutant showed about 70% defect in the rescue of both H3K9 and H3K56 acetylation (Fig. 8C), again comparable to the deficit in its ability to rescue the proliferation block (about 70%).
Given that the H3K9 acetylation mark is important in gene transcription and is known to be present at active gene enhancers/promoters (39), we used chromatin immunoprecipitation (ChIP) to examine the relative rescue of the locus-specific H3K9 acetylation by WT versus acetylation-defective ADA3 mutants in Ada3FL/FL MEFs subjected to deletion of the endogenous Ada3. We chose the c-Myc enhancer to assess the H3K9 acetylation status, as c-Myc is indispensable for cell proliferation and is a known target of ADA3-containing HAT complexes (13, 15, 18). We observed about a 4-fold enrichment in H3K9 acetylation at the c-Myc enhancer in WT ADA3-expressing Ada3-deleted MEFs compared to those expressing the vector control (Fig. 8D). In contrast, H3K9 acetylation at the c-Myc enhancer was substantially less robust in cells reconstituted with ADA3 K418R or 7KR mutants, while acetylation levels in cells expressing the 8KR mutant were essentially comparable to those of vector control cells (Fig. 8D). Taken together, these findings support the notion that ADA3 acetylation is essential for global and gene-specific histone acetylation by ADA3-containing HAT complexes, and that this activity, independent of ADA3's role to facilitate the assembly of HAT complexes, may be key to ADA3's role in cell cycle progression through histone acetylation at proliferation-associated genes such as c-Myc (Fig. 8E).
DISCUSSION
ADA3 is an evolutionarily conserved protein that functions as a transcriptional coactivator and forms a core component of the multisubunit HAT complexes (3). Previous studies by us and others have shown that ADA3 associates with GCN5, PCAF, and p300, HATs found in ADA3-containing complexes (8–10). Thus, ADA3 is ideally positioned to regulate the function of its associated HATs. How ADA3 carries out its function in this regard is unknown. In a previous study (13), we noted that ADA3 could be acetylated by p300 in vitro, raising the possibility that acetylation could regulate ADA3 function in cells. The present study establishes, for the first time, that ADA3 is dynamically regulated by acetylation mediated by its associated HATs GCN5, PCAF, and p300 and deacetylation by SIRT1; that ADA3 acetylation is required for the histone-modifying activity of ADA3-containing HAT complexes; and that ADA3 acetylation is essential for its function in cell cycle progression.
To gain more insights into ADA3 acetylation, we used a mass spectrometry approach to define a single lysine residue, K418, which is acetylated by GCN5 (and PCAF), and seven distinct lysine residues (K109, K122, K124, K147, K194, K222, and K312) that can be acetylated by p300 (Fig. 5A). Using site-directed mutagenesis followed by in vitro acetylation assays or expression in cells, we validated the lysine residues in ADA3 identified through proteomics to be the major sites of acetylation. As p300 is not an integral component of the HAT modules of STAGA or ATAC complexes (3), the seven distinct lysine residues acetylated by p300 are of considerable interest, suggesting that either ADA3 serves to recruit p300 as an accessory HAT in these complexes or that ADA3 functions together with p300 in an STAGA/ATAC-independent manner. Our previous biochemical fractionation analyses (8), which showed that both GCN5 and p300 could be purified as components of ADA3-containing complexes in human cells, support the former possibility, although more in-depth analyses will be needed to determine if one or both of these models are operational in mammalian cells.
The K→R point mutants of the GCN5-targeted, p300-targeted, or both sets of lysine residues allowed us to determine the functional importance of ADA3 acetylation. Our previously established Ada3FL/FL MEFs provided a system where any functional deficits of the K→R mutants could be established by assessing their abilities to rescue the cells from a proliferative block and associated biochemical defects upon Cre-induced deletion of endogenous mouse Ada3 (13). Importantly, analyses of cell proliferation or colony-forming ability showed that mutation of either GCN5-dependent (K418R) or p300-dependent (7KR) acetylation sites led to partial deficits in the ability of the mutants to complement the loss of endogenous ADA3, with mutations at both the GCN5- and p300-mediated acetylation sites, essentially abrogating the ability of ADA3 to sustain cell proliferation (Fig. 7M). These results underscore the critical functional importance of ADA3 acetylation in its function as a component of HAT complexes. The functional importance of ADA3 acetylation by two distinct HATs may reflect the possibility that acetylation by the two HATs occurs at discrete steps during cell proliferation or regulates discrete functional activities that are part of the complex process of cell proliferation. Notably, a previous study showed that ADA3-associated ATAC complex and p300 regulate the expression of distinct set of genes (40).
Previously, we established that deletion of ADA3 causes a dramatic deficit in global H3K9 and H3K56 acetylation on chromatin (13). By further analyzing the K→R mutants of ADA3 in the context of endogenous Ada3 deletion in MEFs, we established that ADA3 acetylation is required for the role of ADA3 in promoting global as well as locus-specific acetylation of chromatin-associated histones. Notably, while mutations of the GCN5-dependent acetylation site (K418R) or p300-dependent acetylation sites (7KR) led to a significant deficit in global H3K9 acetylation compared with wild-type ADA3 (Fig. 8C), these mutants did not affect the global H3K56 acetylation; on the other hand, the 8KR mutant, which eliminates acetylation by both GCN5 and p300, showed about a 70% defect in the rescue of H3K9 and H3K56 acetylation (Fig. 8C). The extent of the deficit in histone acetylation correlated with the extent to which the corresponding mutants were defective in cell cycle rescue in Ada3-deleted MEFs (Fig. 7M). Furthermore, analysis of c-Myc enhancer-associated histone acetylation showed that ADA3 is critical to chromatin modification at key genes that function as master controllers of cell cycle progression and other functions.
That ADA3 acetylation is critical for HAT complexes to promote global and locus-specific histone acetylation arguably arises from a defect in the ability of mutant ADA3 proteins to be assembled into HAT complexes. By examining the association of the ADA3 K→R mutants used in our biochemical and functional analyses with components of HAT complexes, we established that acetylation of ADA3 is dispensable for its association with HATs (p300, GCN5, and PCAF) and other components that define major ADA3-containing HAT complexes, such as STAGA and ATAC (Fig. 8A and B). Thus, the requirement of ADA3 acetylation to promote histone acetylation and cell cycle progression does not reflect the requirement of such acetylation in HAT assembly but must reflect a discrete function. The important question remains of how ADA3 acetylation regulates HAT complex activity despite the fact that it does not affect the overall composition of the complex. One possibility is while the acetylation of ADA3 is not required for its association with HAT complex components, the subunits are not incorporated in correct stoichiometry, leading to a defect in the overall activity of the complex. Alternatively, ADA3 acetylation might play an important role in chromatin recognition. Future biochemical studies are warranted to better understand the role of ADA3 acetylation in this regard.
Notably, detection of ADA3 acetylation in cells was facilitated by the incorporation of HDAC inhibitors (Fig. 1A), suggesting the possibility that ADA3 acetylation status is bidirectionally regulated by the action of HATs and deacetylases. By screening the members of various HDAC families for their interaction with ADA3, we identify SIRT1 as an ADA3 partner and establish that it helps regulate the low steady-state level of ADA3 acetylation (Fig. 3A). That ADA3 acetylation is determined by opposing actions of HATs and a deacetylase strongly supports the potential importance of this posttranslational modification in the functional regulation of ADA3-containing HATs. Our in vitro deacetylation assays (Fig. 4D) showed that SIRT1 was able to deacetylate ADA3 regardless of whether ADA3 was acetylated by p300 or GCN5. Future biochemical and cell-based studies are needed to further establish if SIRT1 is indeed a global deacetylase for ADA3 or if other deacetylases are involved under specific scenarios. The functional consequences of ADA3-SIRT1 interaction could be manifold. A recent study showed that the STAGA complex DUB module component USP22 associates with and is deacetylated by SIRT1 (41). However, the study did not identify any direct SIRT1-binding partner in the STAGA complex. It remains possible that ADA3-mediated recruitment of SIRT1, aside from ADA3 deacetylation, also promotes deacetylation of USP22 in the DUB module, allowing ADA3 to indirectly regulate the function of HAT complexes.
Our studies also suggest that the balance of ADA3 acetylation versus deacetylation in cells is a regulated process, as we show the level of ADA3 acetylation fluctuates during cell cycle progression. ADA3 acetylation increases early upon entry into the cell cycle, followed by a decline in the late G1 phase, and then it reaccumulates as cells enter the S phase, persisting through the G2/M phase. Such dynamic regulation during cell cycle progression further supports the functional role of ADA3 acetylation, suggesting regulation at the levels/activities of HATs/HDACs targeting the acetylation/deacetylation of ADA3. The transient earlier peak and more sustained delayed ADA3 acetylation during cell cycle progression may reflect the relative activities of GCN5 and p300, a possibility consistent with the reported increase in GCN5 levels from G1 to early S phase (33) and the requirement for p300 HAT activity for progression through S phase (34–38). If established in future studies, such a scheme will support distinct roles for GCN5/PCAF- versus p300-mediated acetylation of ADA3. Consistent with our observations, acetylation of yeast ADA3 was found to increase when quiescent cells (comparable to G0 phase of the mammalian cell cycle) transited to growth phase (comparable to S phase of the mammalian cell cycle), with persistent acetylation through the cell division phase (comparable to G2/M phase of the mammalian cell cycle) and a decline as the cells reentered the next cell cycle (24). Future analyses of the dynamics of site-specific ADA3 acetylation during cell cycle progression and in other physiological/pathological scenarios will be of great interest as acetylation site-specific antibodies become available. In conclusion, we establish that mammalian ADA3 is acetylated at distinct sites by its associated HATs, GCN5/PCAF and p300, and deacetylated via a novel interaction with SIRT1, and we further demonstrate that ADA3 acetylation is essential for its physiological function in promoting histone acetylation and cell cycle progression in mammalian cells. These studies should provide a basis for future cell-based and/or animal-based knock-in of acetylation site mutations to examine the in vivo roles of ADA3 acetylation in chromatin modifications and potentially other physiological processes. As we have shown that ADA3 is overexpressed and mislocalized in human cancers, correlating with poor patient survival (42), future studies of ADA3 acetylation in relation to its role in oncogenesis will be of substantial significance.
Supplementary Material
ACKNOWLEDGMENTS
We thank Pawel Ciborowski and Jayme Wiederin of the Mass Spectrometry and Proteomics Core Facility, University of Nebraska Medical Center (UNMC), for help with mass spectrometry studies. We also thank Kishor Bhakat (for providing various constructs) and Andrew Dudley and Gargi Ghosal (for their suggestions in the writing of the manuscript) of UNMC.
We acknowledge support to the UNMC Mass Spectrometry and Proteomics Core Facility and Confocal, Flow Cytometry, and other Core facilities from the NCI Cancer Center Support Grant (P30CA036727) to the Fred & Pamela Buffett Cancer Center and the Nebraska Research Initiative.
We have no conflicts of interest to report.
Funding Statement
S. Mirza was a postdoctoral fellow of the Susan G. Komen Foundation. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00342-16.
REFERENCES
- 1.Baker SP, Grant PA. 2007. The SAGA continues: expanding the cellular role of a transcriptional co-activator complex. Oncogene 26:5329–5340. doi: 10.1038/sj.onc.1210603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koutelou E, Hirsch CL, Dent SY. 2010. Multiple faces of the SAGA complex. Curr Opin Cell Biol 22:374–382. doi: 10.1016/j.ceb.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee KK, Workman JL. 2007. Histone acetyltransferase complexes: one size doesn't fit all. Nat Rev Mol Cell Biol 8:284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 4.Nagy Z, Tora L. 2007. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene 26:5341–5357. doi: 10.1038/sj.onc.1210604. [DOI] [PubMed] [Google Scholar]
- 5.Orpinell M, Fournier M, Riss A, Nagy Z, Krebs AR, Frontini M, Tora L. 2010. The ATAC acetyl transferase complex controls mitotic progression by targeting non-histone substrates. EMBO J 29:2381–2394. doi: 10.1038/emboj.2010.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang YL, Faiola F, Xu M, Pan S, Martinez E. 2008. Human ATAC is a GCN5/PCAF-containing acetylase complex with a novel NC2-like histone fold module that interacts with the TATA-binding protein. J Biol Chem 283:33808–33815. doi: 10.1074/jbc.M806936200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mohibi S, Srivastava S, Band H, Band V. 2014. Role of alteration/deficiency in activation (ADA) complex in cell cycle, genomic instability and cancer, p 33–55. In Kumar R. (ed), Nuclear signaling pathways and targeting transcription in cancer. Springer, New York, NY. [Google Scholar]
- 8.Germaniuk-Kurowska A, Nag A, Zhao X, Dimri M, Band H, Band V. 2007. Ada3 requirement for HAT recruitment to estrogen receptors and estrogen-dependent breast cancer cell proliferation. Cancer Res 67:11789–11797. doi: 10.1158/0008-5472.CAN-07-2721. [DOI] [PubMed] [Google Scholar]
- 9.Wang T, Kobayashi T, Takimoto R, Denes AE, Snyder EL, el-Deiry WS, Brachmann RK. 2001. hADA3 is required for p53 activity. EMBO J 20:6404–6413. doi: 10.1093/emboj/20.22.6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gamper AM, Kim J, Roeder RG. 2009. The STAGA subunit ADA2b is an important regulator of human GCN5 catalysis. Mol Cell Biol 29:266–280. doi: 10.1128/MCB.00315-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonnet J, Wang CY, Baptista T, Vincent SD, Hsiao WC, Stierle M, Kao CF, Tora L, Devys D. 2014. The SAGA coactivator complex acts on the whole transcribed genome and is required for RNA polymerase II transcription. Genes Dev 28:1999–2012. doi: 10.1101/gad.250225.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foulds CE, Feng Q, Ding C, Bailey S, Hunsaker TL, Malovannaya A, Hamilton RA, Gates LA, Zhang Z, Li C, Chan D, Bajaj A, Callaway CG, Edwards DP, Lonard DM, Tsai SY, Tsai MJ, Qin J, O'Malley BW. 2013. Proteomic analysis of coregulators bound to ERalpha on DNA and nucleosomes reveals coregulator dynamics. Mol Cell 51:185–199. doi: 10.1016/j.molcel.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mohibi S, Gurumurthy CB, Nag A, Wang J, Mirza S, Mian Y, Quinn M, Katafiasz B, Eudy J, Pandey S, Guda C, Naramura M, Band H, Band V. 2012. Mammalian alteration/deficiency in activation 3 (Ada3) is essential for embryonic development and cell cycle progression. J Biol Chem 287:29442–29456. doi: 10.1074/jbc.M112.378901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Balasubramanian R, Pray-Grant MG, Selleck W, Grant PA, Tan S. 2002. Role of the Ada2 and Ada3 transcriptional coactivators in histone acetylation. J Biol Chem 277:7989–7995. doi: 10.1074/jbc.M110849200. [DOI] [PubMed] [Google Scholar]
- 15.Yang M, Waterman ML, Brachmann RK. 2008. hADA2a and hADA3 are required for acetylation, transcriptional activity and proliferative effects of beta-catenin. Cancer Biol Ther 7:120–128. doi: 10.4161/cbt.7.1.5197. [DOI] [PubMed] [Google Scholar]
- 16.Nag A, Germaniuk-Kurowska A, Dimri M, Sassack MA, Gurumurthy CB, Gao Q, Dimri G, Band H, Band V. 2007. An essential role of human Ada3 in p53 acetylation. J Biol Chem 282:8812–8820. doi: 10.1074/jbc.M610443200. [DOI] [PubMed] [Google Scholar]
- 17.Kumar A, Zhao Y, Meng G, Zeng M, Srinivasan S, Delmolino LM, Gao Q, Dimri G, Weber GF, Wazer DE, Band H, Band V. 2002. Human papillomavirus oncoprotein E6 inactivates the transcriptional coactivator human ADA3. Mol Cell Biol 22:5801–5812. doi: 10.1128/MCB.22.16.5801-5812.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen J, Luo Q, Yuan Y, Huang X, Cai W, Li C, Wei T, Zhang L, Yang M, Liu Q, Ye G, Dai X, Li B. 2010. Pygo2 associates with MLL2 histone methyltransferase and GCN5 histone acetyltransferase complexes to augment Wnt target gene expression and breast cancer stem-like cell expansion. Mol Cell Biol 30:5621–5635. doi: 10.1128/MCB.00465-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li CW, Ai N, Dinh GK, Welsh WJ, Chen JD. 2010. Human ADA3 regulates RARalpha transcriptional activity through direct contact between LxxLL motifs and the receptor coactivator pocket. Nucleic Acids Res 38:5291–5303. doi: 10.1093/nar/gkq269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meng G, Zhao Y, Nag A, Zeng M, Dimri G, Gao Q, Wazer DE, Kumar R, Band H, Band V. 2004. Human ADA3 binds to estrogen receptor (ER) and functions as a coactivator for ER-mediated transactivation. J Biol Chem 279:54230–54240. doi: 10.1074/jbc.M404482200. [DOI] [PubMed] [Google Scholar]
- 21.Zeng M, Kumar A, Meng G, Gao Q, Dimri G, Wazer D, Band H, Band V. 2002. Human papilloma virus 16 E6 oncoprotein inhibits retinoic X receptor-mediated transactivation by targeting human ADA3 coactivator. J Biol Chem 277:45611–45618. doi: 10.1074/jbc.M208447200. [DOI] [PubMed] [Google Scholar]
- 22.Mirza S, Katafiasz BJ, Kumar R, Wang J, Mohibi S, Jain S, Gurumurthy CB, Pandita TK, Dave BJ, Band H, Band V. 2012. Alteration/deficiency in activation-3 (Ada3) plays a critical role in maintaining genomic stability. Cell Cycle 11:4266–4274. doi: 10.4161/cc.22613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mohibi S, Srivastava S, Wang-France J, Mirza S, Zhao X, Band H, Band V. 2015. Alteration/deficiency in activation 3 (ADA3) protein, a cell cycle regulator, associates with the centromere through CENP-B and regulates chromosome segregation. J Biol Chem 290:28299–28310. doi: 10.1074/jbc.M115.685511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cai L, Sutter BM, Li B, Tu BP. 2011. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell 42:426–437. doi: 10.1016/j.molcel.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Band V, Zajchowski D, Kulesa V, Sager R. 1990. Human papilloma virus DNAs immortalize normal human mammary epithelial cells and reduce their growth factor requirements. Proc Natl Acad Sci U S A 87:463–467. doi: 10.1073/pnas.87.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mir RA, Bele A, Mirza S, Srivastava S, Olou AA, Ammons SA, Kim JH, Gurumurthy CB, Qiu F, Band H, Band V. 2016. A novel interaction of ecdysoneless (ECD) protein with R2TP complex component RUVBL1 is required for the functional role of ECD in cell cycle progression. Mol Cell Biol 36:886–899. doi: 10.1128/MCB.00594-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuo MH, Allis CD. 1998. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 20:615–626. doi:. [DOI] [PubMed] [Google Scholar]
- 28.Yoshida M, Hoshikawa Y, Koseki K, Mori K, Beppu T. 1990. Structural specificity for biological activity of trichostatin A, a specific inhibitor of mammalian cell cycle with potent differentiation-inducing activity in Friend leukemia cells. J Antibiot (Tokyo) 43:1101–1106. doi: 10.7164/antibiotics.43.1101. [DOI] [PubMed] [Google Scholar]
- 29.Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. 2002. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem 277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- 30.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. 2003. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 370:737–749. doi: 10.1042/bj20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gertz M, Fischer F, Nguyen GT, Lakshminarasimhan M, Schutkowski M, Weyand M, Steegborn C. 2013. Ex-527 inhibits Sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc Natl Acad Sci U S A 110:E2772–E2781. doi: 10.1073/pnas.1303628110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bele A, Mirza S, Zhang Y, Ahmad Mir R, Lin S, Kim JH, Gurumurthy CB, West W, Qiu F, Band H, Band V. 2015. The cell cycle regulator ecdysoneless cooperates with H-Ras to promote oncogenic transformation of human mammary epithelial cells. Cell Cycle 14:990–1000. doi: 10.1080/15384101.2015.1006982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paolinelli R, Mendoza-Maldonado R, Cereseto A, Giacca M. 2009. Acetylation by GCN5 regulates CDC6 phosphorylation in the S phase of the cell cycle. Nat Struct Mol Biol 16:412–420. doi: 10.1038/nsmb.1583. [DOI] [PubMed] [Google Scholar]
- 34.Snowden AW, Perkins ND. 1998. Cell cycle regulation of the transcriptional coactivators p300 and CREB binding protein. Biochem Pharmacol 55:1947–1954. doi: 10.1016/S0006-2952(98)00020-3. [DOI] [PubMed] [Google Scholar]
- 35.Yaciuk P, Moran E. 1991. Analysis with specific polyclonal antiserum indicates that the E1A-associated 300-kDa product is a stable nuclear phosphoprotein that undergoes cell cycle phase-specific modification. Mol Cell Biol 11:5389–5397. doi: 10.1128/MCB.11.11.5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ait-Si-Ali S, Polesskaya A, Filleur S, Ferreira R, Duquet A, Robin P, Vervish A, Trouche D, Cabon F, Harel-Bellan A. 2000. CBP/p300 histone acetyl-transferase activity is important for the G1/S transition. Oncogene 19:2430–2437. doi: 10.1038/sj.onc.1203562. [DOI] [PubMed] [Google Scholar]
- 37.Huang WC, Chen CC. 2005. Akt phosphorylation of p300 at Ser-1834 is essential for its histone acetyltransferase and transcriptional activity. Mol Cell Biol 25:6592–6602. doi: 10.1128/MCB.25.15.6592-6602.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan G, Eller MS, Elm C, Larocca CA, Ryu B, Panova IP, Dancy BM, Bowers EM, Meyers D, Lareau L, Cole PA, Taverna SD, Alani RM. 2013. Selective inhibition of p300 HAT blocks cell cycle progression, induces cellular senescence, and inhibits the DNA damage response in melanoma cells. J Investig Dermatol 133:2444–2452. doi: 10.1038/jid.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bannister AJ, Kouzarides T. 2011. Regulation of chromatin by histone modifications. Cell Res 21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krebs AR, Karmodiya K, Lindahl-Allen M, Struhl K, Tora L. 2011. SAGA and ATAC histone acetyl transferase complexes regulate distinct sets of genes and ATAC defines a class of p300-independent enhancers. Mol Cell 44:410–423. doi: 10.1016/j.molcel.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Armour SM, Bennett EJ, Braun CR, Zhang XY, McMahon SB, Gygi SP, Harper JW, Sinclair DA. 2013. A high-confidence interaction map identifies SIRT1 as a mediator of acetylation of USP22 and the SAGA coactivator complex. Mol Cell Biol 33:1487–1502. doi: 10.1128/MCB.00971-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mirza S, Rakha EA, Alshareeda A, Mohibi S, Zhao X, Katafiasz BJ, Wang J, Gurumurthy CB, Bele A, Ellis IO, Green AR, Band H, Band V. 2013. Cytoplasmic localization of alteration/deficiency in activation 3 (ADA3) predicts poor clinical outcome in breast cancer patients. Breast Cancer Res Treat 137:721–731. doi: 10.1007/s10549-012-2363-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.