

Abstract
Poly(ADP-ribosyl)ation of heterogeneous nuclear ribonucleoproteins (hnRNPs) regulates the posttranscriptional fate of RNA during development. Drosophila hnRNP A1, Hrp38, is required for germ line stem cell maintenance and oocyte localization. The mRNA targets regulated by Hrp38 are mostly unknown. We identified 428 Hrp38-associated gene transcripts in the fly ovary, including mRNA of the translational repressor Nanos. We found that Hrp38 binds to the 3′ untranslated region (UTR) of Nanos mRNA, which contains a translation control element. We have demonstrated that translation of the luciferase reporter bearing the Nanos 3′ UTR is enhanced by dsRNA-mediated Hrp38 knockdown as well as by mutating potential Hrp38-binding sites. Our data show that poly(ADP-ribosyl)ation inhibits Hrp38 binding to the Nanos 3′ UTR, increasing the translation in vivo and in vitro. hrp38 and Parg null mutants showed an increased ectopic Nanos translation early in the embryo. We conclude that Hrp38 represses Nanos translation, whereas its poly(ADP-ribosyl)ation relieves the repression effect, allowing restricted Nanos expression in the posterior germ plasm during oogenesis and early embryogenesis.
INTRODUCTION
Defining the mechanisms that control oogenesis has important implications for understanding normal developmental events, such as self-renewal and differentiations of stem cells, determination of cell fate and polarity, and embryonic pattern specification (1–3). Posttranscriptional mechanisms play pivotal roles in controlling these events by regulating mRNA localization and translation during oogenesis (4–6). For example, Drosophila hnRNP A1 homolog Hrb98DE/Hrp38 controls E-cadherin translation by binding to the 5′ untranslated region (UTR) of E-cadherin mRNA for germ line stem cell self-renewal (7). Also, the female-specific RNA-binding protein Sex lethal represses Nanos expression by binding to the 3′ UTR of nanos (nos) during the posttranscriptional process for differentiation of germ line stem cells into cystoblasts (8). The cytoplasmic polyadenylation element binding (CPEB) protein Orb and RNA-transporting protein Bicaudal D (BicD) are specifically expressed in two preoocytes and contribute to determining oocyte identity (9, 10). Several RNA-binding proteins, such as Modulo, PABP, and Smooth, facilitate localization of Bicoid mRNA in the anterior of the oocyte to define the anterior pattern of an embryo (11). Hrp38 also facilitates the enhanced translation of E-cadherin in the oocyte and its surrounding polar cells for localization of the oocyte in the posterior pole (7). An hnRNP A/B family protein, Hrp48, inhibits translation of the posterior determinant oskar mRNA during the transfer from nurse cells to oocytes to establish the posterior pattern (12). hnRNP M homolog Rumpelstiltskin and hnRNP F/H Glorund (Glo) are also involved in localization and translational control of nos mRNA in the posterior for defining the anterior-posterior (A/P) axis of the oocyte (13, 14). In addition, hnRNP proteins Hrp40/squid, Hrp48, and Glorund control localization of gurken mRNA in the anterior-dorsal corner of oocyte to define the dorsal-ventral axis of an embryo (15–17). Together, these studies suggest that hnRNP proteins play crucial roles in regulating temporospatial gene expression during oogenesis.
As the founding member of hnRNP proteins, Drosophila Hrb98DE/Hrp38 regulates splicing and translation of several genes during development (7, 18–20). In addition, posttranslational modification of Hrp38 by poly(ADP-ribosyl)ation results in the regulation of Hrp38-dependent pathways, such as splicing and translation (7, 19, 20). Recent studies have also suggested that mutations of hrp38, or its homolog hrp36, or their abnormal expression, cause several neurodegenerative diseases, such as fragile X syndrome (21, 22), polyglutamine (poly-Q) disorders (23, 24), and amyotrophic lateral sclerosis (ALS) (25, 26). Therefore, fully understanding Hrp38 functions will enable researchers to elucidate the etiology of these diseases at the molecular level. Consistent with its pathological roles, Hrp38 is very important for developmental processes in Drosophila, because a majority of hrp38−/− mutants (about 75%) could not survive to the adult stage (7). Female hrp38−/− mutant escapers have shown defects during oogenesis, including oocyte mislocalizaion from reduced E-cadherin expression (7). Biochemical evidence suggested that Hrp38 binds to the 5′ UTR of E-cadherin mRNA to enhance E-cadherin translation, most likely via the internal ribosome entry site (IRES), a mechanism allowing mid-mRNA sequence translation initiation (7). However, several lines of evidence indicated that E-cadherin is not the only target of Hrp38 during oogenesis. Although the fertility rate of hrp38 female mutants is about 8% of that observed in wild-type flies, only 11% of hrp38−/− mutant eggs show oocyte mislocalization phenotypes (7). This result indicates that more than 80% of hrp38−/− eggs had other defects related to oocyte development for unknown reasons. Therefore, in order to further reveal other genes regulated by Hrp38 during oogenesis, we used RNA immunoprecipitation (IP) coupled with RNA sequencing to identify Hrp38-bound mRNAs at the transcriptome level. This resulted in the identification of 428 Hrp38 targets in the fly ovary, including nos mRNA. Using biochemical and genetic tools, we demonstrated that Hrp38 binding to the 3′ UTR of nos mRNA inhibits nos translation to allow restricted Nos expression in the posterior germ plasm. We also showed that poly(ADP-ribose) disrupts the interaction between Hrp38 and nos 3′ UTR, relieving Hrp38-mediated nos translation repression.
MATERIALS AND METHODS
Drosophila genetics.
Flies were cultured on standard cornmeal-molasses-agar medium at 22°C, unless otherwise indicated. The following stocks were from the Bloomington Stock Center: P{PTT-GC}Hrb98DEZCL0588 (Hrp38:GFP trap line, number 6822); a hrp38 region deficiency line (w1118; Df(3R)Exel6209, P{XP-U}Exel6209/TM6B, Tb1) (number 7687); w[*] ovo[D1] v[24] P{w[+mW.hs]=FRT(w[hs])}101/C [1]DX, y[1] f[1]/Y; P{ry[+t7.2]=hsFLP}38 (number 1813). A hrp38 P-element insertion, w*, P[XP]d05172/TM6B, Tb1, was obtained from the Exelixis Collection at the Harvard Medical School. To generate Parg mutant eggs through the FLP (a yeast recombinase)-DFS (dominant female sterile) method (27), the female FRT-bearing Parg−/− heterozygotes [Parg27.1, P{FRT(whs)101}/FM7a, wa] (7) were crossed with the DFS males (w[*] ovo[D1] v[24] P{w[+mW.hs]=FRT(w[hs])}101/C [1]DX, y[1] f[1]/Y; P{ry[+t7.2]=hsFLP}). Their progeny were treated by heat shock at 37°C for 2 h at the wandering third-instar larva stage for 2 days to induce FLP expression. The enclosed females (Parg27.1, P{FRT(whs)101/w[*] ovo[D1] v[24] P{w[+mW.hs]=FRT(w[hs])}101; P{ry[+t7.2]=hsFLP}/+) were further crossed to the wild-type y, w male to lay eggs for nos mRNA and protein immunostaining.
RNA immunoprecipitation and sequencing.
Fifty pairs of ovaries from 3-day-old wild-type y, w or hrp38 mutant Hrp38d05712/Df(3R)Exel6209 flies (7) were dissected in Grace medium. After ovaries were washed with 1× phosphate-buffered saline (PBS) briefly, they were homogenized with 200 μl of polysome lysis buffer (28) and centrifuged at 14,000 rpm for 10 min at 4°C. One-tenth of the precleared lysates was saved at −20°C as the input. The remaining lysates, brought to a 500-μl volume with polysome lysis buffer, were incubated with 20 μl of rabbit anti-Hrp38 polyclonal antibody (a gift from J. A. Steritz) (18) overnight at 4°C and precipitated with 30 μl of protein A-agarose beads (Invitrogen) for 2 h at 4°C. After agarose beads were washed three times with 500 μl of polysome lysis buffer, RNA-protein complexes were eluted with 200 μl of elution buffer (1% SDS, 50 mM NaCI, 50 mM Tris-HCl [pH 7.0], 5 mM EDTA, and 100 U/ml of RNase inhibitor [Promega]) at 50°C for 30 min. RNAs from elution and input were further extracted with TRIzol (Invitrogen) and cleaned with an RNeasy minikit (Qiagen). All four RNA samples (1/10 wild-type input, immunoprecipitated RNAs from the wild type, 1/10 hrp38 mutant input, and immunoprecipitated RNAs from the hrp38 mutant) were processed with the rRNA depletion protocol. RNA sequencing and analysis of all samples were performed by the Otogenetics Corporation using Illumina HiSeq2000 (paired end; 2 × 100), with 8 million reads after converting all RNA to cDNA by random primers. Expression enrichment of a specific gene was calculated as reads per kilobase transcript per million reads (RPKM) after being normalized with the input. Individual targets were further validated through RNA immunoprecipitation and regular reverse transcription-PCR (RT-PCR) from wild-type (y, w) fly ovaries with anti-Hrp38 antibody (1:25) (10) or a normal rabbit IgG (1:25) as described above. Gene ontology analysis was performed with Gene Ontology Tools developed by the Bioinformatics Group at the Lewis-Sigler Institute of Princeton University (29).
Firefly luciferase reporter construct and assay.
The firefly luciferase reporter vector (pGL3) (Promega) was digested with the restriction enzymes BamHI and XbaI (NewEngland Biolabs) to remove the simian virus 40 (SV40) 3′ UTR of the firefly luciferase gene in the vector. Then, the nos 3′ UTR amplified from a nos cDNA with the primers harboring BamHI and XbaI sites was cloned into the derived pGFL3 vector to make the pGL3:Nos 3′ UTR reporter construct. The pGL3:Nos 3′ UTR vector was used as the template to mutagenize two Hrp38-binding sites from GGG to TTT based on the method supplied with the QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies).
For the luciferase assay, the reporter constructs were transfected to Drosophila S2 cells (S2-DRSC) (DGRC), which were cultured in S2 medium (Sigma) supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin-streptomycin (Invitrogen) at 22°C. Before transfection, 1 ml of cells (0.5 × 106/ml) per well was seeded into a 12-well plate overnight. Two micrograms of the reporter was premixed with 5 ng of the Renilla luciferase reporter (pRL-SV40) (Promega) as the transfection control in 100 μl of Opti-MEM I reduced serum medium (Thermo Fisher Scientific). Two microliters of X-tremeGENE HP DNA transfection reagent (Roche) was added to the mixture and incubated for 30 min at room temperature. After incubation of the transfection mixture with the cells for 72 h, assay of firefly and Renilla luciferase activities was performed using a 96-well plate in triplicate with the dual-luciferase reporter assay system (Promega). The luciferase signals were read by an EnSpire multimode plate reader (PerkinElmer). Firefly luciferase activity was normalized with Renilla activity based on transfection experiments carried out in triplicate. The Student t test was used for statistical analysis to determine significant difference between the different reporters.
dsRNA-mediated RNAi of hrp36, hrp38, and Parg genes.
Knockdown of the expression of hrp36, hrp38, and Parg by RNA interference (RNAi) was done based on double-stranded RNA (dsRNA) treatment of Drosophila S2 cells (30). Briefly, we synthesized around 500-bp-long dsRNA fragments against the hrp36, hrp38, Parg, GFP, and firefly luciferase genes using the RNA MEGAscript kit (Ambion). After 2 days of dsRNA treatment, the firefly reporters were transfected into the cells, as described above, to examine the effect of hnRNP gene knockdown via RNAi on luciferase activity. RNAi efficiency was monitored by Western blotting after measurement of luciferase activity. Basically, cell lysate extracted by luciferase cell lysis buffer (LCLB; Promega) was subjected to SDS-PAGE and transferred to a cellulose membrane (Bio-Rad). The primary antibodies used were rabbit anti-Hrp38 (1:10,000) (18) and mouse anti-Hrp36 (P11; 1:500; a gift from H. Saumweber) (31). For the detection of the pADPr level, the cell lysates extracted from the control and Parg dsRNA-treated cells were immunoprecipitated with rabbit anti-pADPr antibody (Enzo) and probed with mouse anti-pADPr antibody (10H) (Calbiochem) as described before (19). Mouse antitubulin (E7; 1:1,000; DSHB) was used as the loading control.
RNA-protein coimmunoprecipitation in Drosophila S2 cells.
Four milliliters of Drosophila S2 cells (2 × 106/ml) was seeded into a 10-cm culture plate for overnight culture or treated with Parg dsRNA. Four micrograms of pUAST-Hrp38:RFP plasmid (7), pMT (metallothionein promoter)-Gal4 (DGRC), and pGL3:Nos 3′ UTR reporter or pGL3:Nos3 ′ UTR mutants (M1, M2, and M1M2) was cotransfected into S2 cells with X-tremeGENE HP DNA transfection reagent (Roche). After 5 h of culture, CuSO4 (700 μM) was added to the cells for induction of Hrp38:red fluorescent protein (RFP) expression. After 3 days in culture, the cells were treated with lysis buffer (28) and immunoprecipitated with rabbit anti-RFP antibody (MBL International) (1:25) or IgG control (Abcam) (1:25) as described previously (28). The total RNA from the IP elution and 10% input were further extracted with TRIzol (Invitrogen) and purified with an RNeasy minikit (Qiagen) after DNase treatment. The real-time RT-PCR assay was done with Power Sybr green PCR master mix and an ABI 7900 HT instrument (Applied Biosystems). The primers for detecting the firefly luciferase 3′ UTR transcript were as follows: 5′-TTGTGTTTGTGGACGAAGTACC-3′ (forward from the firefly luciferase encoding region) and 5′-AGAGCCTCTGCTCCAGAGCT-3′ (reverse from the nos 3′ UTR). RNA IP was repeated twice for the statistical analysis.
UV cross-linking analysis of RNA-protein interaction.
The interaction of nos mRNA with Hrp38 was confirmed with UV cross-linking analysis based on a previously published protocol (7, 32). Briefly, PCR fragments of the 5′ UTR, coding region, and 3′ UTR of nos mRNA were amplified from a nos cDNA clone (LD32741) (DGRC). pGL3:Nos 3′ UTR mutation constructs (M1, M2, and M1M2) was used as the PCR template to amply the Nos 3′ UTR with the mutated Hrp38-binding sites. A digoxigenin (DIG) RNA labeling kit (Roche) was used to make biotin-labeled nos mRNA probes with the PCR products as the template. Protein lysates were extracted from the ovaries of an Hrp38:GFP trap line (ZCL588) using polysome lysis buffer (28) or S2 cells transfected with pUAST-Hrp38:RFP (7) and pMT (metallothionein promoter)-Gal4 (DGRC) as described above. After incubation of biotin-labeled RNA probes with ovary lysates, UV cross-linking was done as described previously (7). The IP complex was separated by 4 to 12% SDS-PAGE and transferred to the nitrocellulose membrane. An anti-green fluorescent protein (anti-GFP) monoclonal antibody (JL-8) (Clontech) or a rabbit anti-RFP antibody (MBL International) at a 1:25 dilution was used for immunoprecipitation of RNA-protein complexes, along with a mouse or rabbit normal Ig control (Upstate). A chemiluminescence detection kit (Pierce) was applied to measure biotin probes linked to Hrp38:GFP protein after IP. One-tenth of the input was subjected to Western blotting and probed with anti-GFP antibody (JL-8; 1:1,000) or rabbit anti-RFP antibody (1:1,000; MBL) International). The band intensity was measured with NIH ImageJ.
Co-IP.
Coimmunoprecipitation (co-IP) was used to detect the interaction between pADPr and Hrp38 in Drosophila S2 cells by a previously published method (19). Briefly, 1 ml of cells (0.5 × 106/ml) per well was treated with Parg dsRNA (15 μg per well) for 5 days. For the treatment of the cells with a PARP1 inhibitor (olaparib), 5 μmol of olaparib was added into the wild-type cells or, after 1 h of incubation, into Parg dsRNA-treated cells. The cell lysate extracted with radioimmunoprecipitation assay (RIPA) buffer was incubated with rabbit anti-pADPr antibody (Enzo; 1:50) or normal rabbit IgG (Abcam; 1:50) overnight. After incubation with 30 μl of protein A-agarose (Invitrogen) for 2 h at 4°C, the IP complex was subjected to Western blotting with rabbit anti-Hrp38 antibody (1:10,000).
nos RNA in situ hybridization and immunostaining of Drosophila embryos.
Embryos at 0 to 2 h from wild-type fly y, w, hrp38 mutant (Hrp38d05717/Df), and Parg−/− germ line clones generated through the FLP-DFS method were collected for nos RNA fluorescence in situ hybridization (FISH) using a published protocol (33). DIG-labeled nos antisense probe was produced with a DIG RNA labeling kit (SP6/T7; Roche). After hybridization, the signals were detected with biotin-conjugated mouse monoclonal anti-DIG (Jackson ImmunoResearch Laboratories Inc.) and streptavidin-horseradish peroxidase (HRP) conjugate (Thermo Fisher Scientific) coupled with Cy3-tyramide conjugates (PerkinElmer). For immunostaining, the 0- to 2-h embryos were fixed as described previously (34) and stained with anti-rabbit Nanos antibody (1:500; a gift from Akira Nakamura), anti-rabbit pADPr antibody (Enzo; 1:25), or normal rabbit IgG (Abcam; 1:25). The fluorescence intensity of protein and RNA in the individual embryo was measured with NIH ImageJ software.
RESULTS
Identification of Hrp38-associated mRNAs by RIP-Seq.
To identify Hrp38-associated mRNAs during oogenesis, we performed native RNA immunoprecipitation (35) using a rabbit anti-Hrp38 antibody (18) to pull down RNA-protein complexes from younger wild-type and hrp38 mutant ovaries [Fig. 1A]. The Hrp38-bound mRNAs (around 0.1 μg), along with 1/10 of inputs (total RNAs) (3.0 μg) from the wild type and mutant, were extracted and identified by deep RNA sequencing after rRNA depletion (Otogenetics Corp.) (Fig. 1A). Using the input data, we compared the gene expression levels between wild-type and hrp38 mutant ovaries and found that only about 57 genes, including hrp38 itself, showed a significant difference at the transcriptional level. This result suggests that Hrp38 mainly regulates gene expression at the posttranscriptional level. After the normalization of RNA IP data with the input, we identified 428 mRNAs associated with Hrp38 in the wild-type ovary, which have around 2-fold enrichment compared to the Hrp38 mutant ovary. Consistent with our previous study showing that Hrp38 binds to the 5′ UTR of E-cadherin mRNA, our data also confirmed interaction between Hrp38 and E-cadherin mRNA. As expected, 15% of the identified genes with known functions (61/413) are involved in oogenesis based on gene ontology analysis, whereas oogenesis genes usually account for only 8.0% of total genes (1,286 of 16,085 genes) (P = 2 × 10−6, hypergeometric test) (Fig. 1B). Among them, five targets (Nos, Pumilio, Pleota, How, and Shut-down) were shown to be required for maintaining germ line stem cell self-renewal ability (Table 1). Nos and Pumilio are also are also involved in the establishment of the anterior and posterior polarity during oogenesis (36). We have used RIP-RT-PCR to validate that Hrp38 is indeed associated with six targets (E-cadherin, Nos, Pumilio, Pleota, How, and Shut-down) in the wild-type fly ovary (see Fig. S1A in the supplemental material).
FIG 1.

Identification of Hrp38-binding mRNAs at the transcriptome level. (A) Scheme of RNA immunoprecipitation coupled with sequencing (RIP-seq) for identifying Hrp38-associated mRNAs in the Drosophila ovary. (B) Gene ontology analysis of Hrp38-associated mRNAs based on developmental processes. P ≤ 1%. (C) Gene ontology analysis of Hrp38-associated mRNAs based on metabolic processes. P ≤ 1%.
TABLE 1.
Example of mRNAs associated with Hrp38 in the fly ovarya
| Gene name | GenBank accession no. | Protein function |
|---|---|---|
| E-cadherin | NM_057374 | Cell adhesion molecule |
| Nanos | NM_001275794 | Translational repressor |
| Pelota | NM_057634 | Translational release factor |
| Pumilio | X62589 | Translational repressor |
| How | NM_001275893 | RNA-binding protein |
| Shut-down | NM_137993 | piRNA biogenesis |
These six genes have been shown to be required for germ line stem cell maintenance.
Ten percent of the Hrp38-associated transcripts are functional during embryo development, suggesting the maternal effect of Hrp38 protein during embryogenesis (Fig. 1B). Interestingly, Hrp38 is associated with the transcripts of many RNA metabolism genes, which account for 23% of the target genes (95/213), including Hrp38 itself, Hrp48/Hrb27C, and Pabp2 [poly(A)-binding protein 2] (Fig. 1C). In contrast, RNA metabolism genes comprise only 9.3% of total genes in the fly genome (1489/16,085) (P = 1.9 × 10−42, hypergeometric test). This result suggests that Hrp38 specifically regulates the expression of metabolism gene RNA, at least in the ovary, in turn suggesting that coordinated expression of these genes is critical for controlling expression of ovary genes at the posttranscriptional level.
Hrp38 specifically binds to the 3′ UTR of nos mRNA.
Our data for RNA IP coupled with sequencing (RIP-seq) revealed that Hrp38 is associated with nos mRNA in the fly ovary. To validate the specific interaction between Hrp38 and nos mRNA, we used RNA-protein UV cross-linking analysis to determine if Hrp38 would bind to nos mRNA in the fly ovary (Fig. 2A). We made biotin-labeled nos probes from three distinct regions (5′ UTR, coding region, and 3′ UTR) of nos mRNA by in vitro transcription (Fig. 2B). Individual biotin-labeled nos mRNA probes were cross-linked to the total protein lysate of the fly ovary from an Hrp38-GFP trap line (ZCL588) (20). Anti-GFP antibody was used for immunoprecipitation analysis. Interestingly, while UV cross-linking showed that Hrp38 specifically binds to the 3′ UTR of nos mRNA, it was not associated with either the 5′ UTR of nos mRNA or the coding region (Fig. 2C and D). This result not only validated our RIP-seq data but also suggested a potential biological function in relation to the binding of Hrp38 to the 3′ UTR of nos mRNA.
FIG 2.

Hrp38 binding to the 3′ UTR of nos mRNA, as shown by UV cross-linking analysis. (A) Scheme of UV cross-linking analysis using biotin-labeled probes. (B) The structural annotation of nos mRNA. Biotin-labeled probes from three different regions (5′ UTR, coding region, and 3′ UTR) were produced for UV cross-linking analysis. (C) Hrp38 binding to the nos 3′ UTR in the ovary, as revealed by UV cross-linking analysis. (Top) Ovarian lysate from the Hrp38:GFP line was cross-linked to the biotin-labeled Nos RNA probes as indicated. IP was done with mouse anti-GFP antibody or normal mouse IgG as the IP control. (Bottom) The amount of input for IP was shown by Western blotting with anti-GFP antibody. (D) Relative band intensity indicated by the ratio of the immunoprecipitated signal (biotin-Hrp38:GFP) to the input (Hrp38:GFP).
The nos 3′ UTR bears critical Hrp38-binding sites for translational inhibition.
During the late stage of oogenesis, nos mRNA is strictly translated in the posterior pole of the oocyte to establish the A/P body axis and form germ cell plasma (36, 37). The nos 3′ UTR bears a cis-acting translational control element (TCE) to inhibit translation of unlocalized nos mRNA in the cytoplasm of the oocyte (38, 39). A Drosophila hnRNP F/H homolog termed Glorund (Glo) is associated with the nos 3′ UTR TCE as the nos translational repressor (14). Interestingly, a proteomics study showed that Hrp38 interacts with Glo and another hnRNP A1 homolog (Hrp36) in Drosophila embryo (40). Therefore, we hypothesized that the binding of Hrp38 to the 3′ UTR of nos mRNA most likely inhibits nos mRNA translation. To verify this hypothesis, we used the luciferase assay to determine if Hrp38 represses nos 3′ UTR-mediated mRNA translation in Drosophila S2 cells. hnRNP A1/Hrp38 prefers the GGG motif as its binding site (7, 41, 42). The nos 3′ UTR contains two GGG motifs (5′-GAGGG-3′, position 3 of the 3′ UTR, and 5′-CUGGG-3′, position 89 of the 3′ UTR) in the TCE of the nos 3′ UTR, as previously identified (39), and we hypothesized that these are the potential binding sites of Hrp38. Interestingly, these two motifs are localized in the two ends of the TCE.
First, we replaced the 3′ UTR (SV40 3′ UTR) of the firefly luciferase reporter (PL3) with the nos 3′ UTR (Fig. 3A). To test if the two GGG motifs are important for regulating nos translation, we also made three firefly luciferase reporters bearing nos 3′ UTR mutations (GGG to UUU), which have either singly or doubly mutated putative Hrp38-binding sites (Fig. 3A). The individual firefly luciferase reporter was transfected into Drosophila S2 cells, along with the Renilla luciferase reporter pRL-SV40 for normalization of transfection efficiency. Accordingly, we measured the luciferase mRNA levels of all reporters using quantitative RT-PCR, and we found no significant difference between control reporter and the reporter with the normal or mutated 3′ UTR (Fig. 3B and C). This result suggests that replacing the SV40 3′ UTR of the control reporter with either a normal or mutated nos 3′ UTR had no significant effect on the transcription efficiency of the SV40 promoter. However, the luciferase reporter with the normal (wild-type) nos 3′ UTR showed around a 4-fold decrease of luciferase activity compared to that of the control reporter with the SV40 3′ UTR, suggesting that the nos 3′ UTR does, indeed, inhibit translation (Fig. 3B). Furthermore, the reporter construct with mutations (GGG to UUU) of either the G-rich motif in the nos 3′ UTR (5′-GAGGG-3′ and 5′-CUGGG-3′) had increased luciferase activity compared to that of the reporter with the normal nos 3′ UTR (Fig. 3B), indicating that these two Hrp38-binding sites are critical for nos 3′ UTR-mediated translational control. However, double mutations of the G-rich motifs in the nos 3′ UTR did not further abolish translational inhibition compared with the single mutation (Fig. 3B), implying that either of the Hrp38-binding sites is essential for the nos 3′ UTR to perform translational repression.
FIG 3.

Either mutation of Hrp38-binding sites in the nos 3′ UTR or dsRNA-mediated RNAi of hnRNP genes enhances translation of the luciferase reporter. (A) Diagrams showing the different firefly luciferase reporters: the reporter with the SV40 3′ UTR (1), the reporter with the Nos 3′ UTR (WT) (2), the reporter with the Nos 3′ UTR bearing the mutation of the first Hrp38-binding site (GGG to TTT) (Mut-1) (3), the reporter with the Nos 3′ UTR bearing the mutation of the second Hrp38-binding site (GGG to TTT) (Mut-2) (4), and the reporter with the Nos 3′ UTR bearing the mutations of both Hrp38-binding sites (GGG to TTT) (double Mut) (5). (B) Graph showing relative firefly luciferase activity of the reporter with different Nos 3′ UTR constructs compared with the wild-type Nos 3′ UTR reporter. Firefly luciferase activity of these reporters was normalized with the Renilla luciferase reporter (pRL-SV40) for transfection efficiency. *, P ≤ 0.05; **, P ≤ 0.01. (C) Graph showing relative firefly luciferase mRNA abundance compared with the that of the Nos 3′ UTR (WT) reporter. The mRNA level was measured by quantitative RT-PCR with normalization of the Renilla luciferase reporter. N.S., not significant. (D) Western blotting showing the equal expression levels of Hrp38:RFP protein in the transfections of different firefly luciferase reporters in S2 cells. The cell lysates was immunoblotted with the anti-RFP antibody and reprobed with mouse antitubulin antibody for the loading control. (E) Graph showing the mRNA abundance of different firefly luciferase reporters associated with Hrp38:RFP. Quantitative RT-PCR was done to measure mRNA levels after the normalization with the input. RNA IP was performed with rabbit anti-RFP antibody or rabbit IgG as a control after the transfection of UAST>Hrp38:RFP, MT-Gal4, and individual firefly luciferase reporter constructs into S2 cells. **, P < 0.01. (F) UV cross-linking analysis showing that Hrp38 does not bind to the nos 3′ UTR bearing the mutations of the Hrp38-binding sites (M1, M2, and M1M2). An ovarian lysate from the Hrp38:GFP line was used for UV cross-linking analysis with anti-GFP antibody. (G) Western blotting showing the knockdown expression of individual genes upon dsRNA treatment of Drosophila S2 cells. (Top) Immunoblotting of the lysates of hrp38 dsRNA-treated S2 cells with rabbit anti-Hrp38 antibody. (Bottom) Immunoblotting of the lysates of hrp36 dsRNA-treated S2 cells with mouse anti-Hrp36 antibody. All blots were stripped and reprobed with mouse antitubulin antibody for loading control. (H) Graph showing relative firefly luciferase activity of the reporter with the Nos 3′ UTR after RNAi knockdown compared to activity without dsRNA treatment. After 2 days of dsRNA treatment, as indicated, S2 cells were transfected with firefly luciferase reporter having the Nos 3′ UTR, along with the Renilla luciferase reporter. GFP dsRNA treatment was used to show RNAi specificity for enhancing translation, and the firefly luciferase dsRNA was used as the positive control. *, P ≤ 0.05; **, P ≤ 0.01. N.S., not significant.
To further confirm that Hrp38 indeed binds to two GGG motifs in the nos 3′ UTR, we performed RNA IP coupled with quantitative RT-PCR in Drosophila S2 cells (Fig. 3F and G). We transfected pUAST-Hrp38:RFP plasmid (7) and pMT (metallothionein promoter)-Gal4 (43) along with pGL3:Nos 3′ UTR reporter or pGL3:Nos 3′ UTR mutations (M1, M2, and M1M2) into S2 cells. Addition of CuSO4 into the culture medium was used to induce Hrp38:RFP expression. Western blotting showed that equal amounts of Hrp38:RFP were expressed in all the transfections except in the negative control (Fig. 3D). RNA-protein co-IP was done with an anti-RFP antibody to pull down Hrp38:RFP-associated firefly luciferase:Nos 3′ UTR transcripts. Quantitative RT-PCR showed that Hrp38:RFP was associated with the normal nos 3′ UTR but not with either of the nos 3′ UTR mutations with the Hrp38-binding motif (Fig. 3E). In addition, RNA-protein UV cross-linking analysis confirmed that Hrp38 does not bind to the nos 3′ UTR with the mutated Hrp38-binding motif (M1, M2, and M1M2) (Fig. 3F). These results further support the conclusion that either of the GGG motifs in the translational control element (TCE) region of the nos 3′ UTR is required for Hrp38 binding.
Hrp38, along with Hrp36, is a trans-acting factor to inhibit nos 3′ UTR-mediated translation.
To further validate that Hrp38 is a trans-acting factor controlling 3′ UTR-mediated translation, we knocked down the expression of hrp38 and hrp36 (a homolog of Hrp38) in Drosophila S2 cells by double-stranded RNA (dsRNA)-mediated gene interference (31). Western blotting indicated that RNAi is very efficient in knocking down the expression of these genes (Fig. 3G). After 3 days of dsRNA treatment, the luciferase reporter with the normal nos 3′ UTR was transfected into S2 cells to monitor the translational efficiency of luciferase mRNA. Quantitative RT-PCR showed that dsRNA-mediated RNAi had no effect on the mRNA level of the reporter construct (data not shown). However, knockdown of either hrp38 or its homolog hrp36 in S2 cells significantly increased luciferase activity compared to that in the cells without dsRNA treatment (Fig. 3H). In contrast, the luciferase activity of GFP dsRNA-treated cells did not show significant difference from that of untreated cells, and treatment of the cells with firefly luciferase dsRNA almost abolished luciferase activity of the reporter (Fig. 3H), suggesting the specific effect of dsRNA treatment. Together, these results strongly suggest that Hrp38 is a trans-acting factor that inhibits nos 3′ UTR-mediated translation. In addition, it appears that Hrp36 is another translational repressor for controlling nos translation.
hrp38 and the Parg mutant showed Nanos protein misexpression pattern in the fly embryo.
To confirm that Hrp38 controls nos translation during Drosophila oogenesis and embryogenesis, we examined nos expression in the wild type (hrp38) and Parg mutant at both mRNA and protein levels in the early embryo stage (0 to 2 h), in which translation occurs from maternal mRNAs and zygotic expression has still not begun yet. Immunostaining the wild-type embryos with anti-Nanos antibody showed that Nanos protein was exclusively translated in the posterior pole (Fig. 4A, top). Because the female adult escapers of Hrp38 hemizygotes (hrp38d05172/Df) can lay eggs, we also immunostained these hrp38−/− eggs with anti-Nanos antibody (Fig. 4A, middle). We found that hrp38 mutant eggs showed a pattern of Nanos misexpression, with accumulation throughout the whole embryo (Fig. 4A, middle). Quantification of Nanos fluorescence intensity (n = 5) suggested that the Nanos protein level of the Hrp38−/− embryos increased around 2.5-fold compared to that of the wild-type embryos (Fig. 4B and C). However, we did not observe the obvious defects of nos mRNA localization in the hrp38 mutant embryos (Fig. 4A, left side, and E). Therefore, it appears that Hrp38 mutant embryos have the same Nos mRNA localization pattern as that of wild type, in which pronounced nos mRNA can be detected in the posterior pole by RNA in situ hybridization (Fig. 4A, left side). These observations suggest that hrp38 loss of function mainly affects Nanos protein translation but not nos mRNA localization. Taken together, we conclude that Hrp38 is a trans-acting factor that represses nos translation by binding to the nos 3′ UTR during oogenesis and embryogenesis.
FIG 4.

nos mRNA and protein expression patterns of the wild type, hrp38, and Parg mutants. (A) Nos expression pattern of the 0- to 2-h embryos of wild-type (y, w), hrp38−/− (Hrp38do5172/Df), and Parg−/− mutants. (Left) Nos protein immunostaining; (right) Nos mRNA in situ hybridization. (B) Nos protein fluorescence level in the wild type, hrp38, and Parg mutant embryo (0 to 2 h). The mean fluorescence values of five embryos from the WT, hrp38, and Parg mutant were measured using NIH ImageJ software. **, P ≤ 0.01. (C and D) Nos protein fluorescence intensity along the anterior and posterior (AP) axis. (E and F) Nos mRNA fluorescence intensity along the AP axis. Nos protein and RNA expression levels of a typical wild-type, hrp38−/−, or Parg−/− embryo (0 to 2 h) were plotted along the AP axis with NIH ImageJ software.
Our previous studies have shown Hrp38 poly(ADP-ribosyl)ation can disrupt the interaction between Hrp38 and its target mRNAs, regulating Hrp38-dependent processes such as splicing and translation (7). Therefore, we propose that Hrp38 poly(ADP-ribosyl)ated by PARP1 will cause Hrp38 dissociating from the nos 3′ UTR, thus relieving the translation repression of Hrp38. To test his hypothesis, we examined if Parg loss of function can enhance nos translation during oogenesis and embryogenesis. Hrp38 is highly poly(ADP-ribosyl)ated in the Parg mutant due to the failure of degrading poly(ADP-ribose) (pADPr) (20). Because Parg null mutation caused the completely lethality at the late pupa stage (44), we used the FLP-DFS (dominant female sterile) method (27) to generate the Parg mutant embryos. It appears that the Parg−/− eggs had the normal nos mRNA localization (Fig. 4A, bottom). However, we observed that similar to the Nos misexpression pattern of the hrp38 mutant, Parg mutant eggs showed the Nos protein misexpression but not at the mRNA level in the early embryo (0 to 2 h) (Fig. 4A, bottom, and F). Quantification of Nanos fluorescence intensity (n = 5) suggested that the Nanos protein level of the Parg−/− embryos increased around 4.5-fold compared to that of the wild-type embryos (Fig. 4B and D). These results suggest that an increased pADPr level in the Parg mutant enhances Nos translation during embryogenesis.
hnRNP poly(ADP-ribosyl)ation relieves nos 3′ UTR-mediated translation inhibition.
To confirm our hypothesis that poly(ADP-ribose) inhibits Hrp38 binding to the nos 3′ UTR and enhances Nos translation, we used dsRNA-mediated RNAi to knock down the expression of the Parg gene in S2 cells. As expected, Parg RNAi significantly increased the cellular pADPr level, 4.2-fold, compared to that of the control due to the failure of pADPr degradation in S2 cells (Fig. 5A). Accordingly, a co-IP experiment showed that Parg RNAi-treated cells has larger amounts of Hrp38 protein associated with pADPr (around a 2-fold increase) than do cells without dsRNA treatment (Fig. 5B). This result is consistent with our previous finding that Parg loss of function resulted in an increased level of Hrp38 bound to pADPr as shown in the Parg null fly mutant (20). We further examined if Parg RNAi can inhibit Hrp38 binding to the nos 3′ UTR because pADPr can inhibit Hrp38 binding to target mRNAs such as the 5′ UTR of E-cadherin mRNA (7). Using RNA IP coupled with quantitative RT-PCR and RNA-protein UV cross-linking analysis, we found that Parg RNAi significantly inhibited Hrp38 binding to the nos 3′ UTR, around 5-fold (Fig. 5C and D). Indeed, Parg RNAi also significantly enhanced the translation efficiency of the luciferase reporter with the nos 3′ UTR (Fig. 5E), although Parg RNAi did not change the mRNA level of the reporter (data not shown). We further used a PARP1 inhibitor (olaparib) to inhibit PARP1 activity 2.3-fold in the wild-type S2 cells and 2.8-fold in the Parg dsRNA-treated cells (see Fig. S1B in the supplemental material). After transfection of these cells with the luciferase reporter with the nos 3′ UTR, the results showed that inhibition of PARP1 activity in the wild-type cells and Parg-dsRNA-treated cells significantly inhibits the translation of the luciferase reporter (Fig. 5D). These data indicated that pADPr can relieve nos 3′ UTR-mediated translational repression by the inhibition of Hrp38 binding to the nos 3′ UTR. Indeed, we also observed that pADPr specifically accumulated in the posterior pole of the wild-type embryo (see Fig. S1C), where Nos protein is actively translated.
FIG 5.
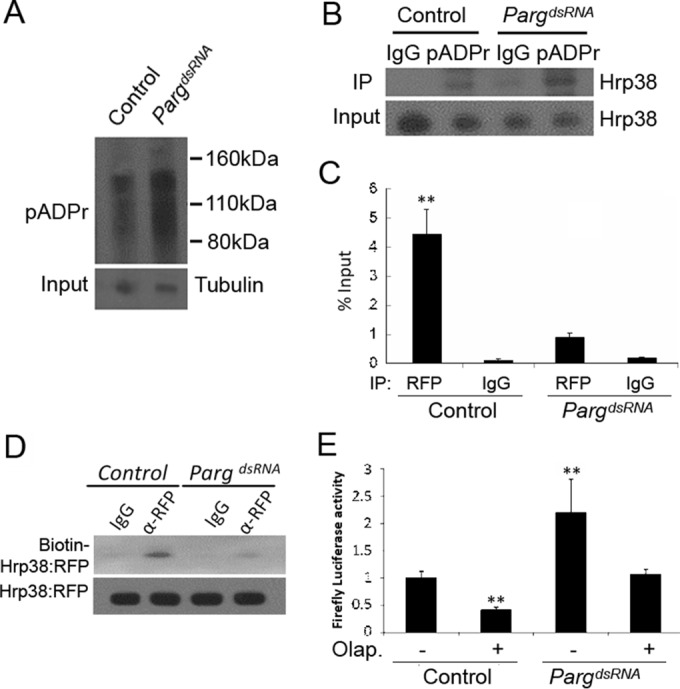
Parg RNAi enhances translation of the luciferase reporter through the inhibition of Hrp38 binding to the nos 3′ UTR. (A) Increased pADPr level in the Parg dsRNA-treated S2 cells compared to the control. Equal amounts of lysates from the control (no treatment) and Parg dsRNA-treated cells were immunoprecipitated with rabbit anti-pADPr antibody and immunoblotted with mouse anti-pADPr antibody (10H). One percent of the input was immunoblotted using antitubulin antibody for the input control. (B) Increased amounts of Hrp38 protein associated with pADPr level in the Parg dsRNA-treated S2 cells compared to the control. Equal amounts of lysates from the control and Parg dsRNA-treated S2 cells were immunoprecipitated with rabbit anti-pADPr antibody or rabbit IgG (control). The immunoprecipitates and 1% of the input were immunoblotted with rabbit anti-Hrp38 antibody. (C) Graph showing the mRNA abundance of the firefly luciferase-Nos 3′ UTR (WT) reporter associated with Hrp38:RFP after Parg dsRNA treatment. After 3 days of Parg dsRNA treatment, UAST>Hrp38:RFP, MT-Gal4, and the firefly luciferase Nos 3′ UTR reporter were transfected into S2 cells for another 3-day incubation. Quantitative RT-PCR was done to measure the mRNA level, with normalization of the input after performing RNA IP with rabbit anti-RFP antibody or rabbit IgG as a control. **, P < 0.01. (D) UV cross-linking analysis showing the decreased amounts of Hrp38:RFP protein binding to the Nos 3′ UTR in the Parg dsRNA-treated S2 cells. The protein lysate from the control or Parg dsRNA-treated cells with the expression of Hrp38:RFP was cross-linked to biotin-labeled Nos 3′ UTR (WT) RNA probe and immunoprecipitated with anti-RFP antibody. (E) Graph showing the luciferase activity of the firefly luciferase Nos 3′ UTR (WT) reporter after Parg dsRNA treatment and/or PARP1 inhibitor (olaparib [Olap.]) treatment. S2 cells were transfected with the firefly luciferase Nos 3′ UTR (WT) and the Renilla luciferase reporter after 3 days of dsRNA treatment. **, P ≤ 0.01.
DISCUSSION
We have used RNA IP coupled with RNA sequencing to identify hundreds of genes (428) whose mRNAs are associated with the RNA-binding protein Hrp38 in the fly ovary. Results showed that nos mRNA, one of the target mRNAs of Hrp38 identified in this study, is regulated by Hrp38 for translational control during ovary development. Biochemical and genetic evidence demonstrated that Hrp38 specifically binds to the nos 3′ UTR to inhibit nos mRNA translation. Strict Nanos accumulation in the posterior pole is critical for the establishment of the A/P body axis and formation of germ cells during oogenesis and the early embryogenesis (36). Because most of nos maternal mRNA (96%) is not localized to the posterior pole, translational repression of nonlocalized maternal nos mRNA is the main mechanism for determining Nos expression pattern in later oogenesis and the early embryo (45). A 90-nucleotide (nt) translational control element (TCE) localized in the nos 3′ UTR, which forms a secondary structure with three stem-loops, has been identified as essential and sufficient for translational repression (39). In addition, Glo, a human hnRNP F/H homolog, has been shown to bind an AU-rich motif in the double-stranded region of TCE stem-loop III to inhibit nos translation (14). However, because the Glo mutant showed only a very low percentage of Nos mislocalization (14), it was speculated that other factors must bind to the TCE or another region of the nos 3′ UTR for translational control (14). Here, we showed that Hrp38, a homolog of human hnRNP A1, is another trans-acting factor that represses nos translation by binding to the 3′ UTR of nos mRNA. Mutagenesis analysis showed that two GGG binding sites in the nos 3′ UTR are essential for Hrp38-mediated inhibition of nos translation. Interestingly, the first GGG motif is located in the 5′ overhang of the TCE, and the second is located in the 3′ end (the double-stranded region of TCE stem-loop I) (Fig. 6A). Hrp38 binding will bring these two otherwise distant GGG motifs into closer proximity, contributing to the formation of a stem-loop structure in the TCE for binding by other repressors, including Smg (40, 41) and Glo (14) (Fig. 6A). Therefore, we propose that Hrp38 binding to these two GGG motifs may facilitate forming or stabilizing the stem-loop structure of TCE by self-interaction of hnRNP proteins (Fig. 6A). This model is similar to the looping-out model by which hnRNP proteins bind to different intron splicing elements that interact with each other for intron definition (42).
FIG 6.
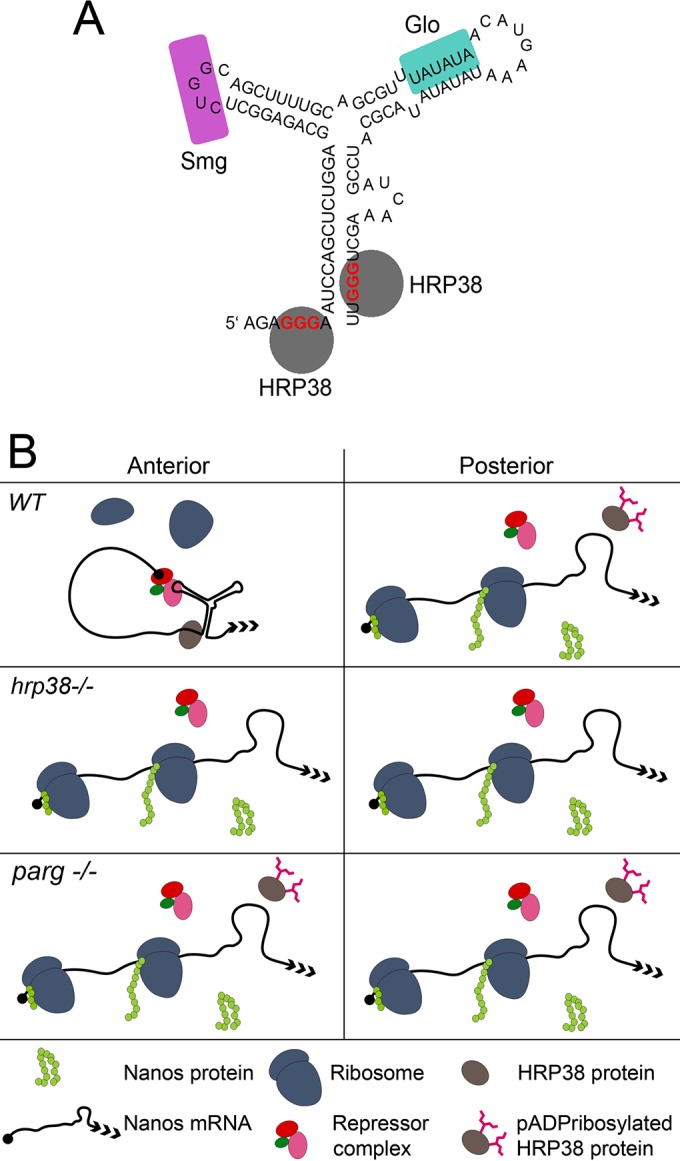
Model showing the mechanisms of Hrp38-dependent nos translational repression and Hrp38 poly(ADP-ribosy)lation-mediated nos translational activation. (A) Stabilization of the TCE stem-loop structure by Hrp38 for nos translational repression. Hrp38-binding sites (GGG motif) (red letters) are localized in the two ends of the TCE (underlined letters). Hrp38 binds to these two GGG motifs to stabilize the stem-loop structure of the TCE. Glo and Smaug (Smg) are additional nos translational repressors which bind to stem-loops II and III of the TCE, respectively. (B) The model controlling nos translation in the fly embryo by Hrp38 and its poly(ADP-ribosyl)ation. In the anterior region (outside the posterior) of the wild-type embryo, Hrp38 binds to the TCE to stabilize the stem-loop structure, facilitating other repressor complex binding to the TCE for translational repression of unlocalized nos mRNA. In the posterior pole, Hrp38 is poly(ADP-ribsoyl)ated by PARP1, resulting in the dissociation of Hrp38 from nos mRNA. The absence of Hrp38 further destabilizes the secondary structure of TCE for alleviation of translational repression acted by other repressor complexes. Therefore, localized nos mRNAs are translated in the posterior pole. In hrp38 and the Parg mutant, the translation repression is abolished in the anterior and posterior, causing Nanos protein accumulation through the embryo.
Although several RNA-binding proteins, such as Smg (46), Ago1(47), Glo (14), Hrp38, and Hrp36 (this study), have been identified to be associated with the nos 3′ UTR for repression of unlocalized nos mRNA, the underlying mechanism is not well understood yet. It is generally believed that nos translation inhibition occurs in the initiation step of translation at the early embryo stage, although postinitiation repression likely has a contribution, too (48, 49). It has been shown that the interaction between Smaug and the Cup protein (an eIF4E binding protein) can inhibit Cup from binding to eIF4G, which is required for recruiting 40S ribosomes to mRNA for cap-dependent translation (48). Indeed, a proteomic analysis of the cap-binding proteins has revealed that Hrp38/Hrb98DE, Hrp36/Hrb87F, and Cup are the components of the cap-binding complex in the fly ovary (50). Therefore, we suspect that Hrp38 and/or Hrp36 also can interact with Cup to block the translation initiation. Another tempting scenario is that Hrp38 binding to the stem-loop structures of the TCE functions as a decoy for recruiting the translational initiation complex to the nos 3′ UTR, thereby skipping the 5′ UTR and the encoding region for translational repression. It is well established that the stem-loop structure in the 5′ UTR of some cellular genes can serve as internal ribosome entry sites (IRESs) to directly recruit the 40S ribosomes for cap-independent initiation (51). Our previous study showed that Hrp38 binds to the 5′ UTR of E-cadherin for promoting E-cadherin translation likely in an IRES-dependent manner, suggesting that Hrp38 is an IRES-transacting factor. In light of the highly structural similarity between the Nos TCE and IRES, it is possible that Hrp38 serves as a decoy factor for nos translational repression.
An important question for nos translation control is how localized nos mRNA in the posterior pole is actively translated. Compared to our understanding the regulation of Nos translational repression, very little is known about the nos translational activation mechanism. It appears that all nos repressors, including Hrp38 and Glo, are fully expressed in the posterior, so there must exist a mechanism to alleviate the repression function of these proteins. It has been proposed that Osk may inhibit Smaug binding to the nos 3′ UTR to prevent the rapid deadenlyation of nos mRNA in the posterior (52). However, it is not clear how the translational machinery in the posterior pole is able to bypass the repression posed by the TCE structure, which is sufficient to inhibit nos translation (39). Recent studies have shown that poly(ADP-ribosyl)ation of the RNA-binding proteins can modulate the RNA-binding ability for controlling the posttranscriptional events (53–56). Indeed, our present data showed that poly(ADP-ribose) can disrupt the interaction between Hrp38 and the nos 3′ UTR, enhancing nos translation (Fig. 4 and 5). Therefore, our data suggest that Hrp38 poly(ADP-ribosyl)ated by PARP1 causes Hrp38 dissociation from the TCE of the nos 3′ UTR in the posterior, thus relieving the translation repression effect of Hrp38 on nos mRNA (Fig. 6B).
A previous study identified 1,219 bound genes of Hrp38 in Drosophila S2 cells using RNA IP coupled with microarray, with emphasis on identifying an alternative splicing pattern (57). The difference between the present analysis and the previous study may indicate that Hrp38, as a regulator of posttranscriptional events of different sets of genes during development, acts in a development-specific manner, in particular since S2 cells were originally isolated from the late stage (20 to 24 h old) of fly embryos (58). Indeed, the mRNAs we identified appear to be biased toward ovary development. We also found that 11% of Hrp38-associated mRNA is involved in neurological processes and morphogenesis, including memory (2.8%), dendrite morphogenesis (4.6%) and synaptic growth at neuromuscular junction (3.7%). Recent studies have also suggested that either Hrp38 loss of function or human hnRNP A1 mutation is involved in the pathogenesis of many neurodegenerative diseases (59). Therefore, it will be interesting to further investigate the etiology of these diseases in the context of hnRNP A1 mutations.
Supplementary Material
ACKNOWLEDGMENTS
We thank H. Saumweber, J. A. Steitz, and A. Nakamura for providing reagents. F. Roegiers and R. Katz contributed valuable comments on the manuscript. We also thank Yinfei Tan for assistance with the luciferase assay.
We have no conflicts of interest with regard to the subject of this paper.
Y.J. and A.V.T. designed and performed the experiments and analyzed the results. Both authors reviewed the results and approved the final version of the manuscript.
Y.J. is supported by a Dr. David A. Hungerford Fellowship in Chromosome Research.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00207-16.
REFERENCES
- 1.Losick VP, Morris LX, Fox DT, Spradling A. 2011. Drosophila stem cell niches: a decade of discovery suggests a unified view of stem cell regulation. Dev Cell 21:159–171. doi: 10.1016/j.devcel.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huynh JR, St Johnston D. 2004. The origin of asymmetry: early polarization of the Drosophila germline cyst and oocyte. Curr Biol 14:438–449. [DOI] [PubMed] [Google Scholar]
- 3.Perrimon N, Pitsouli C, Shilo BZ. 2012. Signaling mechanisms controlling cell fate and embryonic patterning. Cold Spring Harb Perspect Biol 4:a005975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lasko P. 2011. Posttranscriptional regulation in Drosophila oocytes and early embryos. Wiley Interdiscip Rev RNA 2:408–416. doi: 10.1002/wrna.70. [DOI] [PubMed] [Google Scholar]
- 5.Lasko P. 2012. mRNA localization and translational control in Drosophila oogenesis. Cold Spring Harbor Perspect Biol 4:a012294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weil TT. 2014. mRNA localization in the Drosophila germline. RNA Biol 11:1010–1018. doi: 10.4161/rna.36097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ji Y, Tulin AV. 2012. Poly(ADP-ribose) controls DE-cadherin-dependent stem cell maintenance and oocyte localization. Nat Commun 3:760. doi: 10.1038/ncomms1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chau J, Kulnane LS, Salz HK. 2012. Sex-lethal enables germline stem cell differentiation by down-regulating Nanos protein levels during Drosophila oogenesis. Proc Natl Acad Sci U S A 109:9465–9470. doi: 10.1073/pnas.1120473109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lantz V, Chang JS, Horabin JI, Bopp D, Schedl P. 1994. The Drosophila orb RNA-binding protein is required for the formation of the egg chamber and establishment of polarity. Genes Dev 94:598–613. [DOI] [PubMed] [Google Scholar]
- 10.Oh J, Steward R. 2001. Bicaudal-D is essential for egg chamber formation and cytoskeletal organization in Drosophila oogenesis. Dev Biol 232:91–104. doi: 10.1006/dbio.2001.0170. [DOI] [PubMed] [Google Scholar]
- 11.Arn EA, Cha BJ, Theurkauf WE, Macdonald PM. 2003. Recognition of a bicoid mRNA localization signal by a protein complex containing Swallow, Nod, and RNA binding proteins. Dev Cell 4:41–51. doi: 10.1016/S1534-5807(02)00397-0. [DOI] [PubMed] [Google Scholar]
- 12.Yano T, de Quinto SL, Matsui Y, Shevchenko A, Shevchenko A, Ephrussi A. 2004. Hrp48, a Drosophila hnRNPA/B homolog, binds and regulates translation of oskar mRNA. Dev Cell 6:637–648. doi: 10.1016/S1534-5807(04)00132-7. [DOI] [PubMed] [Google Scholar]
- 13.Jain RA, Gavis ERER. 2008. The Drosophila hnRNP M homolog Rumpelstiltskin regulates nanos mRNA localization. Development 135:973–982. doi: 10.1242/dev.015438. [DOI] [PubMed] [Google Scholar]
- 14.Kalifa Y, Huang T, Rosen LN, Chatterjee S, Gavis ER. 2006. Glorund, a Drosophila hnRNP F/H homolog, is an ovarian repressor of nanos translation. Dev Cell 10:291–301. doi: 10.1016/j.devcel.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 15.Norvell A, Kelley RL, Wehr K, Schüpbach T. 1999. Specific isoforms of squid, a Drosophila hnRNP, perform distinct roles in Gurken localization during oogenesis. Genes Dev 13:864–876. doi: 10.1101/gad.13.7.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goodrich JS, Clouse KN, Schüpbach T. 2004. Hrb27C, Sqd and Otu cooperatively regulate gurken RNA localization and mediate nurse cell chromosome dispersion in Drosophila oogenesis. Development 131:1949–1958. doi: 10.1242/dev.01078. [DOI] [PubMed] [Google Scholar]
- 17.Kalifa Y, Armenti ST, Gavis ER. 2009. Glorund interactions in the regulation of gurken and oskar mRNAs. Dev Biol 326:68–74. doi: 10.1016/j.ydbio.2008.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borah S, Wong A, Steitz JA. 2009. Drosophila hnRNP A1 homologs Hrp36/Hrp38 enhance U2-type versus U12-type splicing to regulate alternative splicing of the prospero twintron. Proc Natl Acad Sci U S A 106:2577–2582. doi: 10.1073/pnas.0812826106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji Y, Jarnik M, Tulin AV. 2013. Poly(ADP-ribose) glycohydrolase and poly(ADP-ribose)-interacting protein Hrp38 regulate pattern formation during Drosophila eye development. Gene 526:187–194. doi: 10.1016/j.gene.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ji Y, Tulin AV. 2009. Poly(ADP-ribosyl)ation of heterogeneous nuclear ribonucleoproteins modulates splicing. Nucleic Acids Res 37:3501–3513. doi: 10.1093/nar/gkp218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sofola OA, Jin P, Qin Y, Duan R, Liu H, de Haro M, Nelson DL, Botas J. 2007. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron 55:565–571. doi: 10.1016/j.neuron.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin P, Duan R, Qurashi A, Qin Y, Tian D, Rosser TC, Liu H, Feng Y, Warren ST. 2007. Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron 55:556–564. doi: 10.1016/j.neuron.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sengupta S, Lakhotia SC. 2006. Altered expressions of the noncoding hsromega gene enhances poly-Q-induced neurotoxicity in Drosophila. RNA Biol 3:e1–e8. [DOI] [PubMed] [Google Scholar]
- 24.Mallik M, Lakhotia SC. 2010. Improved activities of CREB binding protein, heterogeneous nuclear ribonucleoproteins and proteasome following downregulation of noncoding hsrω transcripts help suppress poly(Q) pathogenesis in fly models. Genetics 184:927–945. doi: 10.1534/genetics.109.113696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP. 2013. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Romano M, Buratti E, Romano G, Klima R, Belluz LDB, Stuani C, Baralle F, Feiguin F. 2014. Evolutionarily conserved heterogeneous nuclear ribonucleoprotein (hnRNP) A/B Proteins functionally interact with human and Drosophila TAR DNA-binding protein 43 (TDP-43). J Biol Chem 289:7121–7130. doi: 10.1074/jbc.M114.548859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chou TB, Perrimon N. 1992. Use of a yeast site-specific recombinase to produce female germline chimeras in Drosophila. Genetics 131:643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peritz T, Zeng F, Kannanayakal TJ, Kilk K, Eiríksdóttir E, Langel U, Eberwine J. 2006. Immunoprecipitation of mRNA-protein complexes. Nat Protoc 1:577–580. doi: 10.1038/nprot.2006.82. [DOI] [PubMed] [Google Scholar]
- 29.Sealfon RS, Hibbs MA, Huttenhower C, Myers CL, Troyanskaya OG. 2006. GOLEM: an interactive graph-based gene-ontology navigation and analysis tool. BMC Bioinformatics 7:443. doi: 10.1186/1471-2105-7-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kao LR, Megraw TL. 2004. RNAi in cultured Drosophila cells. Methods Mol Biol 247:443–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saumweber H, Symmons P, Kabisch R, Will H, Bonhoeffer F. 1980. Monoclonal antibodies against chromosomal proteins of Drosophila melanogaster. Chromosoma 80:253–275. doi: 10.1007/BF00292684. [DOI] [PubMed] [Google Scholar]
- 32.Walker J, de Melo Neto O, Standart N. 1998. Gel retardation and UV-crosslinking assays to detect specific RNA-protein interactions in the 5′ or 3′ UTRs of translationally regulated mRNAs. Methods Mol Biol 77:365–378. [DOI] [PubMed] [Google Scholar]
- 33.Legendre F, Cody N, Iampietro C, Bergalet J, Lefebvre FA, Moquin-Beaudry G, Zhang O, Wang X, Lécuyer E. 2013. Whole mount RNA fluorescent in situ hybridization of Drosophila embryos. J Vis Exp 71:e50057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Müller HAJ. 2008. Immunolabeling of embryos. Methods Mol Biol 420:207–218. doi: 10.1007/978-1-59745-583-1_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, Lee JT. 2010. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell 40:939–953. doi: 10.1016/j.molcel.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lehmann R, Nusslein-Volhard C. 1991. The maternal gene nanos has a central role in posterior pattern formation of the Drosophila embryo. Development 112:679–691. [DOI] [PubMed] [Google Scholar]
- 37.Clark IE, Wyckoff D, Gavis ER. 2000. Synthesis of the posterior determinant Nos is spatially restricted by a novel cotranslational regulatory mechanism. Curr Biol 10:1311–1314. doi: 10.1016/S0960-9822(00)00754-5. [DOI] [PubMed] [Google Scholar]
- 38.Dahanukar A, Wharton RP. 1996. The Nanos gradient in Drosophila embryos is generated by translational regulation. Genes Dev 10:2610–2621. doi: 10.1101/gad.10.20.2610. [DOI] [PubMed] [Google Scholar]
- 39.Forrest KM, Clark IE, Jain RA, Gavis ER. 2004. Temporal complexity within a translational control element in the nanos mRNA. Development 131:5849–5857. doi: 10.1242/dev.01460. [DOI] [PubMed] [Google Scholar]
- 40.Guruharsha KG, Rual JF, Zhai B, Mintseris J, Vaidya P, Vaidya N, Beekman C, Wong C, Rhee DY, Cenaj O, McKillip E. 2011. A protein complex network of Drosophila melanogaster. Cell 147:690–703. doi: 10.1016/j.cell.2011.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burd CG, Dreyfuss G. 1994. RNA binding specificity of hnRNP A1: significance of hnRNP A1 high-affinity binding sites in pre-mRNA splicing. EMBO J 13:1197–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martinez-Contreras R, Fisette JF, Nasim F, Madden R, Cordeau M, Chabot B. 2006. Intronic binding sites for hnRNP A/B and hnRNP F/H proteins stimulate pre-mRNA splicing. PLoS Biol 4:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klueg KM, Alvarado D, Muskavitch MAT, Duffy JB. 2002. Creation of a GAL4/UAS-coupled inducible gene expression system for use in Drosophila cultured cell lines. Genesis 34:119–122. doi: 10.1002/gene.10148. [DOI] [PubMed] [Google Scholar]
- 44.Hanai S, Kanai M, Ohashi S, Okamoto K, Yamada M, Takahashi H, Miwa M. 2004. Loss of poly(ADP-ribose) glycohydrolase causes progressive neurodegeneration in Drosophila melanogaster. Proc Natl Acad Sci U S A 101:82–86. doi: 10.1073/pnas.2237114100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bergsten SE, Gavis ER. 1999. Role for mRNA localization in translational activation but not spatial restriction of nanos RNA. Development 126:659–669. [DOI] [PubMed] [Google Scholar]
- 46.Smibert CA, Wilson JE, Kerr K, Macdonald PM. 1996. Smaug protein represses translation of unlocalized nanos mRNA in the Drosophila embryo. Genes Dev 10:2600–2609. doi: 10.1101/gad.10.20.2600. [DOI] [PubMed] [Google Scholar]
- 47.Pinder BD, Smibert CA. 2013. MicroRNA-independent recruitment of Argonaute 1 to nanos mRNA through the Smaug RNA-binding protein. EMBO Rep 14:80–86. doi: 10.1038/embor.2012.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nelson MR, Leidal AM, Smibert CA. 2004. Drosophila Cup is an eIF4E binding protein that functions in Smaug-mediated translational repression. EMBO J 23:150–159. doi: 10.1038/sj.emboj.7600026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andrews S, Snowflack DR, Clark IE, Gavis ER. 2011. Multiple mechanisms collaborate to repress nanos translation in the Drosophila ovary and embryo. RNA 17:967–977. doi: 10.1261/rna.2478611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pisa V, Cozzolino M, Gargiulo S, Ottone C, Piccioni F, Monti M, Gigliotti S, Talamo F, Graziani F, Pucci P, Verrotti AC. 2009. The molecular chaperone Hsp90 is a component of the cap-binding complex and interacts with the translational repressor Cup during Drosophilaoogenesis. Gene 432:67–74. doi: 10.1016/j.gene.2008.11.025. [DOI] [PubMed] [Google Scholar]
- 51.Komar AA, Hatzoglou M. 2011. Cellular IRES-mediated translation: the war of ITAFs in pathophysiological states. Cell Cycle 10:229–240. doi: 10.4161/cc.10.2.14472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zaessinger S, Busseau I, Simonelig M. 2006. Oskar allows mRNA translation in Drosophila embryos by preventing its deadenylation by Smaug/CCR4. Development 133:4573–4583. doi: 10.1242/dev.02649. [DOI] [PubMed] [Google Scholar]
- 53.Leung AK, Vyas S, Rood JE, Bhutkar A, Sharp PA, Chang P. 2011. Poly (ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Mol Cell 42:489–499. doi: 10.1016/j.molcel.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Di Giammartino DC, Shi Y, Manley JL. 2013. PARP1 represses PAP and inhibits polyadenylation during heat shock. Mol Cell 49:7–17. doi: 10.1016/j.molcel.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ji Y, Tulin AV. 2013. Post-transcriptional regulation by poly(ADP-ribosyl) ation of the RNA-binding proteins. Int J Mol Sci 14:16168–16183. doi: 10.3390/ijms140816168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bock FJ, Todorova TT, Chang P. 2015. RNA regulation by poly(ADP-ribose) polymerases. Mol Cell 58:959–969. doi: 10.1016/j.molcel.2015.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Blanchette M, Green RE, MacArthur S, Brooks AN, Brenner SE, Eisen MB, Rio DC. 2009. Genome-wide analysis of alternative pre-mRNA splicing and RNA-binding specificities of the Drosophila hnRNP A/B family members. Mol Cell 33:438–449. doi: 10.1016/j.molcel.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schneider I. 1972. Cell lines derived from late embryonic stages of Drosophila melanogaster. J Embryol Exp Morph 27:363–365. [PubMed] [Google Scholar]
- 59.Bekenstein U, Soreq H. 2013. Heterogeneous nuclear ribonucleoprotein A1 in health and neurodegenerative disease: from structural insights to posttranscriptional regulatory roles. Mol Cell Neurosci 56:436–446. doi: 10.1016/j.mcn.2012.12.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.